Modeling of Phase Equilibria in Ni-H: Bridging the Atomistic with the Continuum Scale


Abstract
:1. Introduction
2. Methods
3. Results
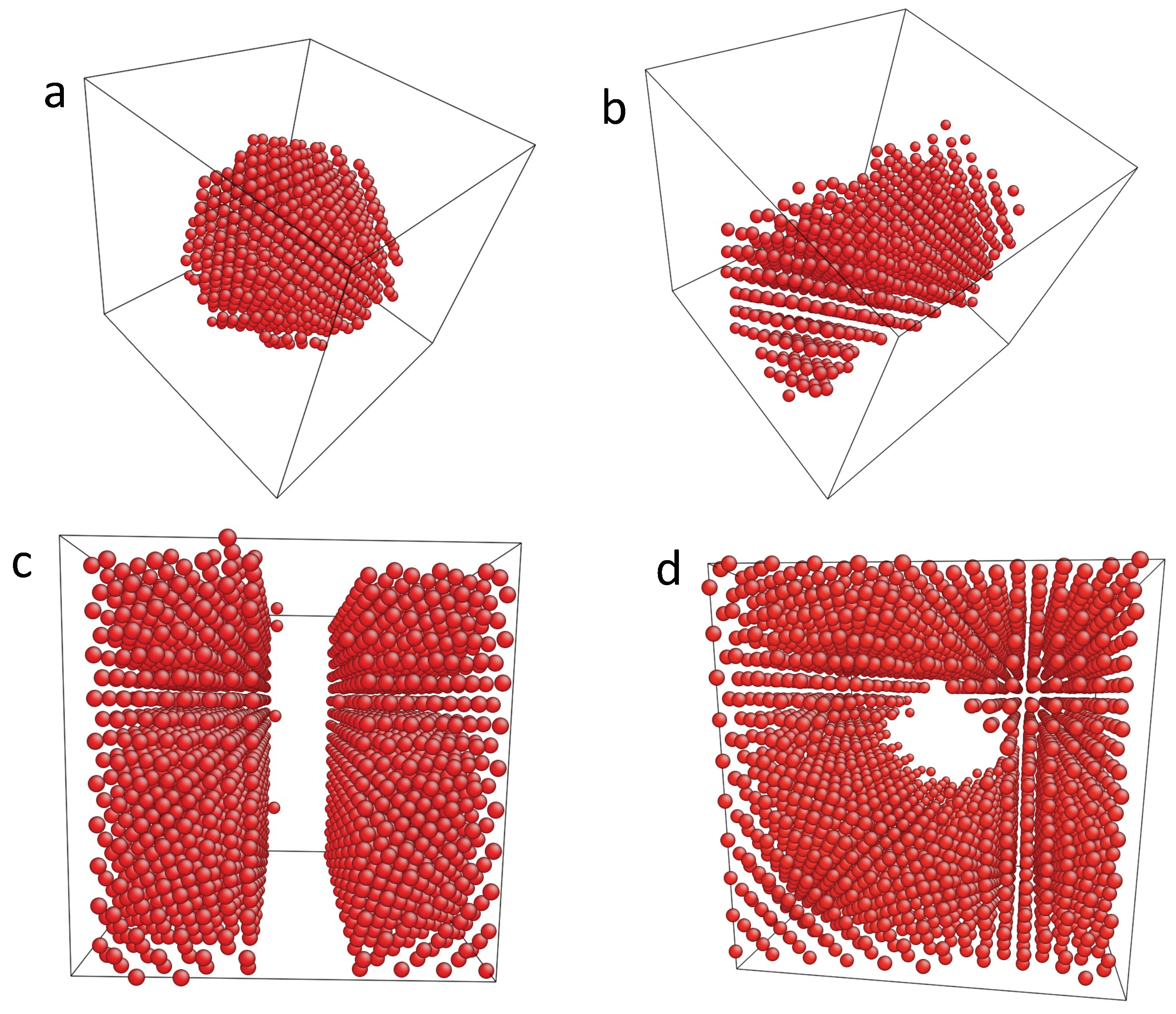
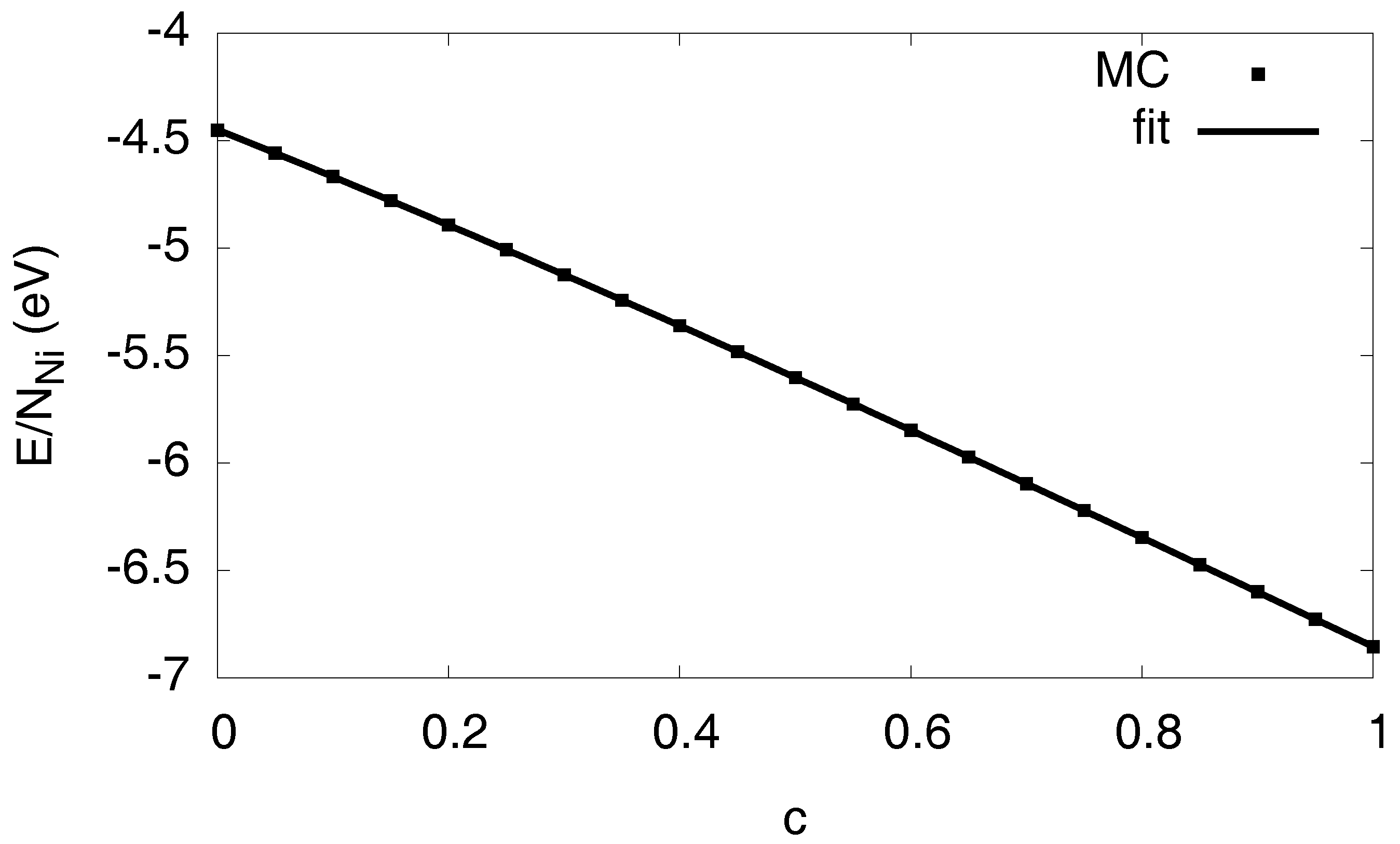
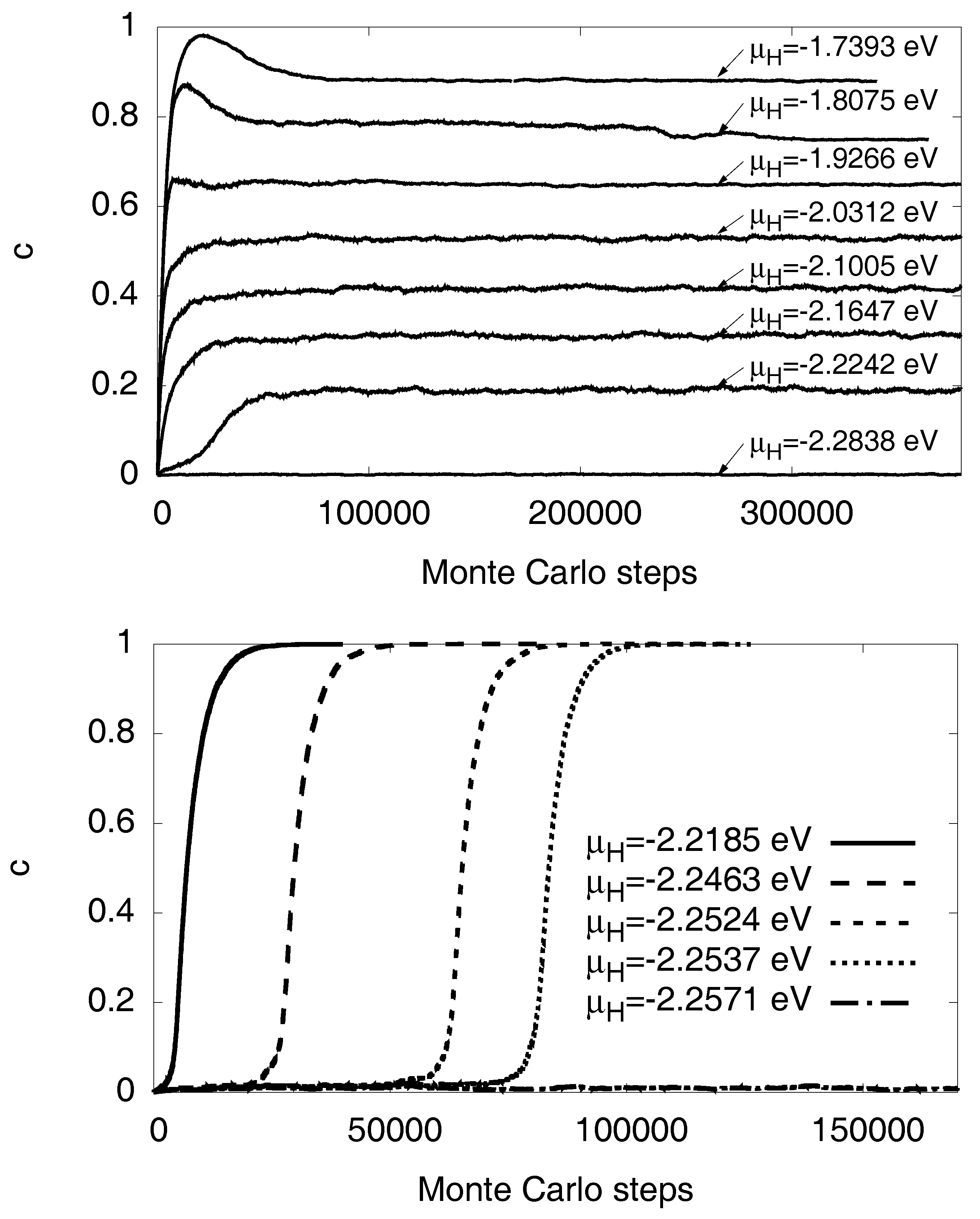
3.1. Monte Carlo Modeling
