In Vitro Corrosion Properties of Mg Matrix In Situ Composites Fabricated by Spark Plasma Sintering

Abstract
:1. Introduction
2. Experimental Procedure
2.1. Sample Preparation
2.2. Composition and Microstructure Analysis
2.3. Immersion Tests
2.4. Polarization and Impedance Tests
3. Results and Discussion
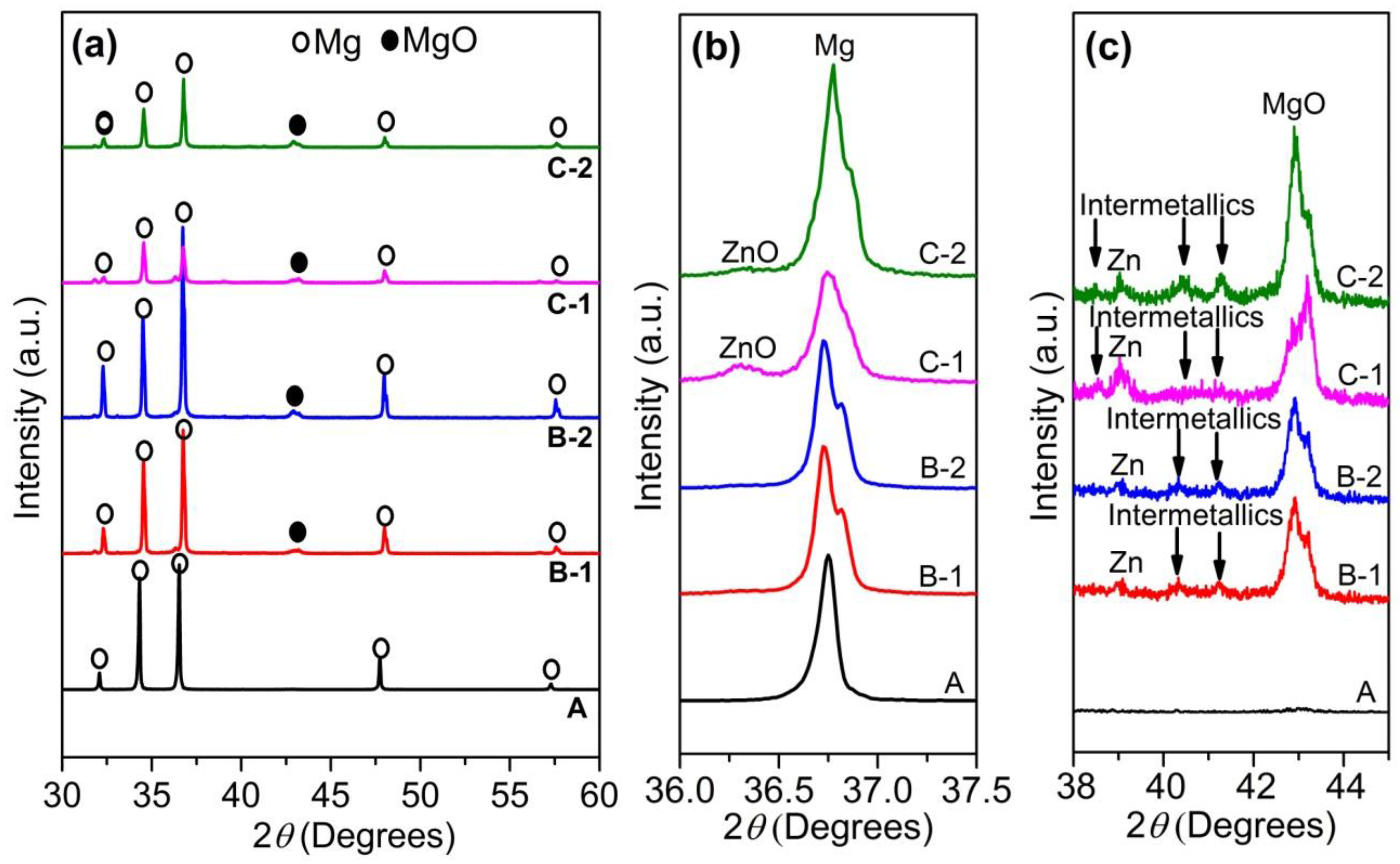
3.1. Composition and Microstructure of Composites
