Preparation of Vanadium Nitride Using a Thermally Processed Precursor with Coating Structure

Abstract
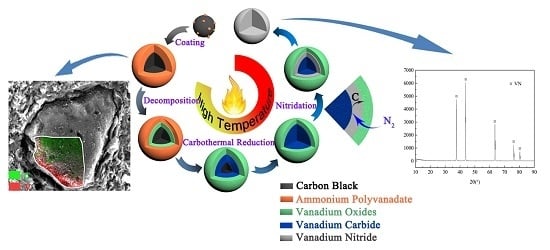
:
1. Introduction
2. Experimental Section
2.1. Materials
2.2. Typical Procedure for the Preparation of VN
2.3. Material Characterization
3. Results and Discussion
3.1. Effect of the Main Reaction Conditions on the N Content in VN Product
3.1.1. Effect of C/V2O5 Mass Ratio
3.1.2. Effect of Reaction Temperature
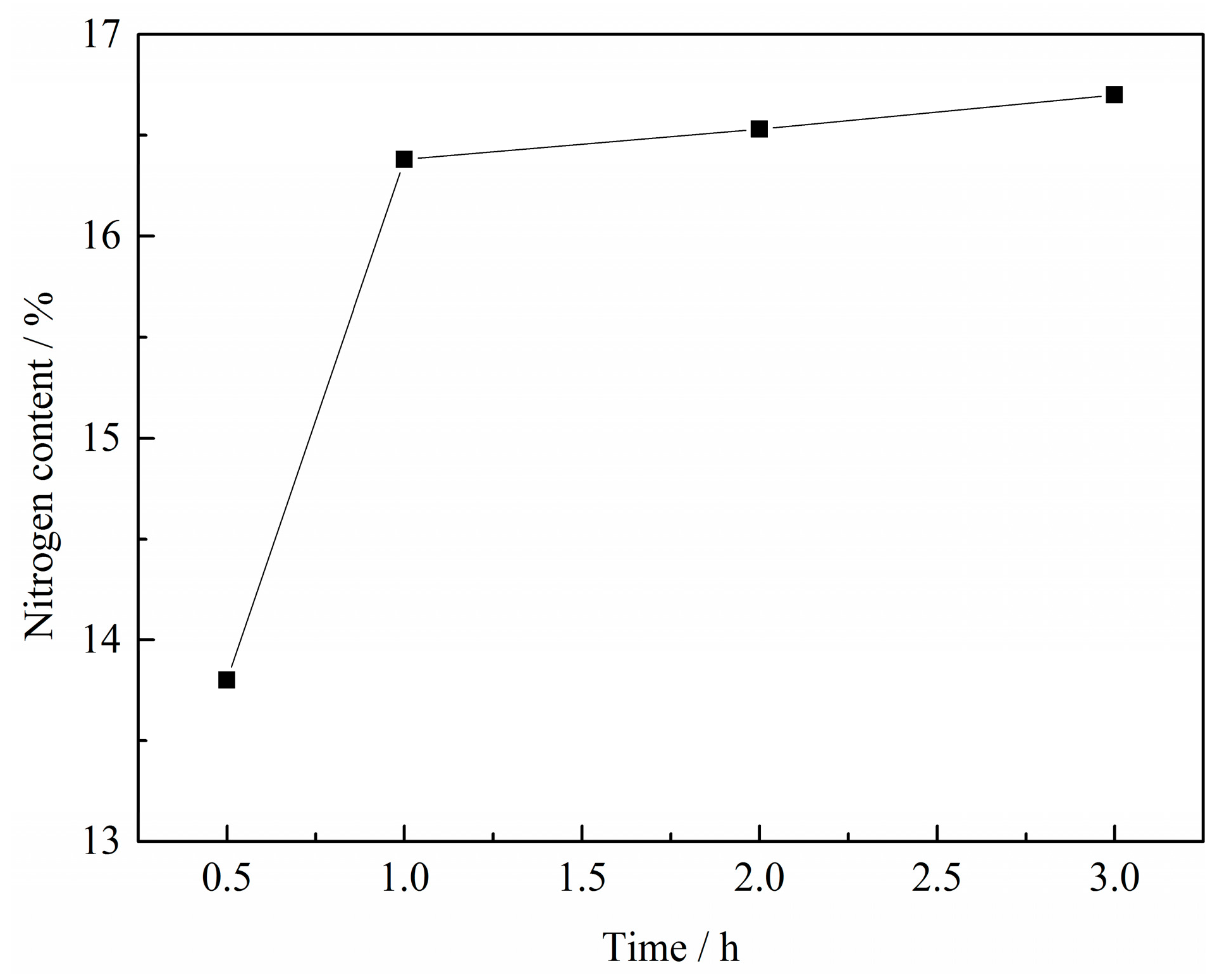
3.1.3. Effect of Reaction Time
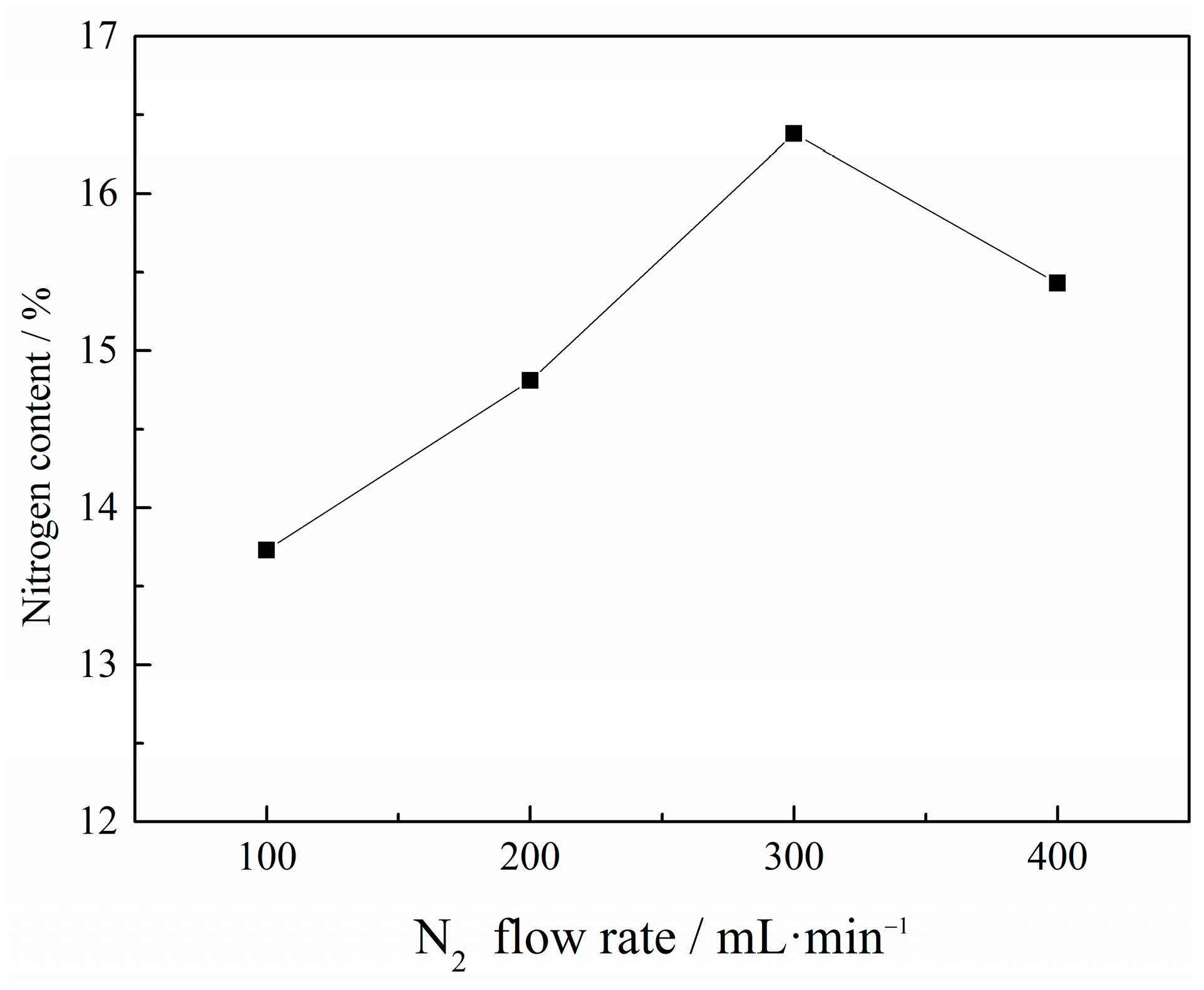
3.1.4. Effect of N2 Flow Rate
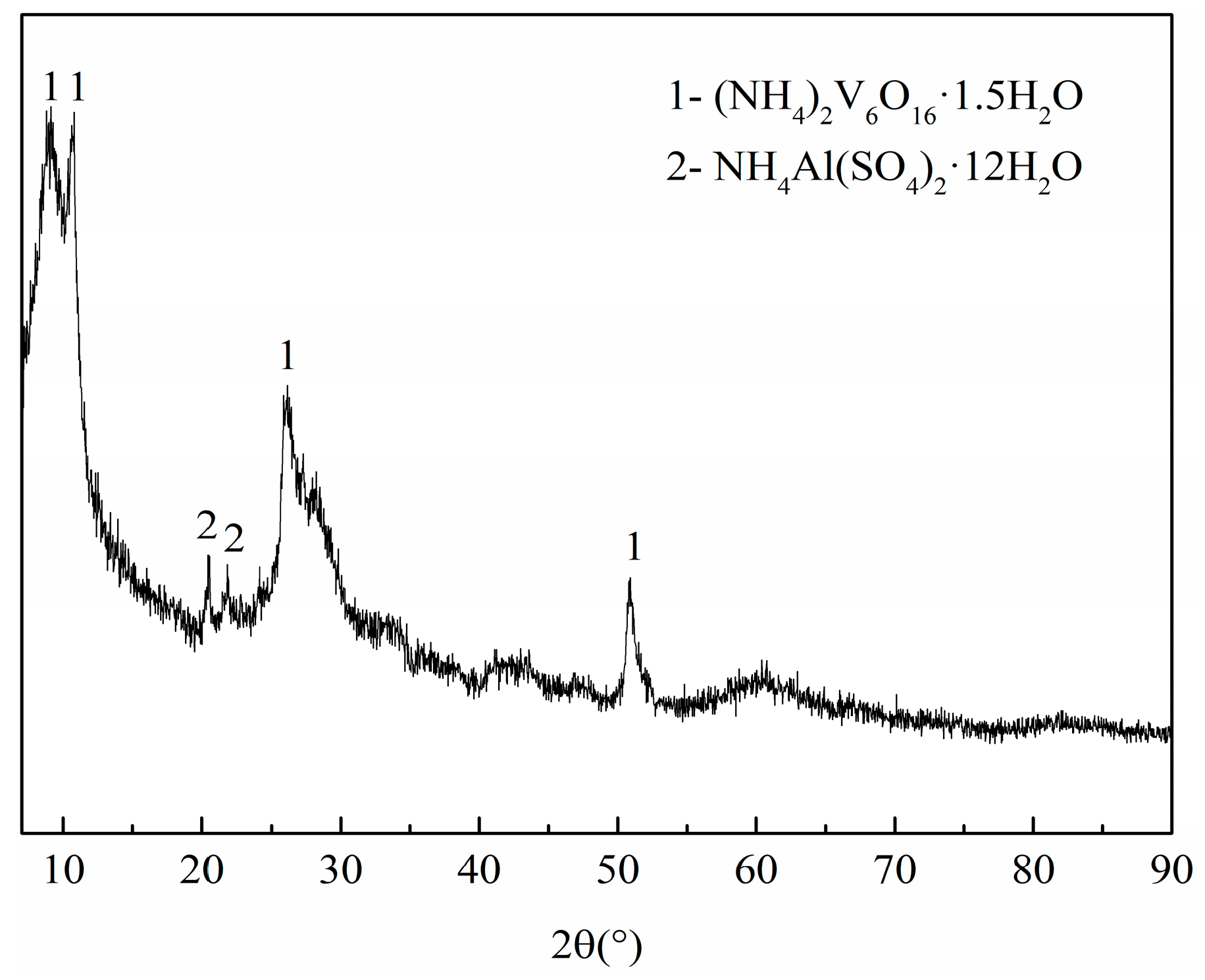
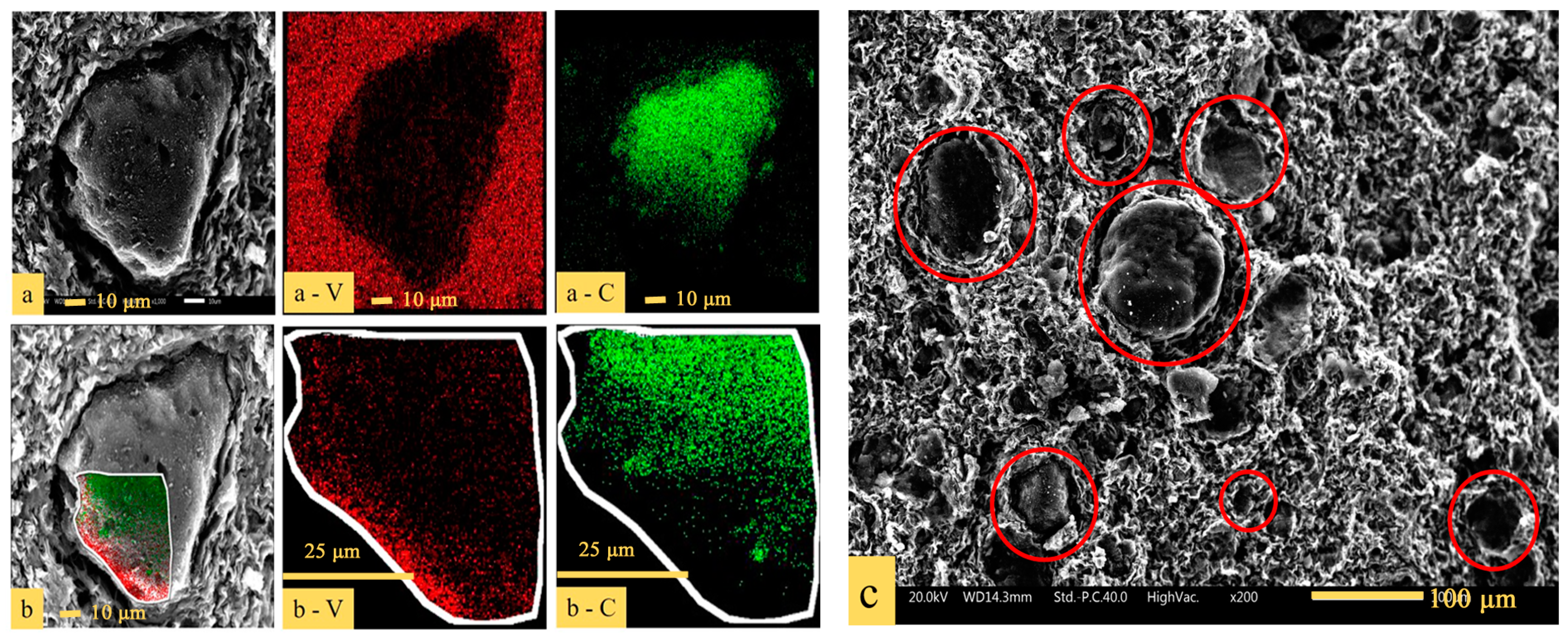
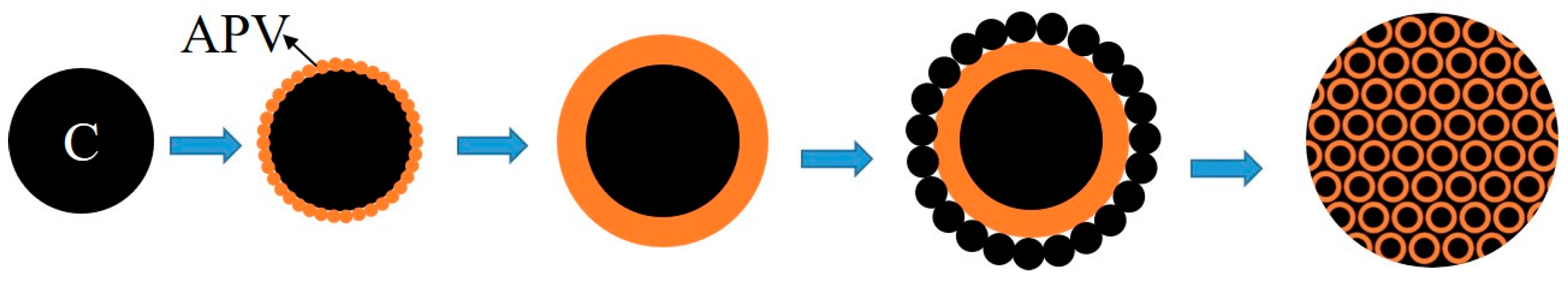
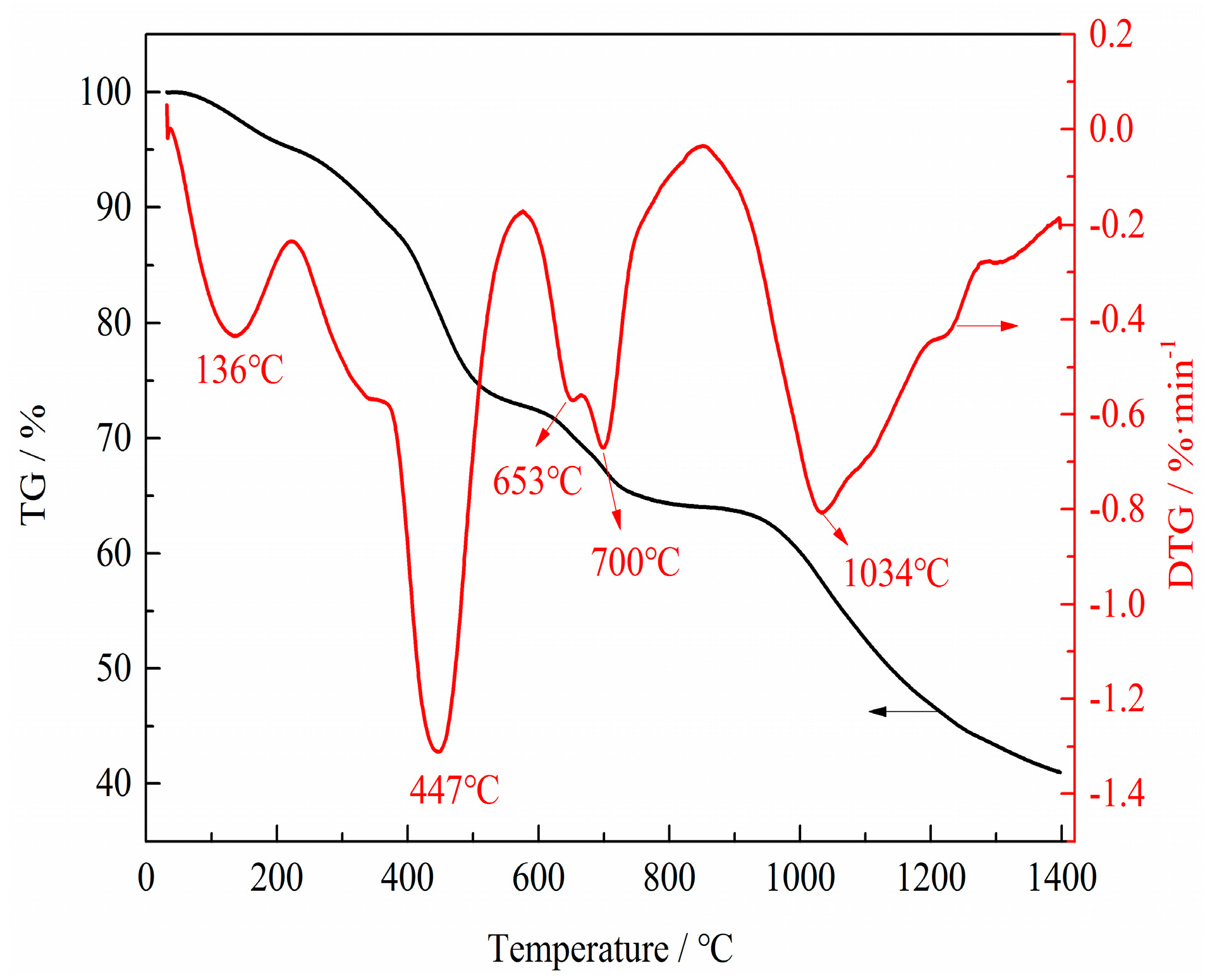
3.2. Phase and Microstructure Analyses of the Precursor
3.3. Phase Evolution of Preparing VN from the Precursor
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Huang, J.; Peng, H.; Xia, G. Microwave synthesis of vanadium nitride for industrial applications. Ironmak. Steelmak. 2009, 36, 110–114. [Google Scholar] [CrossRef]
