2. Scale Bridging for Strain Dependent Solution Enthalpies in Fe-C
In this section, we demonstrate how data, which is obtained from ab initio or empirical potential calculations, can be transferred to the continuum level. Usually, this transfer is not straightforward, and we use here a particularly simple case to demonstrate this.
The starting point is published data on the solution enthalpy of carbon in bcc iron [
4]. This paper contains a rich collection of strain dependent energies for different interatomic potential descriptions, and it is useful to compare the predictions to a continuum perspective, which allows using the computed data also for larger scale modeling.
First, we note that C in bcc iron occupies octahedral positions in equilibrium, and induces a tetragonal distortion. This effect is considered in the careful ab initio based computations of Hristova et al. [
4]. They additionally considered the situation where they neglect this anisotropy and assumed a cubic symmetry. It turns out that the results do not change significantly, if the full relaxation is considered. For the present purpose, it has the advantage that we can use the simpler cubic symmetry for the comparison, which leads to more compact analytical expressions on the continuum scale. In the end, we briefly discuss the transfer to the more general situation. In addition, we note that hydrogen in austenite, which is discussed in the other parts of this manuscript from the perspective of a scale bridging, does not exhibit such tetragonal distortions, and therefore the expressions can directly be transferred to this case.
For the calculation of hydrogen solution enthalpies, the authors of [
4] use a
supercell consisting of 54 iron atoms. For the numerical parameters related to the ab initio calculations, we refer to the description in [
4]. One of the octahedral positions is occupied by a carbon atom. From the arising energy differences, the solution enthalpy is defined as
Here, all energies are
bare energies from the underlying simulations.
is the volume dependent energy of the system with an additional carbon atom and
the same without the carbon. Notice that both expressions are taken at the same volume. We note that carbon wants to expand the lattice, therefore the first situation has a higher compressive strain as the latter. For a handshaking to continuum methods, it is important to take into account such effects, as we will see below. Finally,
is a reference potential from a “carbon reservoir”. For the data related to density functional theory calculations (DFT), Hristova et al. use diamond as carbon reservoir [
4]. As we are mainly interested here in the elastic rather than the chemical contributions, this reference term turns out to be of minor importance, but in general it is important to consider it e.g., for thermodynamic considerations.
On the continuum level, a natural approach for the total energy of the system is an additive decomposition into chemical and elastic contributions,
where
c is the carbon concentration, and
the strain tensor components, as defined below. A central difference between the discrete and continuum energies is that for large scale applications it is often more convenient to normalize the energy per volume rather than per particle. The reason is that, in more complex situations, where inhomogeneous strain and concentration distributions occur in non-equilibrium states, e.g., phase field approaches can be a versatile approach for modeling the time dependent microstructural evolution. For that, a generating energy functional is written as space-integral over an energy density, which is therefore defined with dimension energy per volume.
For the isotropic expansion (under the constraints mentioned above), the elastic energy can be expressed as
using the linear theory of elasticity and an isotropic volume expansion, as explained in detail below. Here,
is the undeformed volume in the Lagrangian reference state,
B the bulk modulus, and
are the linear elastic strain tensor components
expressed through the displacement field components
. Here, the strain is diagonal with
and
for
and 0 otherwise. The eigenstrain
expresses the stress-free expansion of the lattice through the presence of a finite carbon concentration
c. As long as the concentration is not too high, it is appropriate to assume Vegard’s law, hence the eigenstrain scales linearly with
c. In addition, if we neglect the tetragonal distortion, the expansion is uniform, hence the eigenstrain takes the tensorial form
with the Vegard coefficient
. For the definition of the concentration, we can use
c being the ratio of populated and total number of octahedral positions, hence
c is in the range from 0 to 1.
In general, the bulk modulus is also concentration dependent. However, for low concentrations, this effect can typically be neglected. This is supported by the DFT data in [
4]. Both with and without one carbon atom the bulk modulus is reported to be
. Conceptually, a concentration dependence of the elastic constants leads to a higher order correction of the leading terms.
According to [
4], the single carbon atom in the 54 atom host matrix leads to a volume increase of 1.7%, hence the strain in each direction is
of this value,
. The volume of the reference cell is
per iron atom.