3.2. Corrosion Behavior under Static Immersion
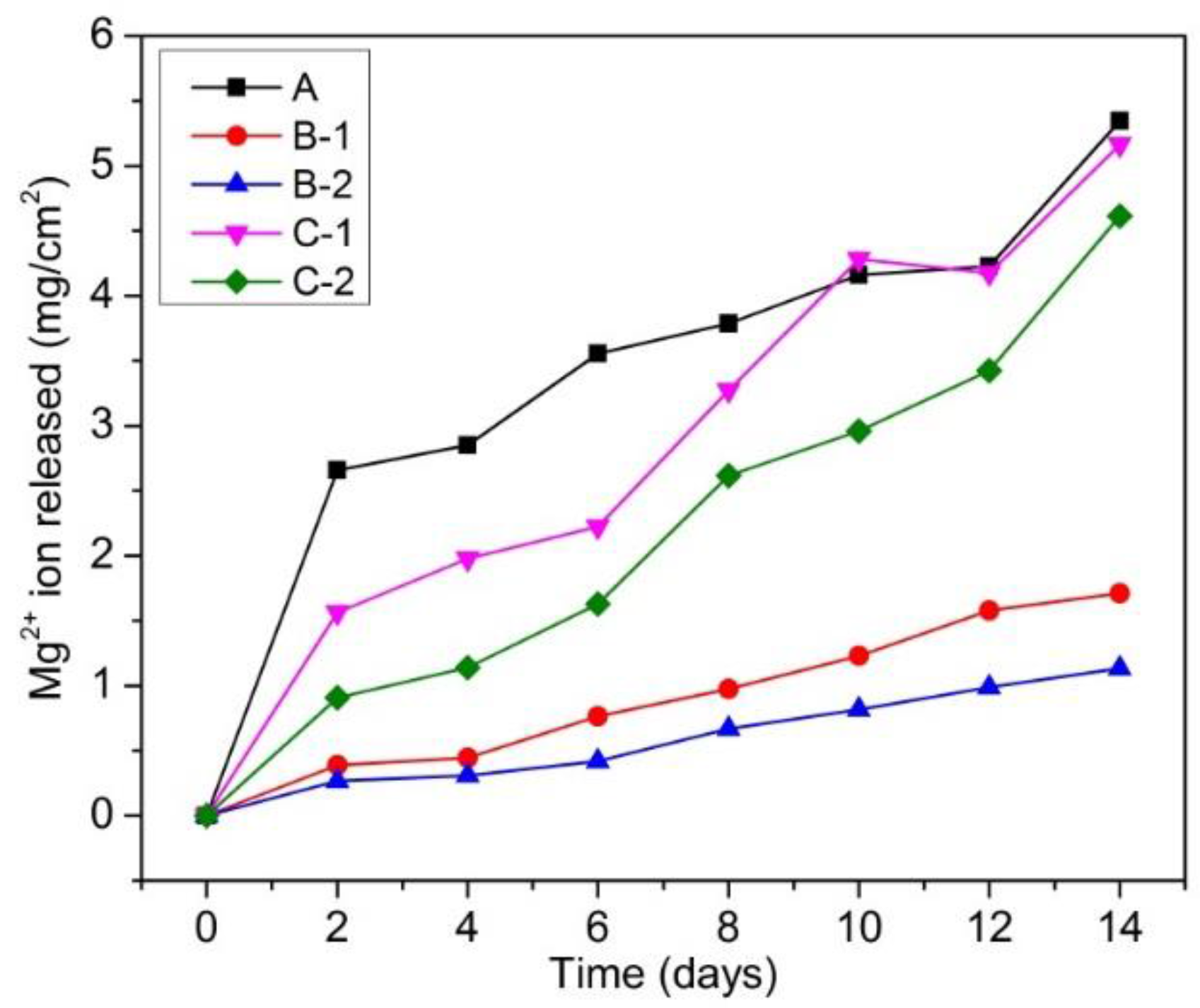
3.2.1. Mg2+ Ion Release Behavior
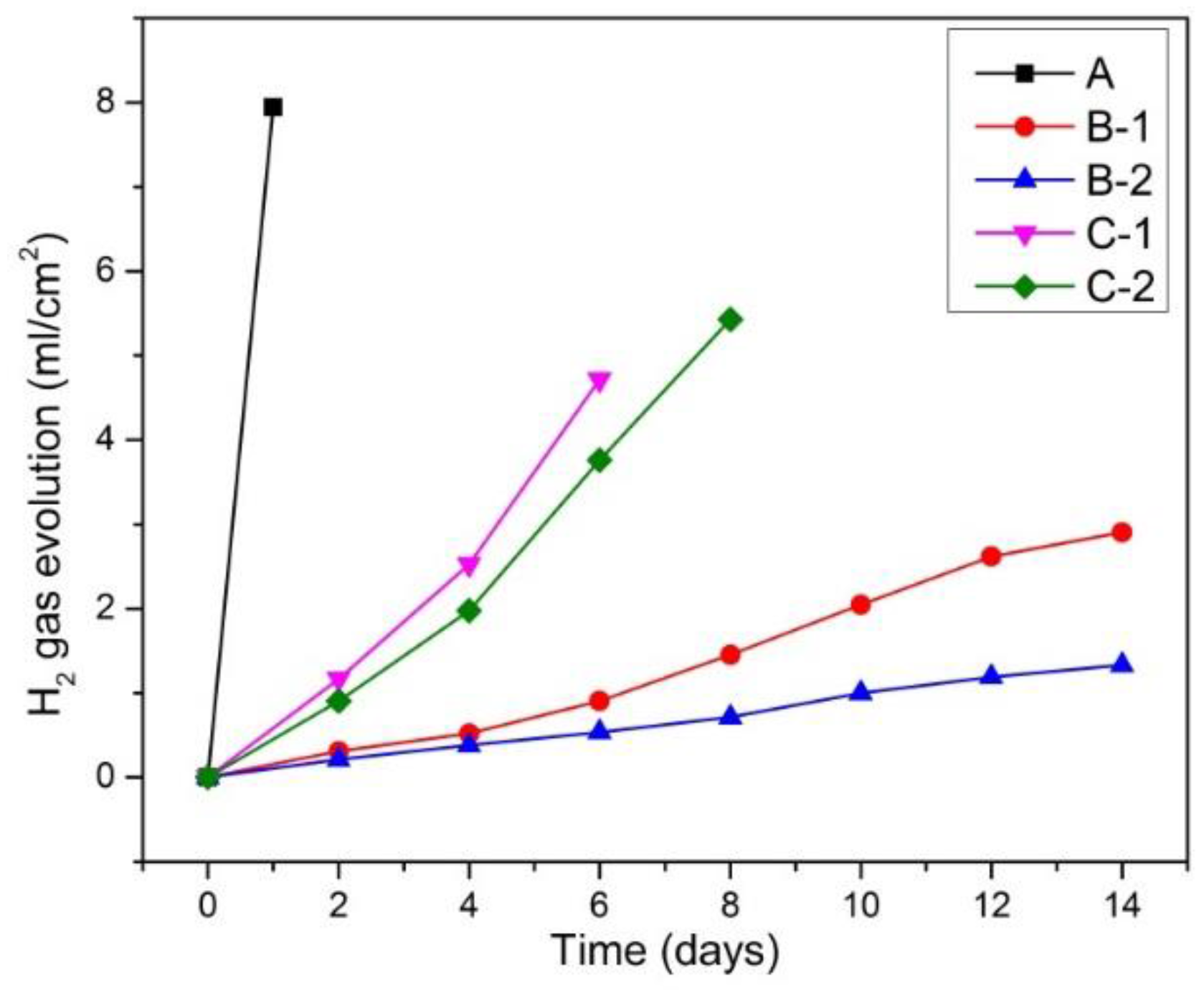
3.2.2. H2 Gas Evolution Behavior