- Yu, S.; Li, W.; Ji, Z.; Fu, N.; Sui, Z. Effect of technical parameters on preparing for vanadium nitride. Adv. Mater. Res. 2010, 150–151, 480–483. [Google Scholar] [CrossRef]
- Morel, A.; Piron, Y.B.; Porto, R.L.; Brousse, T.; Belanger, D. Suitable conditions for the use of vanadium nitride as an electrode for electrochemical capacitor. J. Electrochem. Soc. 2016, 163, A1077–A1082. [Google Scholar] [CrossRef]
- Zhou, X.; Chen, H.; Shu, D.; He, C.; Nan, J. Study on the electrochemical behavior of vanadium nitride as a promising supercapacitor material. J. Phys. Chem. Solids 2009, 70, 495–500. [Google Scholar] [CrossRef]
- Wu, M.; Guo, H.; Lin, Y.; Wu, K.; Ma, T.; Hagfeldt, A. Synthesis of highly effective vanadium nitride (VN) peas as a counter electrode catalyst in dye-sensitized solar cells. J. Phys. Chem. C 2014, 118, 12625–12631. [Google Scholar] [CrossRef]
- Krawiec, P.; Cola, P.L.D.; Gläser, R.; Weitkamp, J.; Weidenthaler, C.; Kaskel, S. Oxide foams for the synthesis of high-surface-area vanadium nitride catalysts. Adv. Mater. 2006, 18, 506–508. [Google Scholar] [CrossRef]
- Gajbhiye, N.S.; Ningthoujam, R.S. Low temperature synthesis, crystal structure and thermal stability studies of nanocrystalline VN particles. Mater. Res. Bull. 2006, 41, 1612–1621. [Google Scholar] [CrossRef]