The corresponding expression to the solution enthalpy (
1) is in continuum representation
after Taylor expansion. This gives
Under isotropic volume change the strain becomes
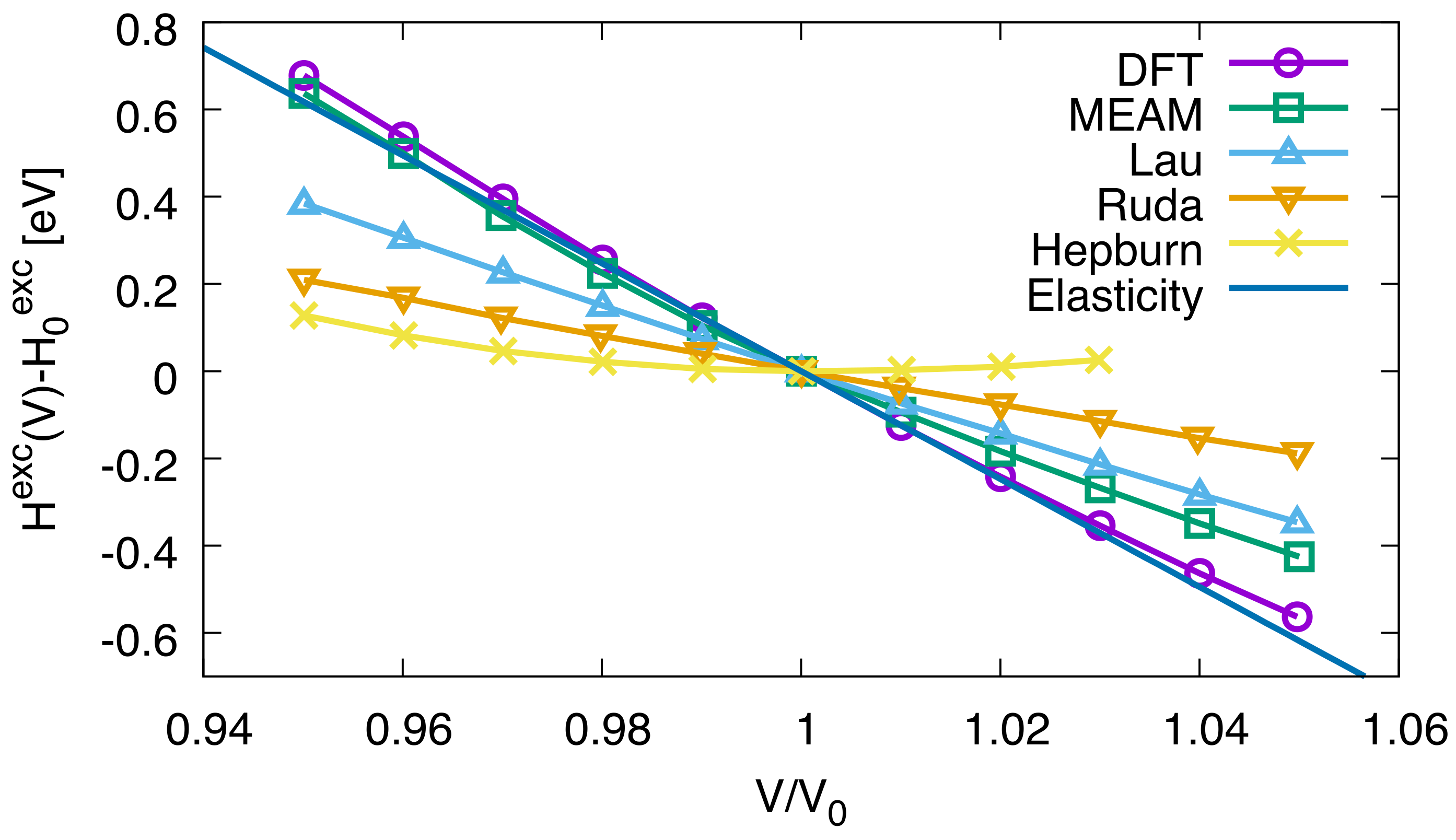
, and, according to the above expression, the solution enthalpy should be a linear function of the strain or volume and decay with increasing volume. Interestingly, this behavior is qualitatively satisfied by the DFT data in [
4], but not at all by several of the used empirical potentials (see
Figure 1). The authors argue that these potentials are therefore unsuitable for describing the strain effects of hydrogen in iron. Here, we confirm this statement from a continuum perspective, as apparently these potentials disagree with predictions from elasticity theory. We can furthermore perform a quantitative comparison using the slope
, which in the continuum picture becomes
Inserting the material parameters given above gives the prediction
, which is in perfect agreement with the DFT value (identical to the given accuracy, see Table 3 in [
4]), and deviates from the empirical potentials, as expected. The continuum result for the strain dependence of the solution enthalpy is depicted in
Figure 1. Only for large strains, where linear theory of elasticity is no longer applicable [
5], deviations occur with the DFT results.