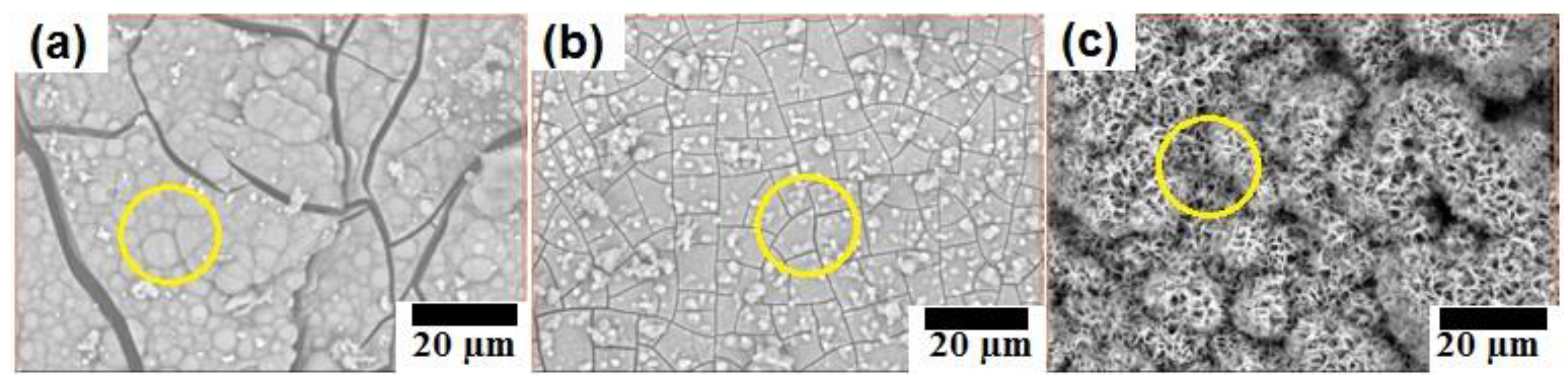
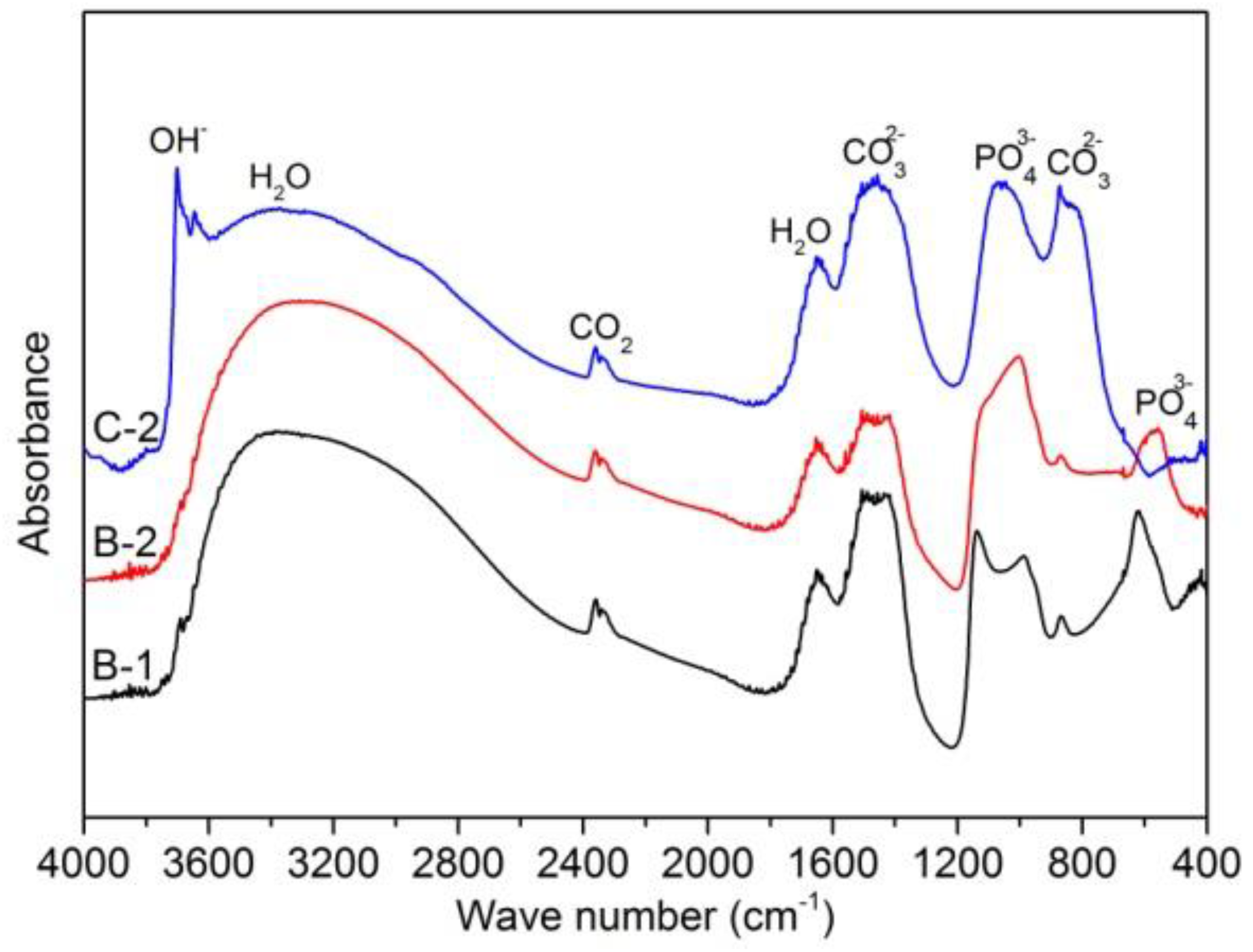
3.2.3. Surface Morphology and Composition of Immersed Sample Surface
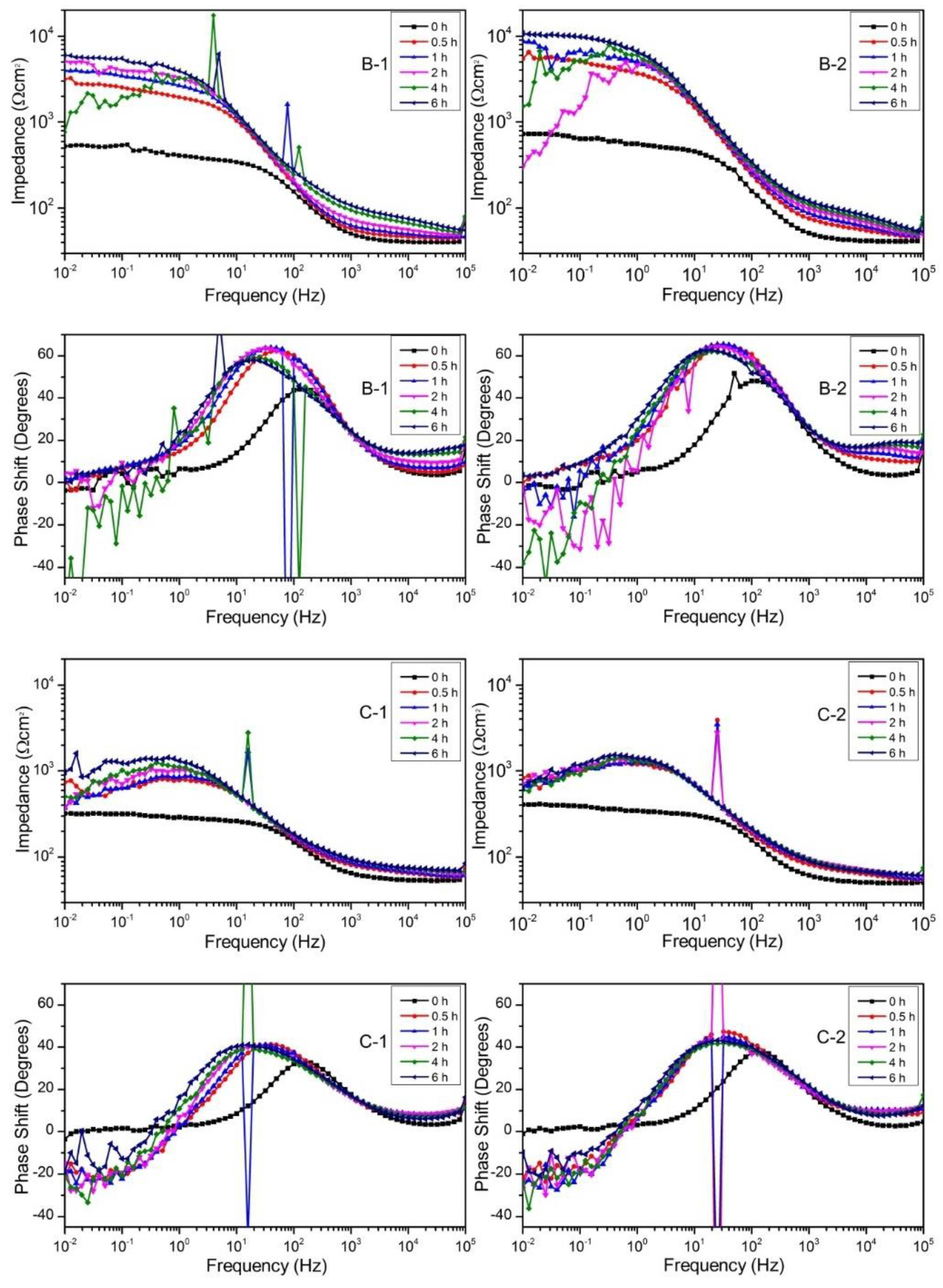
3.3. Polarization and Impedance Behavior
4. Conclusions
- (1)
- Several in situ reactions occurred during sintering process from room temperature to 550 °C to form Zn, MgO and Mg-Zn intermetallic compounds.