- Smolik, J.; Mazurkiewicz, A.; Słomka, Z.; Bujak, J.; Gołacka, J.K.; Garbacz, H.; Wieciński, P. Nanomultilayer coatings based on vanadium nitride. Solid State Phenom. 2015, 237, 15–20. [Google Scholar] [CrossRef]
- Caicedo, J.C.; Zambrano, G.; Aperador, W.; Escobar-Alarcon, L.; Camps, E. Mechanical and electrochemical characterization of vanadium nitride (VN) thin films. Appl. Surf. Sci. 2011, 258, 312–320. [Google Scholar] [CrossRef]
- Duan, X.; Srinivasakannan, C.; Zhang, H.; Zhang, Y. Process optimization of the preparation of vanadium nitride from vanadium pentoxide. Arab. J. Sci. Eng. 2015, 40, 2133–2139. [Google Scholar]
- Tripathy, P.K.; Sehra, J.C.; Kulkarni, A.V. On the carbonitrothermic reduction of vanadium pentoxide. J. Mater. Chem. 2001, 11, 691–695. [Google Scholar] [CrossRef]
- Pan, H.; Zhang, Z.; Peng, J.; Zhang, L.; Li, W. Densification of vanadium nitride by microwave-assisted carbothermal nitridation. Adv. Mater. Res. 2011, 201–203, 1787–1792. [Google Scholar] [CrossRef]
- Chen, Z.; Xue, Z.; Wang, W.; Yu, Y.; Liu, Q.; Li, P. One-step method of carbon thermal reduction and nitride to produce vanadium nitrogen alloy. Adv. Mater. Res. 2012, 476–478, 194–198. [Google Scholar] [CrossRef]