Finally, we mention that consideration of tetragonality is a rather straightforward extension of the preceding discussion. For that, one uses the more general expression
for the elastic energy, and the eigenstrain depends then on the carbon concentrations in the different sublattices. In practise,
becomes a superposition of the tetragonal expansions in the different directions.
Overall, this example shows how the transfer from discrete atomistic descriptions to continuum pictures involving elastic effects can be done. From this, one can obtain a closed analytical expression with all material parameters, which can subsequently be used for higher scale modeling.
3. Hydrogen in Fe-Mn-Al Alloys
In this section, we discuss certain thermodynamic aspects of hydrogen in austenitic Fe-Mn-Al alloys. The starting point is the work by von Appen et al. on hydrogen solubility in Fe-Mn alloys [
6].
Figure 2 shows a collection of the solution enthalpy of hydrogen. Similarly to the discussion above, it is defined as
where the reference chemical potential
is half of the energy of an isolated H
molecule. Notice that the calculations were obtained for fully relaxed configurations, hence in particular the pressure vanishes, and consequently the volumes of the two energy terms with and without hydrogen differ. However, from the considerations above, we can conclude that the Legendre transformation between fixed volume and fixed pressure can be performed using theory of elasticity, without computationally expensive additional ab initio simulations. Obviously, the energy essentially depends linearly on the number of manganese neighbors around the H occupied octahedral site (see
Figure 2). We note that the same linear energy trend is even more pronounced if the solution enthalpy is plotted versus the Voronoi volume. We can therefore conclude that the leading contribution is due to elastic effects.
We can use this information to obtain some insights into the equilibrium occupation of the different octahedral sites by hydrogen. For an alloy with the composition
, we assume a random distribution of Fe and Mn atoms, which ignores potential near ordering effects. Therefore, the probability for an octahedral site to have exactly
n manganese neighbors is given by
The occupation of a single octahedral site follows a Fermi-Dirac distribution,
where
is the probability of the site to be populated with hydrogen,
is the inverse temperature,
the equilibrium chemical potential of hydrogen (not to be confused with
above), and
E is the solution enthalpy for the given atomic environment. Therefore, the equilibrium hydrogen concentration in the Fe-Mn system becomes
As the concentrations are typically low, the additional consideration of more than one hydrogen atom in the supercell should not be necessary. With the linear approximation
(see
Figure 2, using
and
), we can relate the average concentration
c to the chemical potential. If we consider (here, for example, for
)
at room temperature, which is a concentration that may already trigger hydrogen embrittlement, the equilibrium chemical potential is around
. An octahedron with 8 Mn neighbors is rare within such an alloy, but energetically most favorable. Its average population with hydrogen is around
, which is two orders of magnitude higher than the average concentration
c. Similarly, the energetically most unfavorable configuration, where hydrogen is surrounded by Fe only, has an expected H occupation of
, which is one order of magnitude lower than the average value. The octahedral positions with a large number of Mn atoms may therefore be considered as hydrogen traps, in particular as their energy is about 130 meV lower than the average site. However, in equilibrium, even these sites have only a minute H occupation, and therefore these sites cannot trap a large amount of hydrogen. This is a general feature of hydrogen with such low concentrations. The chemical potential is very low, and hence all site occupations are in the tail
of the Fermi-Dirac distribution. Typical trap depths are not sufficient to provide a significant increase of the hydrogen concentration beyond a dilute limit, which would require reaching the regime
.