3.2. Free Energy Formulation
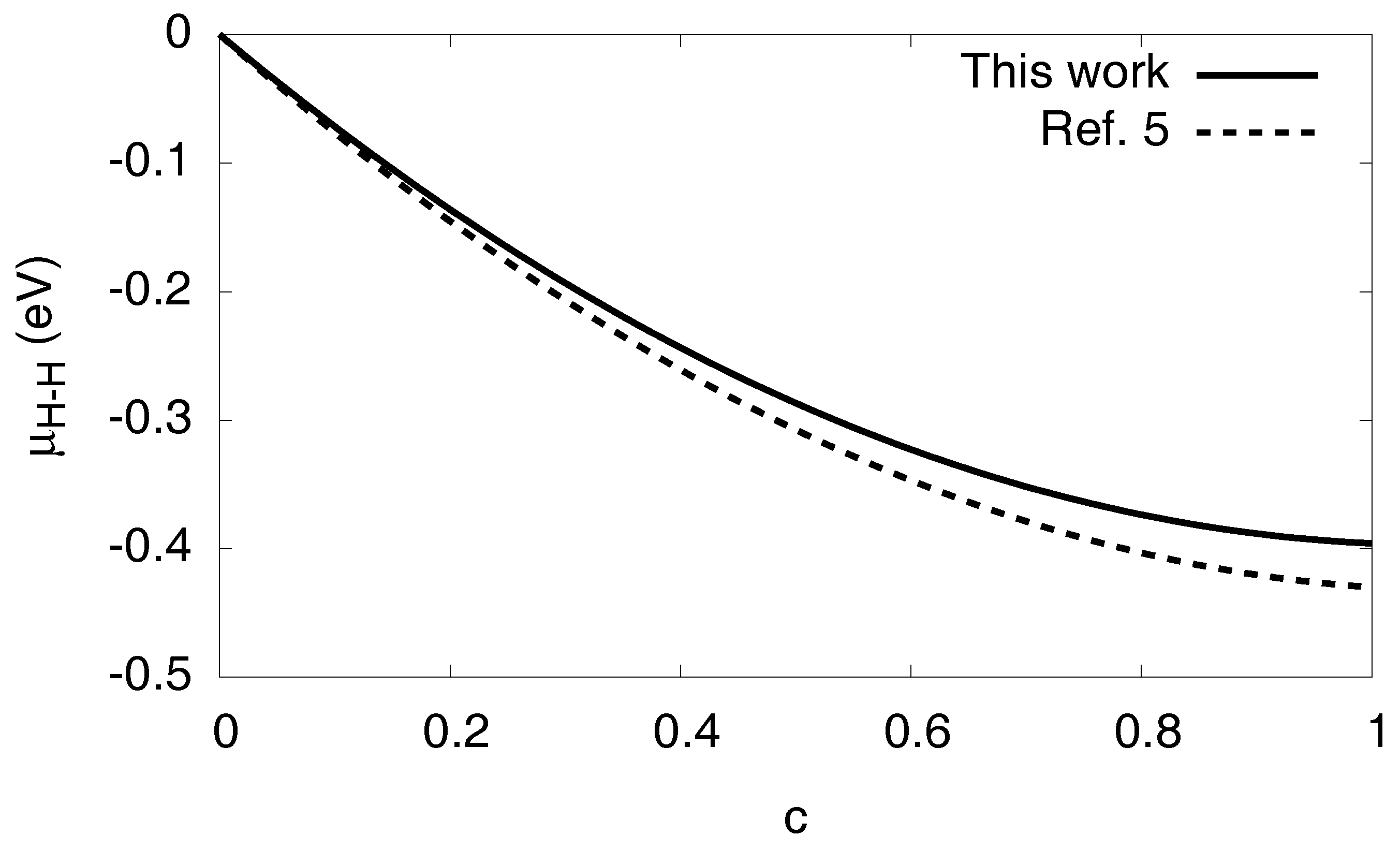
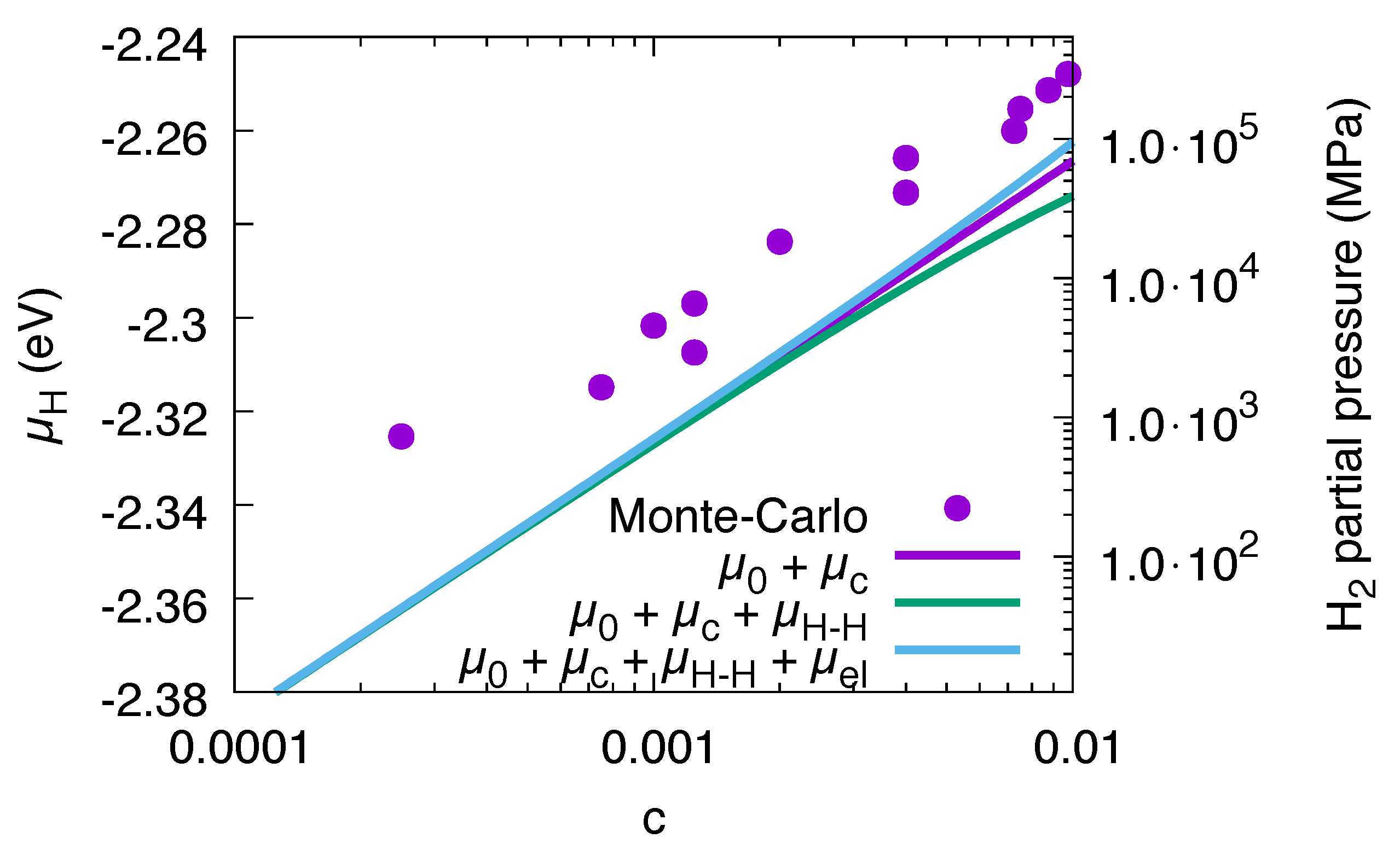
3.3. Solvation Energy and H–H Interaction