- Cai, P.; Yang, Z.; Wang, C.; Xia, P.; Qian, Y. Synthesis of nanocrystalline VN via thermal liquid-solid reaction. Mater. Lett. 2006, 60, 410–413. [Google Scholar] [CrossRef]
- Yeh, C.L.; Chen, Y.D. Combustion synthesis of vanadium carbonitride from V-C powder compacts under nitrogen pressure. Ceram. Int. 2007, 33, 365–371. [Google Scholar] [CrossRef]
- Cheng, F.; He, C.; Shu, D.; Chen, H.; Zhang, J.; Tang, S.; Finlow, D.E. Preparation of nanocrystalline VN by the melamine reduction of V2O5 xerogel and its supercapacitive behavior. Mater. Chem. Phys. 2011, 131, 268–273. [Google Scholar] [CrossRef]
- Azargohar, R.; Dalai, A.K. Production of activated carbon from luscar char: Experimental and modeling studies. Microporous Mesoporous Mater. 2005, 85, 219–225. [Google Scholar] [CrossRef]
- Zhang, B.; Li, Z. Synthesis of vanadium carbide by mechanical alloying. J. Alloys Compd. 2005, 392, 183–186. [Google Scholar] [CrossRef]
- Yao, W.; Makowski, P.; Giordano, C.; Goettmann, F. Synthesis of early-transition-metal carbide and nitride nanoparticles through the urea route and their use as alkylation catalysts. Chem. Eur. J. 2009, 15, 11999–12004. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Gu, Y.; Shi, L.; Yang, Z.; Ma, J.; Qian, Y. A room-temperature synthesis of nanocrystalline vanadium nitride. J. Eur. Ceram. Soc. 2010, 30, 2099–2107. [Google Scholar] [CrossRef]
- Zhao, Z.; Liu, Y.; Cao, H.; Ye, J.; Gao, S.; Tu, M. Synthesis of VN nanopowders by thermal nitridation of the precursor and their characterization. J. Alloys Compd. 2008, 464, 75–80. [Google Scholar] [CrossRef]
- Liu, A.; Liu, Y.; Ma, S.; Qiu, Y.; Rong, P.; Ye, J. Synthesis of (Cr,V)2(C,N) solid solution powders by thermal processing precursors. Mater. Chem. Phys. 2017, 193, 196–202. [Google Scholar] [CrossRef]
- Cai, Z.; Zhang, Y.; Liu, T.; Huang, J. Mechanisms of vanadium recovery from stone coal by novel BaCO3/BaO composite additive roasting and acid leaching technology. Minerals 2016, 6, 26. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, K.; Tian, X.; Qin, W. Vanadium leaching from carbonaceous shale using fluosilicic acid. Int. J. Miner. Process. 2011, 100, 184–187. [Google Scholar] [CrossRef]