Aluminum changes the energy landscape rather strongly, as worked out for specific atomic arrangements in [
7,
8]. Both author groups use different concentrations of their alloys, which is captured by a 32 atom fcc supercell. Song et al. use Fe
Mn
Al
, Hüter et al. Fe
Mn
Al
, and both groups come to similar conclusions despite the different stoichiometry, which suggests that the situation is more general. We follow the notation in [
8], where
x-
y-
z denotes an octahedral environment with
x iron,
y manganese and
z aluminum atoms; this notation, however, cannot distinguish between different atomic arrangements, but we stay with it for convenience. In the following discussion, we focus on the Fe
Mn
Al supercell. This system allows for distinguishing 6-0-0, 5-0-1, 4-2-0 and 3-2-1 neighbourhood configurations for the hydrogen atom.
The starting point is the trans (high energy) 4-2-0 configuration in
Figure 2 (notice that the simple notation 4-2-0 does not reflect whether Al is present in a next-to-nearest neighbor position, therefore this information needs to be provided additionally). In contrast to the Fe-Mn alloys discussed above, the energy is here not only determined by the nearest neighbor configuration. A 4-2-0 neighborhood, where an Al atom occupies a next-to-nearest neighbor position, diminishes the hydrogen solution enthalpy by about 60 meV [
7]. It is therefore speculated that this octahedral configuration may act as an H trap; in view of our discussion above, we stress that this effect is however rather weak.
More pronounced, however, is the effect that a direct neighborhood of Al to an H atom is energetically highly unfavorable. Configurations like 3-2-1 or 5-0-1 are about 130–140 meV less favorable than the potential 4-2-0 “trap” site. These octahedra are populated with a concentration that is about two orders of magnitude lower than the average concentration at room temperature; we may therefore effectively say that such sites are blocked.
Compared to the Fe-Mn system, Al therefore leads to strong alternations of the entire energy landscape. Changes of atomic neighborhoods by replacing one Fe atom by Al raises the energy in several cases by around 80 meV. In contrast, only one extreme case in the Fe-Mn system, for a transition between the trans 4-2-0 to the mere 3-3-0 environment, leads to a comparable shift of about −80 meV. In all other cases, the energetic shifts are much milder in the Fe-Mn system. Altogether, we can therefore conclude that the entire solution enthalpy landscape becomes significantly rougher in the Fe-Mn-Al case by the addition of rather small amounts of Al. Already for about 3 at% of Al almost 20% of all octahedral cages are neighboring an aluminum atom and are therefore energetically unfavorable for hydrogen.
4. Ferritic Grain Boundary Hydrogen Embrittlement: Estimates From Ab Initio Datasets
In this section, we discuss a possible phase separation into hydrogen enriched regions and hydrogen depleted regions at ferritic grain boundaries, and the function of aluminum as protective agent against grain boundary related hydrogen embrittlement.
An important contribution to the macroscopic interpretation of hydrogen embrittlement is the phase formation based on available ab initio data. Especially in the case of hydrogen in steel, this approach becomes complex, as at least carbon, hydrogen and iron need to be considered, and the required amount of ab initio data quickly becomes large. Especially for grain boundaries, which require rather expensive electronic scale simulations, the data set is sparse. Therefore, the development of possible approximative schemes can be helpful in this context.
Our first approach employs a mean field approximation for grain boundary energies, based on data published in [
4,
9,
10] for varying carbon and hydrogen populations. The most investigated grain boundary in these publications is
in bcc iron, and the authors present—amongst other results—segregation and grain boundary energies for carbon and hydrogen at grain boundaries, where the most attractive type of grain boundary site is populated. In the considered supercell, four of these sites exist per grain boundary, i.e., any combination of altogether four hydrogen or carbon atoms per grain boundary corresponds to a fully saturated grain boundary with respect to this site. For details about the determination of the most attractive sites in a grain boundary state of minimal energy, we refer to [
4,
9,
10,
11]. The grain boundary energies reported in [
9] are defined as
where
is the energy of a grain boundary with interfacial area
A and
n atoms of type
X in it.
is the absolute energy of the used simulation cell, which includes two grain boundaries of the simulated type due to periodicity constraints. By
the absolute energy of a simulation cell with the same number of iron atoms is defined. Note that all simulations were relaxed using zero stress boundary conditions for the supercell, i.e., the only elastic contributions of the resulting energies are those confined to atomistic length scales around the grain boundary.
is the reference chemical potential, for hydrogen that corresponds to H
molecules, for carbon to diamond, as discussed also in the preceding sections.