3.4. Configurational Entropy
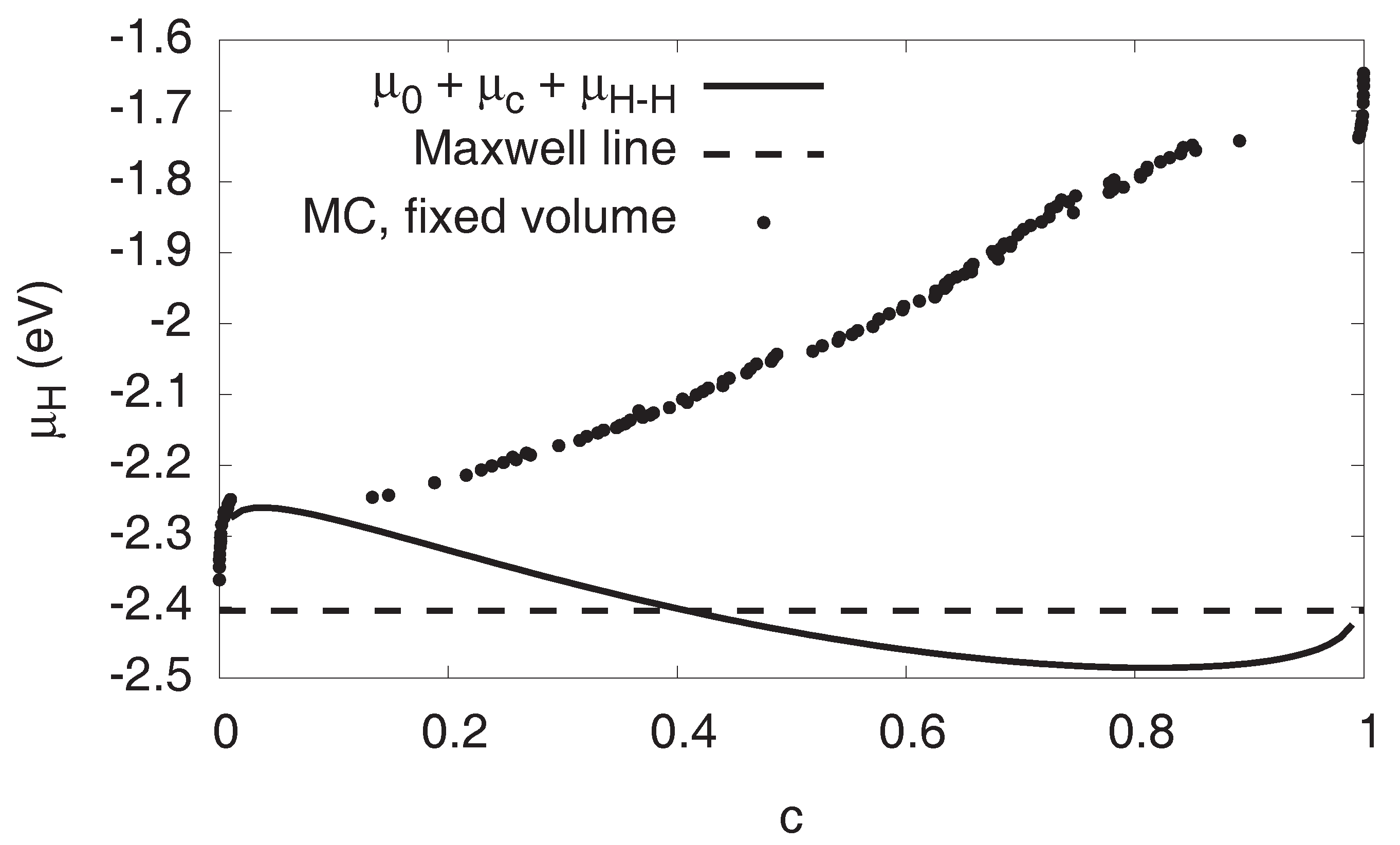
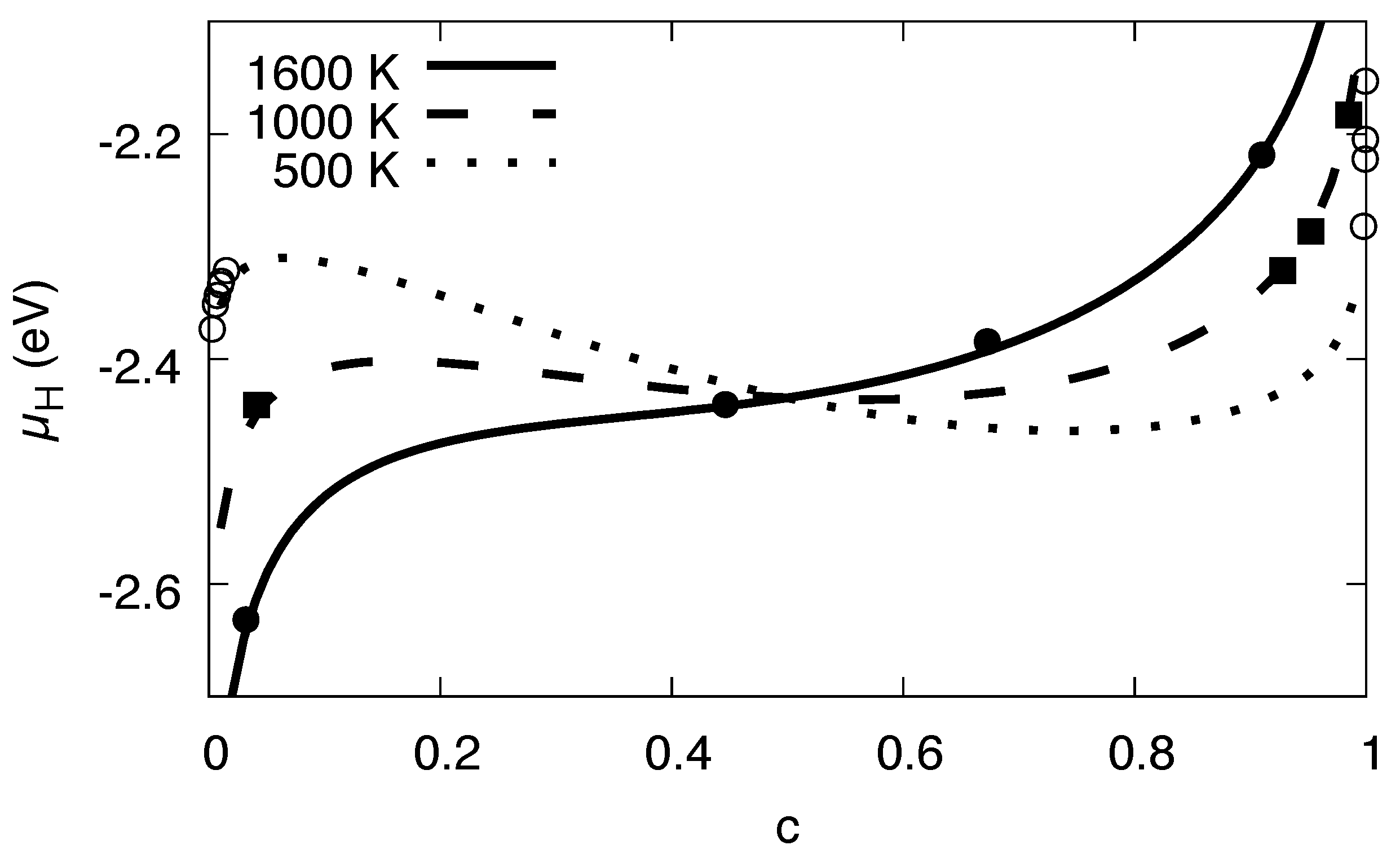
3.5. Maxwell Construction