- Zhang, Y.; Bao, S.; Liu, T.; Huang, J. The technology of extracting vanadium from stone coal in China: History, current status and future prospects. Hydrometallurgy 2011, 109, 116–124. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, C.; Li, P.; Li, S. A new process of extracting vanadium from stone coal. Int. J. Miner. Metall. Mater. 2010, 17, 381–388. [Google Scholar] [CrossRef]
- Nguyen, T.; Lee, M. Solvent extraction of vanadium (V) from sulfate solutions using LIX 63 and PC 88A. J. Ind. Eng. Chem. 2015, 31, 118–123. [Google Scholar] [CrossRef]
- Li, X.; Wei, C.; Deng, Z.; Li, M.; Li, C.; Fan, G. Selective solvent extraction of vanadium over iron from a stone coal/black shale acid leach solution by D2EHPA/TBP. Hydrometallurgy 2011, 105, 359–363. [Google Scholar] [CrossRef]
- Li, X.; Wei, C.; Deng, Z.; Li, C.; Fan, G.; Li, M.; Huang, H. Recovery of vanadium from H2SO4-HF acidic leaching solution of black shale by solvent extraction and precipitation. Metals 2016, 6, 63. [Google Scholar] [CrossRef]
- Gu, H.M.; Shih, W.Y.; Shih, W.H. Single-calcination synthesis of pyrochlore-free 0.9Pb(Mg1/3Nb2/3)O3–0.1PbTiO3 and Pb(Mg1/3Nb2/3)O3 Ceramics Using a Coating Method. J. Am. Ceram. Soc. 2003, 86, 217–221. [Google Scholar] [CrossRef]