The scheme to estimate grain boundary energies for various hydrogen and carbon populations of the reported
grain boundaries is a summation of the averaged energetic contributions per atom relative to the fully saturated grain boundary for each pure population, which can be understood as a mean field approach. This can be expressed as
where the required grain boundary energies reported in [
9] are
,
, and
. Besides the reported grain boundary energies for zero to four sites occupied by carbon or hydrogen, also a co-segregated grain boundary energy is provided, for two hydrogen atoms and one carbon atom. This co-segregated grain boundary energy is an interesting test data point for the approximative scheme, which would predict the grain boundary energy for the reported population as 1/4 of a carbon saturated grain boundary, 2/4 of a hydrogen saturated grain boundary and 1/4 of an empty grain boundary. In
Table 1 below, we see the comparison of predicted mean field grain boundary energies and ab initio calculated grain boundary energies for the testable data points in [
9].
Apparently, the mean field description works excellently for hydrogen, while the discrepancies for carbon can reach up to 0.09 J/m
. This has to be put in context of the variance in reported ab initio results for grain boundary energies that correspond to identical definitions and therefore should give identical numbers. We provide here the grain boundary energies reported for two grain boundaries. The reporting references include [
9,
10,
11,
12,
13,
14,
15], and the reported grain boundary energies range from
to
(
5(310)) and from
to
(
3(112)).
While the approximative scheme can be tested for the grain boundary in the especially interesting regime of co-segregated grain boundaries, we are currently not aware of grain boundary energies for co-segregated sites for any other grain boundary. Consequently, we continue with a focus on the grain boundary.
As the next step, we obtain the energies required for the determination of grain boundary phase separation. From the findings reported in [
9], we know that the ground state of the grain boundary is carbon saturated with respect to the most attractive sites. These sites are also most attractive to hydrogen, thus the phase equilibrium in the ternary Fe-C-H system at the grain boundary reduces to the coexistence of regions in the grain boundary with varying hydrogen and carbon fractions, where all attractive sites are occupied by one of those species.
When the mixture of carbon saturated (hydrogen free) phase (
) and hydrogen saturated phase (
) forms, the averaged Gibbs energy per host atom is given as
where
is the averaged Gibbs energy per host atom in the hydrogen-free phase,
is the averaged Gibbs energy per host atom in the hydride phase,
are the averaged hydrogen concentration, hydrogen concentration in the carbon-saturated phase and hydrogen concentration in the hydride phase. We assume that we have an ideal statistical distribution of the hydrogen atoms along the available sites, and restrict the description to the configurational entropy contribution. The Gibbs energies of pure iron (
),
and a pure hydride phase (
, fully saturated,
) are
where, in the low concentration regime,
is the dominant contribution. We require equal chemical potentials for the heterogenous mixture of hydrogen saturated and carbon saturated grain boundary phases and the
-phase, so we obtain for low temperatures
where we used
, with
denoting the respective phase equilibrium concentration. For the solulibility limit, we get
with
. The derivative of the Gibbs energy at zero concentration of hydrogen,
, is obtained as finite difference approximation from the available ab initio data. The expression for the enthalpy difference
becomes more familiar when we rewrite it in terms of classical defect energies
, and we identify the relevant contributions for
,
In the last line, we took advantage of the scaling behaviour of the energy with respect to the particle numbers. By we define the total energy of the grain boundary simulation cell including n carbon atoms and m hydrogen atoms at the attractive sites at each of the two grain boundaries, and host atoms. To calculate the differences in energies per atom from the differences in grain boundary energies, holds for the given grain boundary area of . The values obtained here for a separation in the considered grain boundary into fully carbon saturated and fully hydrogen saturated phases lead to a hydrogen solubility at the interface governed by the enthalpy difference (, where the reference chemical potentials correspond to diamond and .