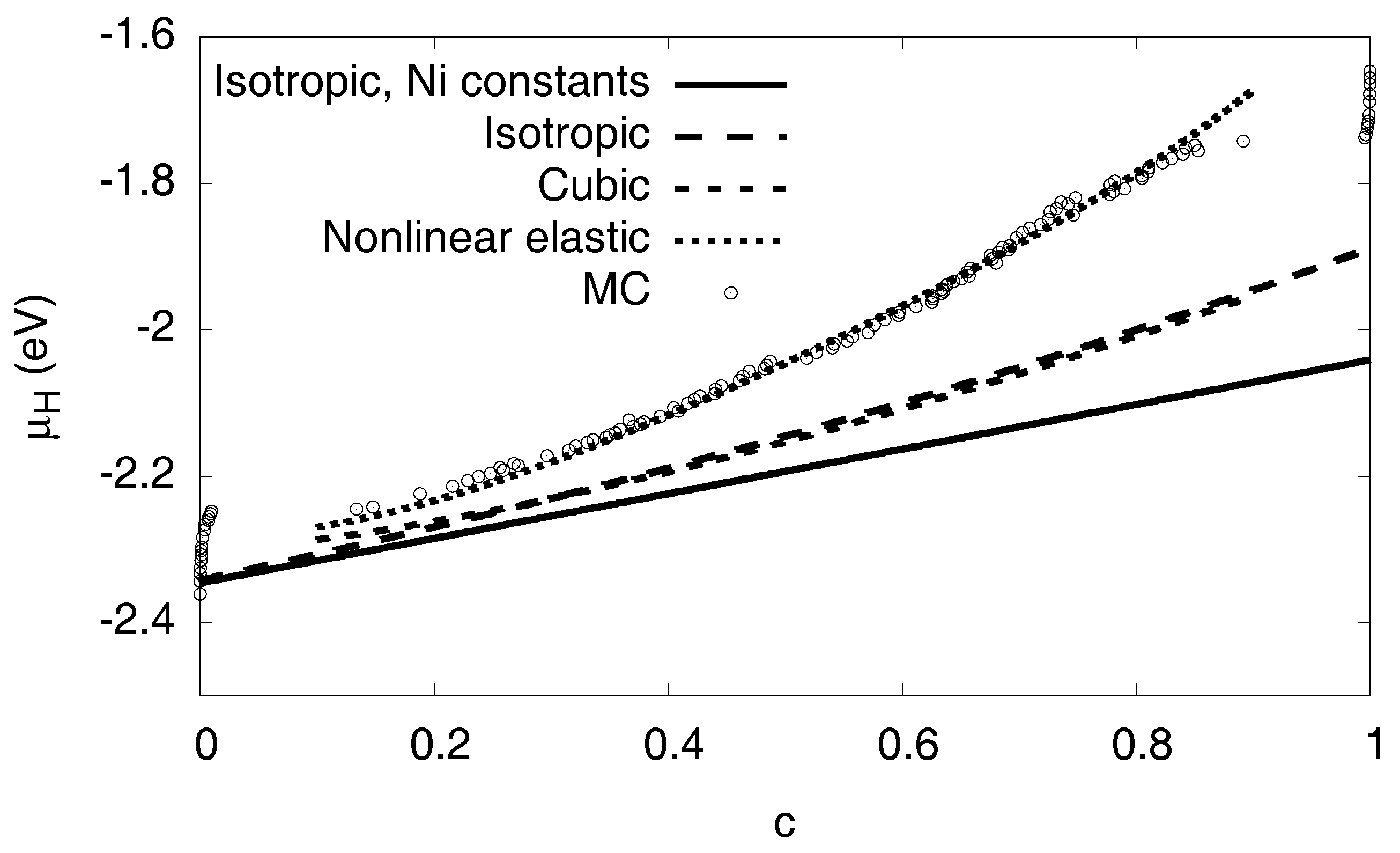
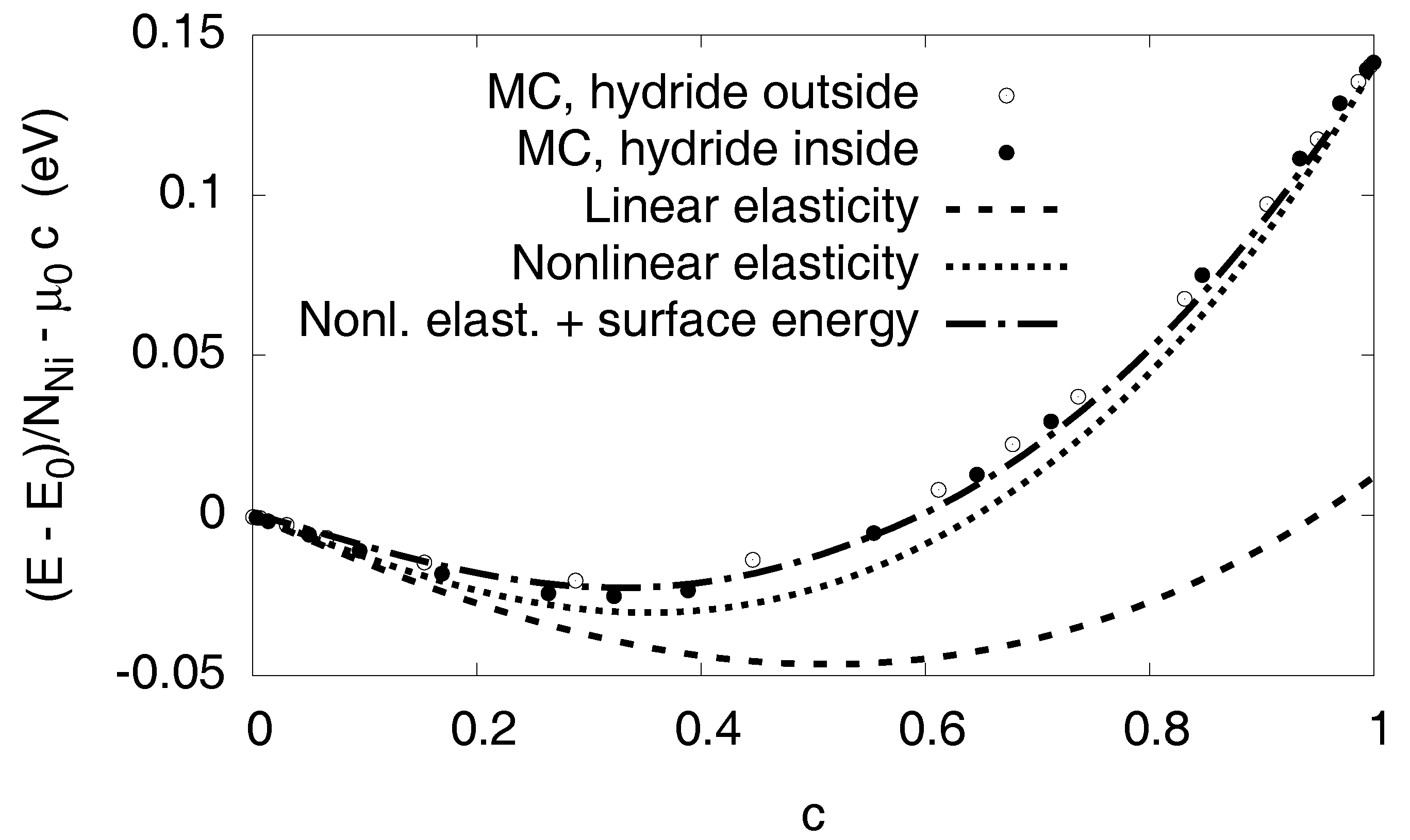
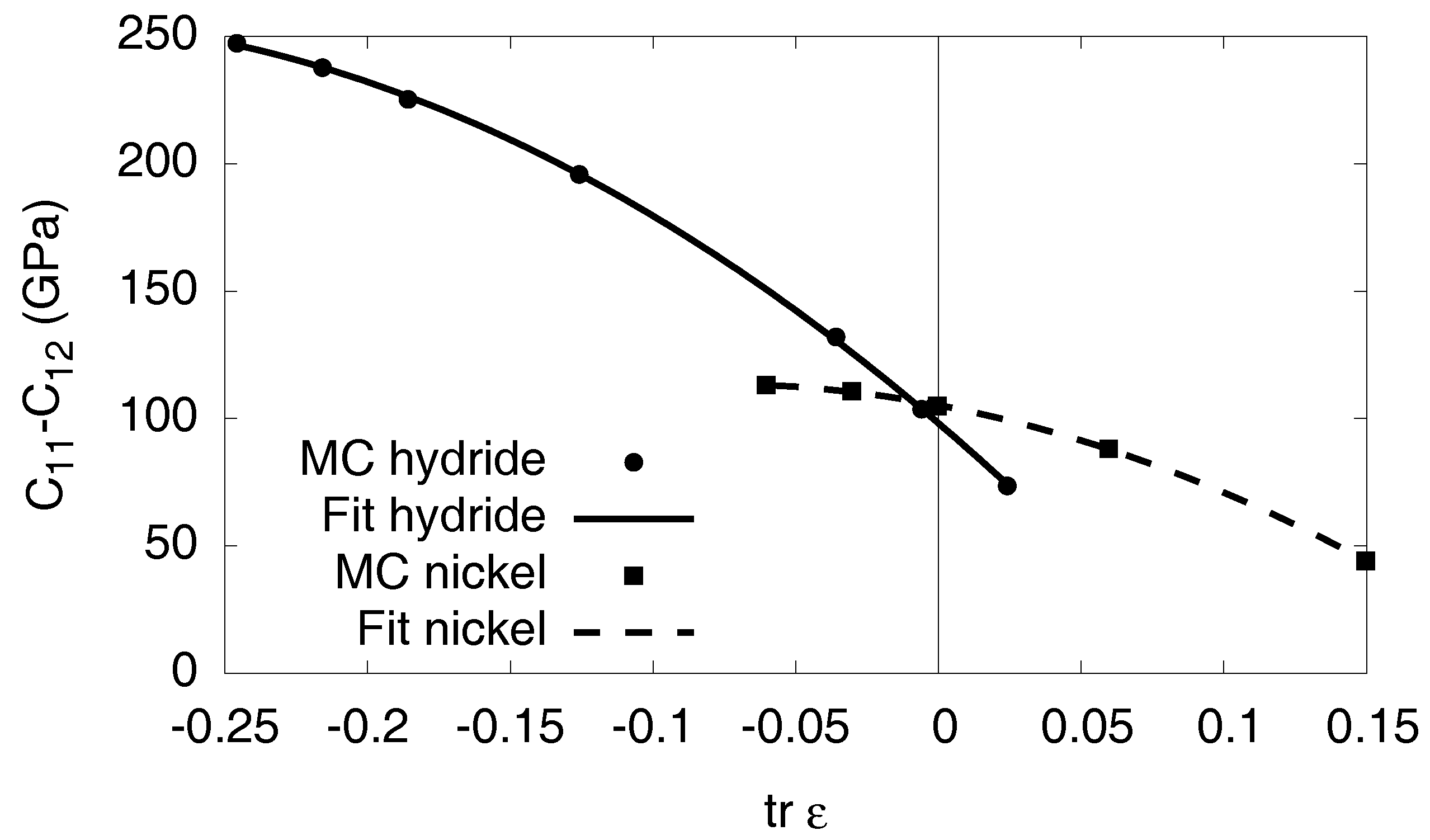
3.6. Elastic Effects