- Ivashchenko, V.I.; Turhci, P.E.A. Phonon softening and the phase transition in VN. Phys. Rev. B 2008, 78, 224113. [Google Scholar] [CrossRef]
- Vernardou, D.; Apostolopoulou, M.; Louloudakis, D.; Spanakis, E.; Katsarakis, N.; Koudoumas, E.; McGrath, J.; Pemble, M.E. Electrochemical properties of opal-V6O13 composites. J. Alloys Compd. 2014, 586, 621–626. [Google Scholar] [CrossRef]
- Vernardou, D.; Pemble, M.E.; Sheel, D.W. In-situ FTIR studies of the growth of vanadium dioxide coatings on glass by atmospheric pressure chemical vapour deposition for VCl4 and H2O system. Thin Solid Films 2007, 515, 8768–8770. [Google Scholar] [CrossRef]
- Vernardou, D.; Louloudakis, D.; Spanakis, E.; Katsarakis, N.; Koudoumas, E. Functional properties of APCVD VO2 layers. Int. J. Thin Films Sci. Technol. 2015, 4, 187–191. [Google Scholar]
- Vernardou, D.; Bei, A.; Louloudakis, D.; Katsarakis, N.; Koudoumas, E. Oxygen source-oriented control of atmospheric pressure chemical vapor deposition of VO2 for capacitive applications. J. Electrochem. Sci. Eng. 2016, 6, 165–173. [Google Scholar]