- (2)
- Newly formed Zn and Mg-Zn intermetallic compounds have positive effects while the remained ZnO has negative effects on the corrosion resistance of as fabricated composites.
- (3)
- Mg-20 wt % ZnO composites promoted the formation of Mg(OH)2 on the surface of the sample during immersion. This Mg(OH)2 film plays as a weak protective layer resulting in slight improvement of corrosion resistance.
- (4)
- Mg-10 wt % ZnO composites promoted the formation of calcium phosphate on the surface of samples. The calcium phosphate layer is possible to not only significantly increase the corrosion resistance of the composites but also is beneficial for osseointegration. These results suggested Mg-10 wt % ZnO composites become very promising candidates for temporary implant applications.
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Basu, B.; Katti, D.S.; Kumar, A. Advanced Biomaterials: Fundamentals, Processing, and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2010. [Google Scholar]
- Niinomi, M. Recent metallic materials for biomedical applications. Metall. Mater. Trans. A 2002, 33, 477–486. [Google Scholar] [CrossRef]
- Yu, K.; Chen, L.; Zhao, J.; Li, S.; Dai, Y.; Huang, Q.; Yu, Z. In vitro corrosion behavior and in vivo biodegradation of biomedical β-Ca3(PO4)2/Mg–Zn composites. Acta Biomater. 2012, 8, 2845–2855. [Google Scholar] [CrossRef] [PubMed]
- Zreiqat, H.; Howlett, C.R.; Zannettino, A.; Evans, P.; Schulze-Tanzil, G.; Knabe, C.; Shakibaei, M. Mechanisms of magnesium-stimulated adhesion of osteoblastic cells to commonly used orthopaedic implants. J. Biomed. Mater. Res. 2002, 62, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Gao, J.; Wang, Y. Evaluation of cyto-toxicity and corrosion behavior of alkali-heat-treated magnesium in simulated body fluid. Surf. Coat. Technol. 2004, 185, 92–98. [Google Scholar] [CrossRef]
- Staiger, M.P.; Pietak, A.M.; Huadmai, J.; Dias, G. Magnesium and its alloys as orthopedic biomaterials: A review. Biomaterials 2006, 27, 1728–1734. [Google Scholar] [CrossRef] [PubMed]
- Salahshoor, M.; Guo, Y. Biodegradable Orthopedic Magnesium-Calcium (MgCa) Alloys, Processing, and Corrosion Performance. Materials 2012, 5, 135–155. [Google Scholar] [CrossRef] [PubMed]
- Witte, F.; Hort, N.; Vogt, C.; Cohen, S.; Kainer, K.U.; Willumeit, R.; Feyerabend, F. Degradable biomaterials based on magnesium corrosion. Curr. Opin. Solid State Mater. Sci. 2008, 12, 63–72. [Google Scholar] [CrossRef]
- Seong, J.W.; Kim, W.J. Development of biodegradable Mg-Ca alloy sheets with enhanced strength and corrosion properties through the refinement and uniform dispersion of the Mg2Ca phase by high-ratio differential speed rolling. Acta Biomater. 2015, 11, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Zhou, W.; Zheng, Y.; Dong, L.; Xi, Y.; Chai, D. Microstructure, mechanical property, bio-corrosion and cytotoxicity evaluations of Mg/HA composites. Mater. Sci. Eng. C 2010, 30, 827–832. [Google Scholar] [CrossRef]