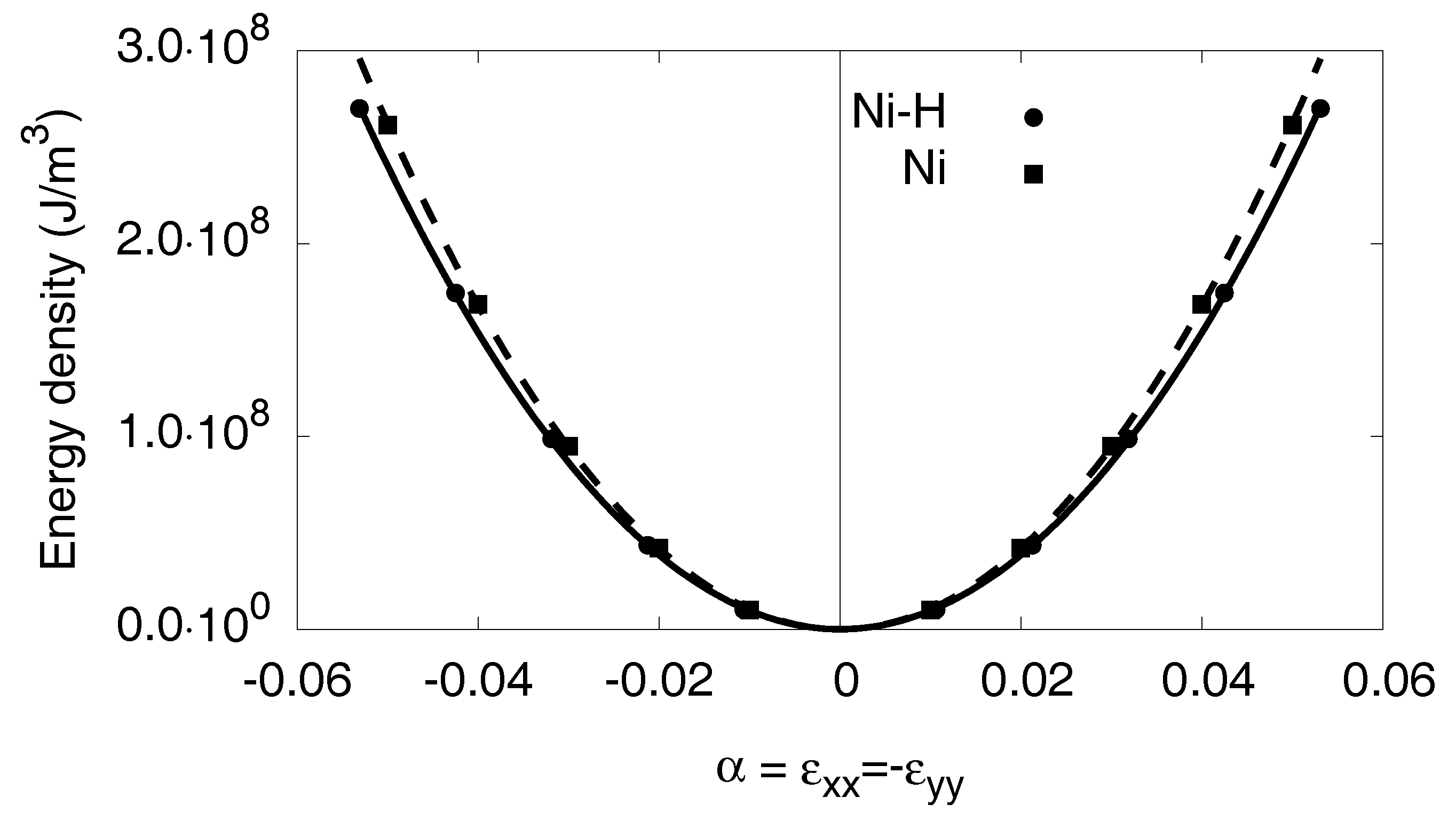
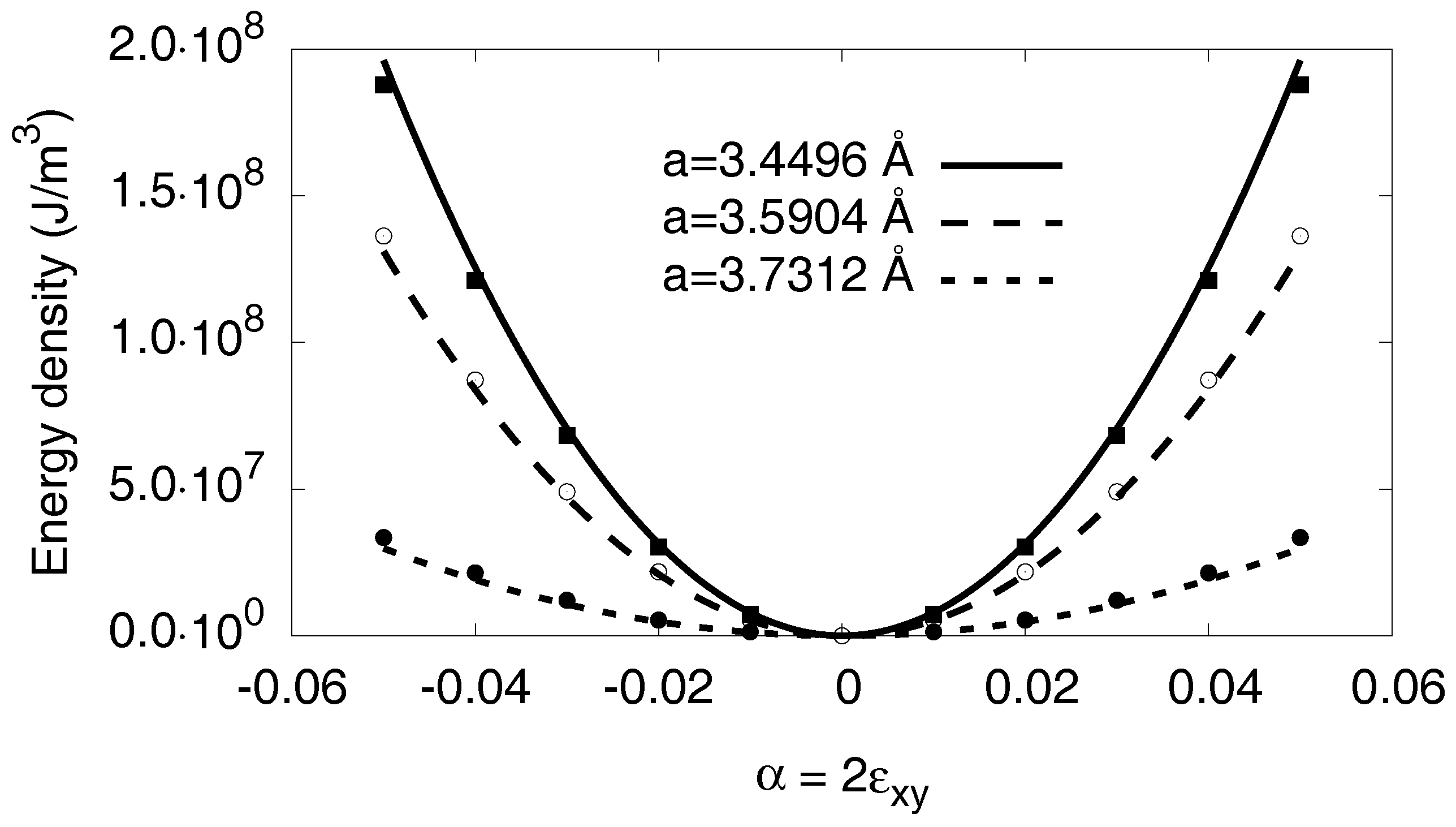
3.6.1. Linear Elastic Effects
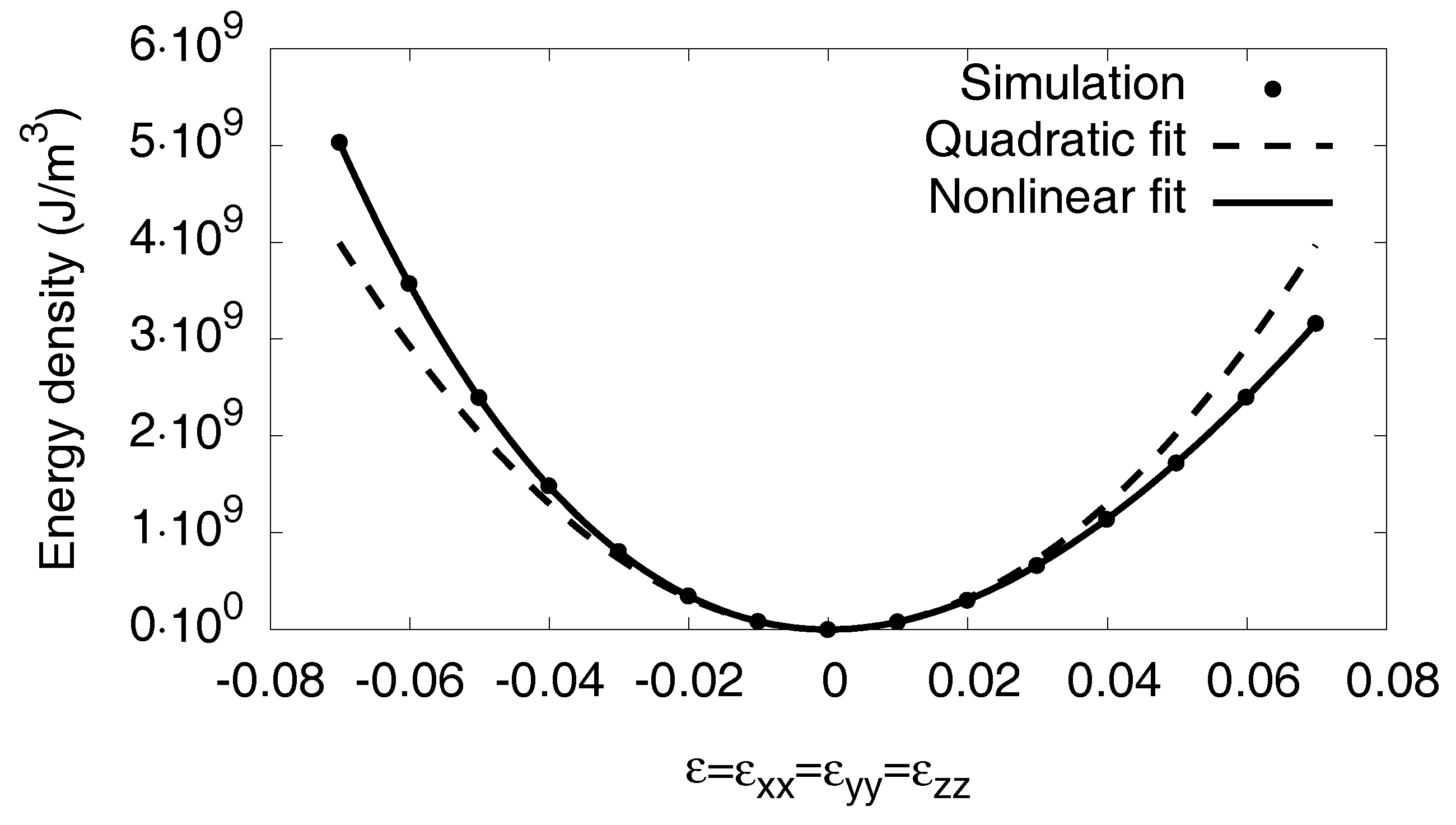