When we relate the enthalpy difference to the bulk solution energies in the preferred octahedral site for carbon, at 0.74 eV [
12], and the preferred tetrahedral site for hydrogen, at 0.25 eV [
11], we can estimate that the grain boundary will be co-segregated by hydrogen and carbon without separation into hydrogen-free and carbon-free regions. The reason is a relative shift in the chemical potentials of about 0.49 eV in favor of a partial substitution of carbon with hydrogen at the grain boundary.
From this perspective, it is interesting to consider the influence of aluminum, which is known as protective agent against hydrogen embrittlement. The ab initio data we use to estimate the influence of aluminum has been published in [
16]. In this publication, there were no grain boundary or bulk solution energies for hydrogen in Fe-Al reported, but the diffusion barriers of hydrogen in carbide-free bainite have been calculated. Since Fe-(Al)-H was chosen as a representative system, we use the reported results as the upper limit for the repulsive interaction of Al with hydrogen in the direct neighborhood. Depending on the Al fraction in the supercell, the shift in the hydrogen solution energy at the transition point along the tetrahedral-tetrahedral path varies and reaches up to 350 meV.
We choose an intermediate value of 150 meV that reflects two aspects: the transition point shift due to Al alloying—at least in fcc Fe—is around twice as high as the shift of the low energy occupied sites solution energies due the Al alloying [
8], and even with Al segregating to ferritic grain boundaries [
17], we do not assume a local peaking to 50 atomic percent of Al. However, even with 150 meV shift per hydrogen atom, the preference of hydrogen to segregate to the grain boundaries is suppressed.
Besides this thermodynamic picture, we briefly consider possible kinetic scenarios for the substitution of carbon with hydrogen in grain boundaries. While the carbon atoms act as a strengthening element in grain boundaries, as reported in detail in [
4], they also increase the local electronic density. Specifically, as reported also in [
9], the carbon atom in the grain boundary shows overlapping bands with its adjacent Fe atoms and a spherically symmetric local increase of the charge density distribution (see
Figure 3 in [
9]). It is possible that the hydrogen atoms in the metallic matrix, which are partially negatively polarised, lead to a local charge redistribution at these spots of increased electronic density, providing access to a substitution of the carbon by hydrogen. We note that this mechanism will require a more detailed quantum mechanical study of the dynamic bonding behavior at this site.
5. Hydride Precipitation near Grain Boundaries
As described in
Section 2, the application of a tensile strain can lower the solution enthalpy of interstitial elements and therefore favor segregation. If the material is homogeneously deformed, also the shift of the solution enthalpy is the same everywhere. For a grand canonical ensemble, the hydrogen concentration raises uniformly. In contrast, for a canonical ensemble with a fixed total amount of hydrogen, the concentration distribution is unaffected. This is different for non-uniform deformation, e.g., localized tensile strains near crack tips or dislocations, where the equilibrium hydrogen concentration can strongly increase and eventually lead to localized hydride formation.
As we have worked out recently, solubility limits can also be affected by the proximity of surfaces and interfaces even in the absence of external stresses [
18]. At the solubility limit, a secondary phase (hydride) forms, which typically has an elastic mismatch with the parent phase. This induces long-ranged coherency stresses, which tend to suppress the phase separation. If free surfaces are close to the potentially forming precipitate, the stresses can partially be released, and therefore the elastic energy penalty for two-phase situations is reduced. Consequently, there is a preference for phase separation near surfaces, or, in other words, the solubility limit for hydrogen is reduced in these regions.
We have worked out a scale bridging description of these processes, where all relevant material parameters are derived from density functional theory, and the long-ranged elastic distortions are treated on the continuum level. Both descriptions are linked by a statistical mechanics and phase field perspective for the prediction of stress and surface affected solubility limits. A particular finding is that, in the low concentration and low temperature regime, which is most relevant for hydrogen in metals and steel, the solubility limits between bulk and surface differ by a factor
This expression is independent of chemical bonding effects and only depends on elastic parameters. It contains the bulk elastic energy
with Young’s modulus
E, Poisson ratio
, Vegard coefficient
and low temperature hydrogen concentration
in the precipitate phase. In Equation (22),
is a dimensionless geometrical factor of order unity, which describes the near surface relaxation (for
), which can be obtained e.g., by finite element simulations [
19]. We note that this prediction is based in particular on a strain dependent energy including hydrogen, analogous to carbon in Fe as analyzed in
Section 2. For a detailed discussion, we refer to [
18].