- Zheng, Y.F.; Gu, X.N.; Xi, Y.L.; Chai, D.L. In vitro degradation and cytotoxicity of Mg/Ca composites produced by powder metallurgy. Acta Biomater. 2010, 6, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- Witte, F.; Feyerabend, F.; Maier, P.; Fischer, J.; Störmer, M.; Blawert, C.; Dietzel, W.; Hort, N. Biodegradable magnesium-hydroxyapatite metal matrix composites. Biomaterials 2007, 28, 2163–2174. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Liu, D.; Anguilano, L.; You, C.; Chen, M. Fabrication and Characterization of a Biodegradable Mg-2Zn-0.5Ca/1β-Tcp Composite. Mater. Sci. Eng. C 2015, 54, 120–132. [Google Scholar] [CrossRef] [PubMed]
- Khanra, A.K.; Jung, H.C.; Hong, K.S.; Shin, K.S. Comparative property study on extruded Mg-HAP and ZM61-HAP composites. Mater. Sci. Eng. A 2010, 527, 6283–6288. [Google Scholar] [CrossRef]
- Kwon, S.H.; Jun, Y.K.; Hong, S.H.; Kim, H.E. Synthesis and dissolution behavior of β-TCP and HA/β-TCP composite powders. J. Eur. Ceram. Soc. 2003, 23, 1039–1045. [Google Scholar] [CrossRef]
- Deng, C.J.; Wong, M.L.; Ho, M.W.; Yu, P.; Dickon, H.L.N. Formation of MgO and Mg-Zn intermetallics in an Mg-based composite by in situ reactions. Compos. Part A Appl. Sci. Manuf. 2005, 36, 551–557. [Google Scholar] [CrossRef]
- Lei, T.; Tang, W.; Cai, S.H.; Feng, F.F.; Li, N.F. On the corrosion behaviour of newly developed biodegradable Mg-based metal matrix composites produced by in situ reaction. Corros. Sci. 2012, 54, 270–277. [Google Scholar] [CrossRef]
- Cao, N.Q.; Narita, K.; Kobayashi, E.; Sato, T. Evolution of the microstructure and mechanical properties of Mg-matrix in situ composites during spark plasma sintering. Powder Metall. 2016, 59, 302–307. [Google Scholar] [CrossRef]
- Cai, S.; Lei, T.; Li, N.; Feng, F. Effects of Zn on microstructure, mechanical properties and corrosion behavior of Mg-Zn alloys. Mater. Sci. Eng. C 2013, 32, 2570–2577. [Google Scholar] [CrossRef]
- Gu, X.; Zheng, Y.; Cheng, Y.; Zhong, S.; Xi, T. In vitro corrosion and biocompatibility of binary magnesium alloys. Biomaterials 2009, 30, 484–498. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhang, X.; Zhao, C.; Li, J.; Song, Y.; Xie, C.; Tao, H.; Zhang, Y.; He, Y.; Jiang, Y.; et al. Research on an Mg-Zn alloy as a degradable biomaterial. Acta Biomater. 2010, 6, 626–640. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Li, J.; Song, Y.; Zhao, C.; Zhang, X.; Xie, C.; Zhang, Y.; Tao, H.; He, Y.; Jiang, Y.; et al. In vitro degradation, hemolysis and MC3T3-E1 cell adhesion of biodegradable Mg-Zn alloy. Mater. Sci. Eng. C 2009, 29, 1907–1912. [Google Scholar] [CrossRef]
- Trumbo, P.; Yates, A.A.; Schlicker, S.; Poos, M. Dietary reference intakes: Vitamin A, vitamin K, arsenic, boron, chromium, copper, iodine, iron, manganese, molybdenum, nickel, silicon, vanadium, and zinc. J. Am. Diet. Assoc. 2001, 101, 294–301. [Google Scholar] [CrossRef]
- Tapiero, H.; Tew, K.D. Trace elements in human physiology and pathology: Zinc and metallothioneins. Biomed. Pharmacother. 2003, 57, 399–411. [Google Scholar] [CrossRef]
- Patrick, K.B.; Emily, R.S.; Shan, Z.; Roger, J.G.; Feng, Z.; Jeremy, G.; Jaroslaw, W.D. Biodegradable Metals for Cardiovascular Stents: From Clinical Concerns to Recent Zn-Alloys. Adv. Healthc. Mater. 2016, 5, 1121–1140. [Google Scholar]
- Omori, M. Sintering, consolidation, reaction and crystal growth by the spark plasma system (SPS). Mater. Sci. Eng. A 2000, 287, 183–188. [Google Scholar] [CrossRef]