3.6.2. Nonlinear Elastic Effects
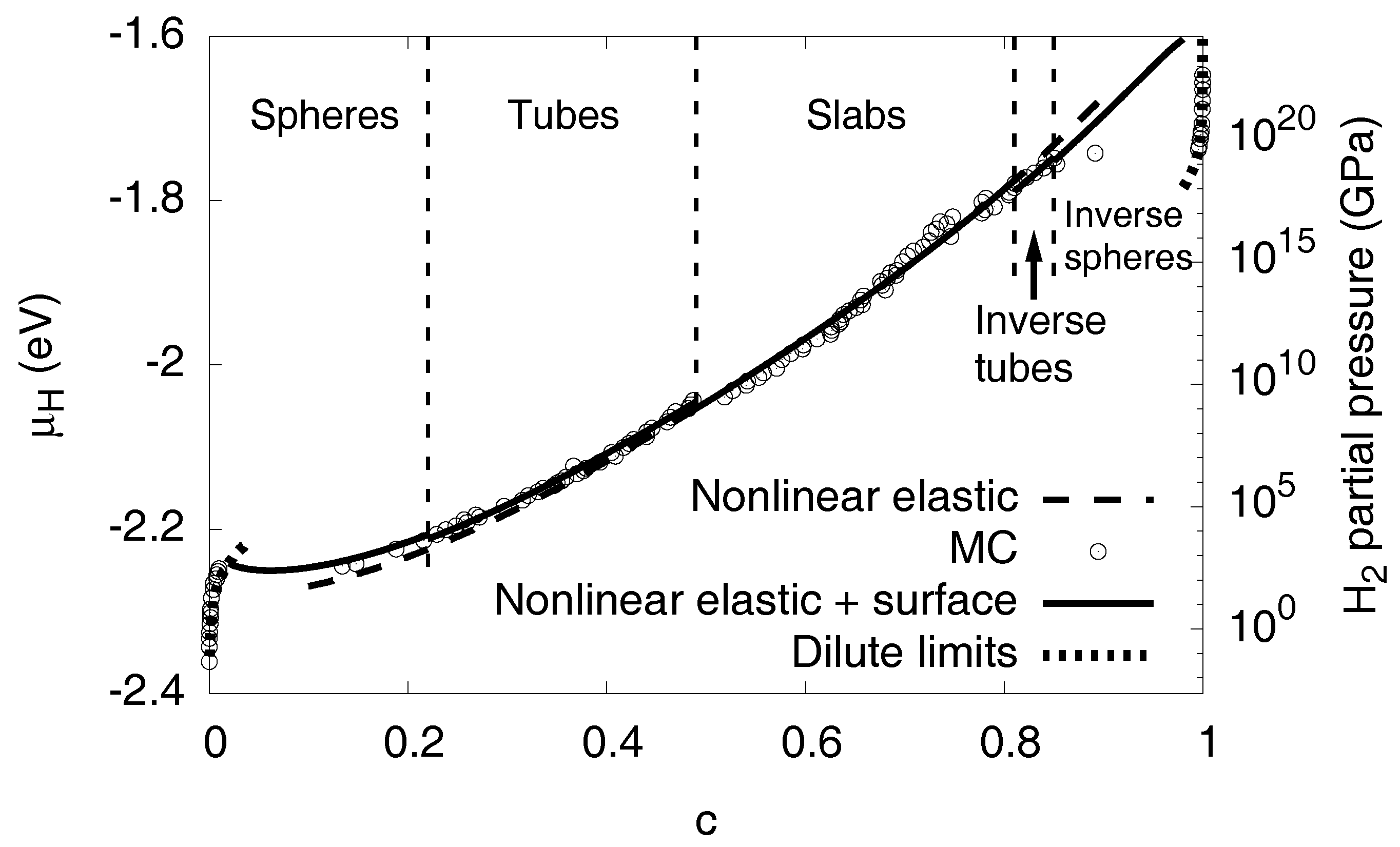
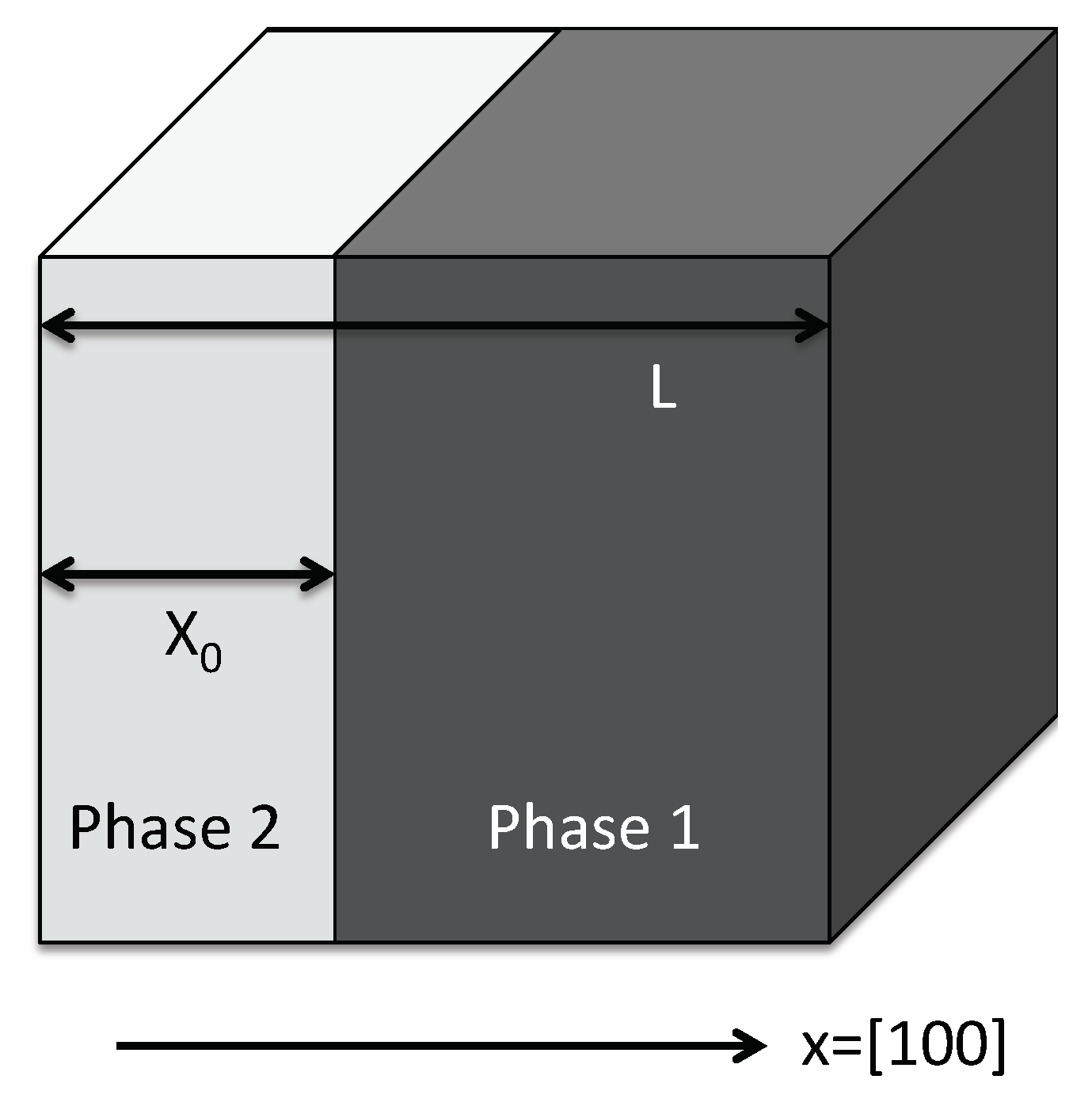
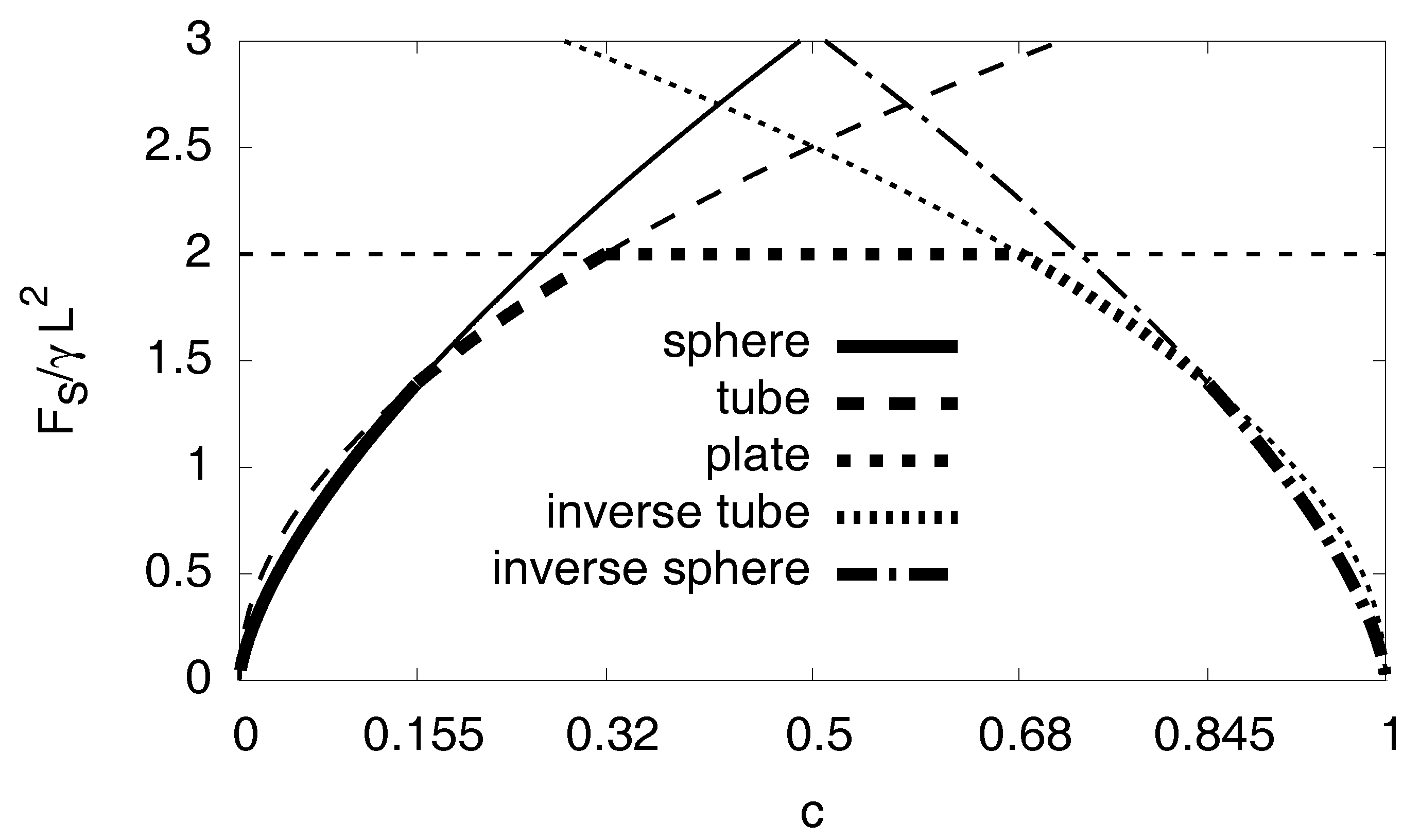