We can use these results to discuss the possibility of precipitation, in particular hydride formation, near grain boundaries. For that, we assume that the grain boundary appears on mesoscopic scales as a layer with a locally different mechanical strength. The inclusion is assumed to have a diagonal eigenstrain relative to the matrix phase, and the elastic constants are taken as equal. A sketch of the geometry is shown in
Figure 3.
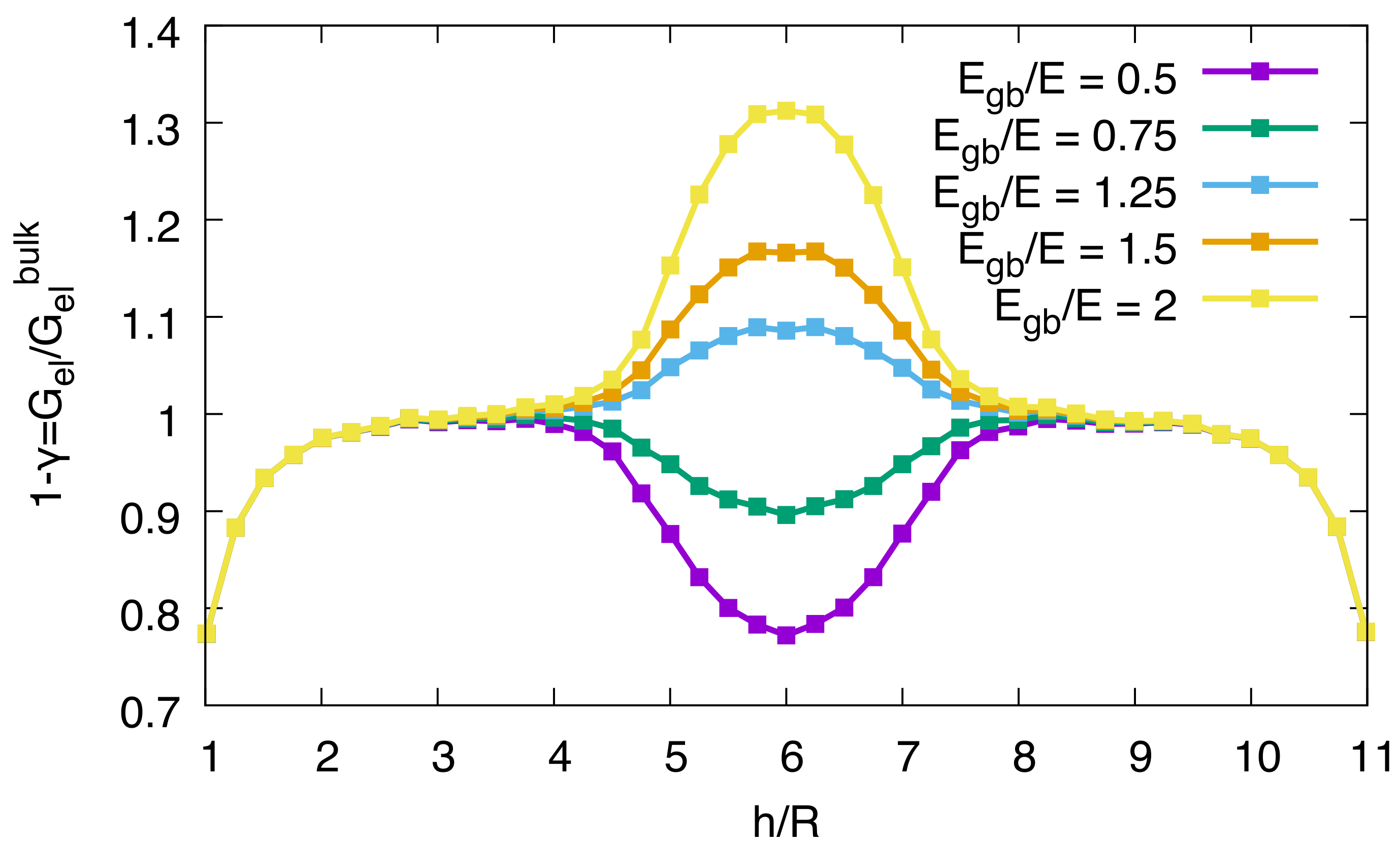
A spherical precipitate is placed at a certain distance from either a free surface or a grain boundary. If the precipitate is neither close to the free surface nor to the grain boundary, the elastic energy reaches the bulk value, which can be calculated analytically under the given conditions (see [
18] for details). For different distances of the precipitate to a free surface or the grain boundary and different elastic properties of the grain boundary, the elastic energy of the system is calculated with the finite element method. Close to the free surface, the elastic energy drops with the distance of the precipitate’s center of mass to the surface
h (see
Figure 4).