- Chartier, T.; Badev, A. Handbook of Advanced Ceramics; Elsevier Inc.: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Mann, C.K.; Yoe, J.H. Spectrophotometric determination of magnesium with sodium 1-azo-2-hydroxy-3-(2,4-dimethylcarboxanilido)-naphthalene-1-(2-hydroxybenzene-5-sulfonate). Anal. Chem. 1956, 28, 202–205. [Google Scholar] [CrossRef]
- Watanabe, H.; Tanaka, H. Dual-wavelength spectrophotometric determination of magnesium with xylidyl blue I and nonionic surfactant. Bunseki Kagaku 1977, 26, 635–639. [Google Scholar] [CrossRef]
- Kumar, M.; Dasarathy, H.; Riley, C. Electrodeposition of brushite coatings and their transformation to hydroxyapatite in aqueous solutions. J. Biomed. Mater. Res. Part A 1999, 45, 302. [Google Scholar] [CrossRef]
- Shadanbaz, S.; Dias, G.J. Calcium phosphate coatings on magnesium alloys for biomedical applications: A review. Acta Biomater. 2012, 8, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Ren, M.; Cai, S.; Liu, T.; Huang, K.; Wang, X.; Zhao, H.; Niu, S.; Zhang, R.; Wu, X. Calcium phosphate glass/MgF2 double layered composite coating for improving the corrosion resistance of magnesium alloy. J. Alloys Compd. 2014, 591, 34–40. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Composition | Sintering in Vacuum Furnace Time/Temperature/Pressure | Sintering by SPS Time/Temperature/Pressure |
|---|---|---|---|
| A | Mg-0 wt % ZnO | - | 10 min/550 °C/50 MPa |
| B-1 | Mg-10 wt % ZnO | - | 10 min/550 °C/50 MPa |
| B-2 | Mg-10 wt % ZnO | 2.5 h/550 °C/0 MPa | 10 min/550 °C/50 MPa |
| C-1 | Mg-20 wt % ZnO | - | 10 min/550 °C/50 MPa |
| C-2 | Mg-20 wt % ZnO | 2.5 h/550 °C/0 MPa | 10 min/550 °C/50 MPa |
| Reagent | NaCl | KCl | Na2HPO4·H2O | KH2PO4 | MgSO4·7H2O | NaHCO3 | CaCl2 |
|---|---|---|---|---|---|---|---|
| Concentration (g/L) | 8.00 | 0.40 | 0.06 | 0.06 | 0.20 | 0.35 | 0.14 |
| Element | Sample B-1 | Sample B-2 | Sample C-2 |
|---|---|---|---|
| Oxygen (at %) | 70 | 62 | 68 |
| Calcium (at %) | 14 | 15 | 0 |
| Phosphorus (at %) | 10 | 12 | 1 |
| Magnesium (at %) | 6 | 11 | 31 |
| Sample | Ecorr (V vs. SCE) | Icorr (A cm−2) |
|---|---|---|
| A | −1.63 | 2.7 × 10−5 |
| B-1 | −1.50 | 3.0 × 10−6 |
| B-2 | −1.47 | 2.2 × 10−6 |
| C-1 | −1.59 | 3.0 × 10−5 |
| C-2 | −1.57 | 8.0 × 10−6 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, N.Q.; Pham, D.N.; Kai, N.; Dinh, H.V.; Hiromoto, S.; Kobayashi, E. In Vitro Corrosion Properties of Mg Matrix In Situ Composites Fabricated by Spark Plasma Sintering. Metals 2017, 7, 358. https://doi.org/10.3390/met7090358
Cao NQ, Pham DN, Kai N, Dinh HV, Hiromoto S, Kobayashi E. In Vitro Corrosion Properties of Mg Matrix In Situ Composites Fabricated by Spark Plasma Sintering. Metals. 2017; 7(9):358. https://doi.org/10.3390/met7090358
Chicago/Turabian StyleCao, Nguyen Q., Dinh N. Pham, Narita Kai, Hai V. Dinh, Sachiko Hiromoto, and Equo Kobayashi. 2017. "In Vitro Corrosion Properties of Mg Matrix In Situ Composites Fabricated by Spark Plasma Sintering" Metals 7, no. 9: 358. https://doi.org/10.3390/met7090358
APA StyleCao, N. Q., Pham, D. N., Kai, N., Dinh, H. V., Hiromoto, S., & Kobayashi, E. (2017). In Vitro Corrosion Properties of Mg Matrix In Situ Composites Fabricated by Spark Plasma Sintering. Metals, 7(9), 358. https://doi.org/10.3390/met7090358