- Bennett, L.H.; Massalski, T.B.; Giessen, B.C. Alloy Phase Diagrams. Mater. Res. Soc. Symp. Proc. 1983, 19. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Element | V | Al | Fe | K | Na | P |
|---|---|---|---|---|---|---|
| Concentration (g·L−1) | 20.63 | 5.78 | 0.06 | 0.34 | 0.42 | 0.1 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, J.; Zhang, Y.; Liu, T.; Huang, J.; Xue, N.; Hu, P. Preparation of Vanadium Nitride Using a Thermally Processed Precursor with Coating Structure. Metals 2017, 7, 360. https://doi.org/10.3390/met7090360
Han J, Zhang Y, Liu T, Huang J, Xue N, Hu P. Preparation of Vanadium Nitride Using a Thermally Processed Precursor with Coating Structure. Metals. 2017; 7(9):360. https://doi.org/10.3390/met7090360
Chicago/Turabian StyleHan, Jingli, Yimin Zhang, Tao Liu, Jing Huang, Nannan Xue, and Pengcheng Hu. 2017. "Preparation of Vanadium Nitride Using a Thermally Processed Precursor with Coating Structure" Metals 7, no. 9: 360. https://doi.org/10.3390/met7090360
APA StyleHan, J., Zhang, Y., Liu, T., Huang, J., Xue, N., & Hu, P. (2017). Preparation of Vanadium Nitride Using a Thermally Processed Precursor with Coating Structure. Metals, 7(9), 360. https://doi.org/10.3390/met7090360