When the precipitate approaches the grain boundary, the elastic energy can either increase or decrease, depending on whether the elastic constant
in the grain boundary layer is effectively higher or lower than in the bulk. In the limit that
tends to zero, the grain boundary acts effectively as a free surface, and we recover the same elastic energy relaxation. On the contrary, for
the grain boundary becomes rigid, and we receive the same situation as for nucleation near a fully confined surface, as discussed in detail in [
18].
We note that, due to the computational expense, limited datasets of usable mechanical data for grain boundaries are available. In [
9], the dependence of the theoretical strength
of the
grain boundary on the hydrogen and carbon population is reported. By varying the carbon and hydrogen content of this grain boundary,
varies by 10–50 percent when the empty grain boundary is populated with hydrogen or carbon. In particular, an energetic preference is reported in [
9] for a segregation of hydrogen to the carbon saturated grain boundary to replace carbon, and an associated decrease of the theoretical strength by about 20 percent is determined. We note that this value is subject to variations arising due to orientational changes and elastic anisotropy in real samples. We assume that these effects lead effectively to a local alternation of the ratio
in the spirit of our continuum picture.
The energy modification can then be inserted into Equation (22) to obtain the increase or decrease of the solubility limit. Near a stiff grain boundary, hydride formation is less likely.
In conjunction with the role of Al as a protective agent against grain boundary embrittlement discussed in the previous section, the findings in this section suggest the possibility of an increased weakness against hydride formation close to grain boundaries when the segregation of Al to grain boundaries prevents the substitution of carbon by hydrogen. However, we mention that a detailed understanding of the role of Al on carbon population of attractive grain boundary sites is presently lacking to give a more complete estimate here. In particular, the possible configurations of Al that range from a disperse distribution to highly localised concentrations require further investigations on the quantum mechanical scale.
We remark that one can interpret the Boltzmann factor in Equation (22) as a kinetic modification of the energy barrier for a hydride nucleation process. Apart from phase separation triggered by nucleation events, for higher concentrations, also barrierless transformations driven by spinodal decomposition are conceivable. The mixing enthalpy (per unit volume) is given by
as a function of the hydrogen concentration
c, using the number of metal atoms
per unit cell of volume
and the interaction strength parameter
that can be obtained by ab initio simulations. The dominant configurational contribution to the lattice gas entropy is in the low concentration regime
According to the seminal work by Cahn and Hilliard [
20,
21], the chemical spinodal is determined by the condition
. Asymptotically for
and for the low concentration branch,
In contrast, the coherent spinodal, which is comparable to the bulk elastic solubility limit, is determined by the condition [
20]
In the low temperature limit for the low concentration branch, we get
Surface spinodal modes have been discussed [
22], and they can appear in between the coherent and the surface spinodal in the vicinity of a free surface. From these expressions, we can conclude that, for hydrogen related problems in steel, the required concentrations for spinodal decomposition are typically too high, and phase separation should therefore be initiated by nucleation, as discussed above.
Within this section, we have focused on the influence of mechanical characteristics of grain boundaries on the hydride formation in the vicinity of a grain boundary. Analogously to the thermodynamic description of the influence of elastic relaxation close to free surfaces in [
18], the stiffness of the grain boundary can lead to a local decrease or increase of the hydrogen solubility limit, hence favoring or suppressing hydride precipitation from the host matrix.







{kind=link}
{kind=link}
{kind=link}
{kind=link}