3.7. Interfacial Effects
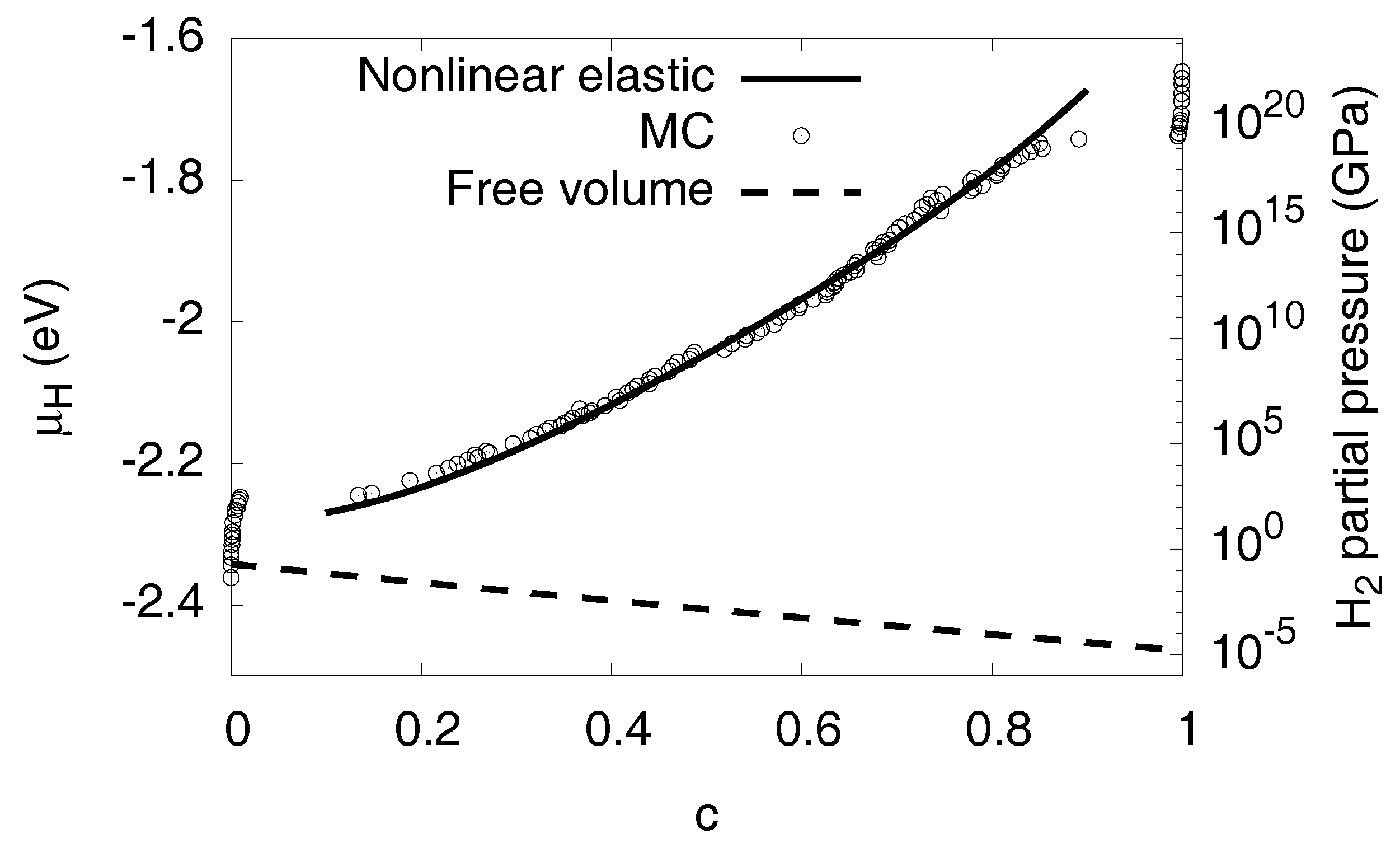
3.8. High Concentration Limit
3.9. Conversion to Partial Pressures
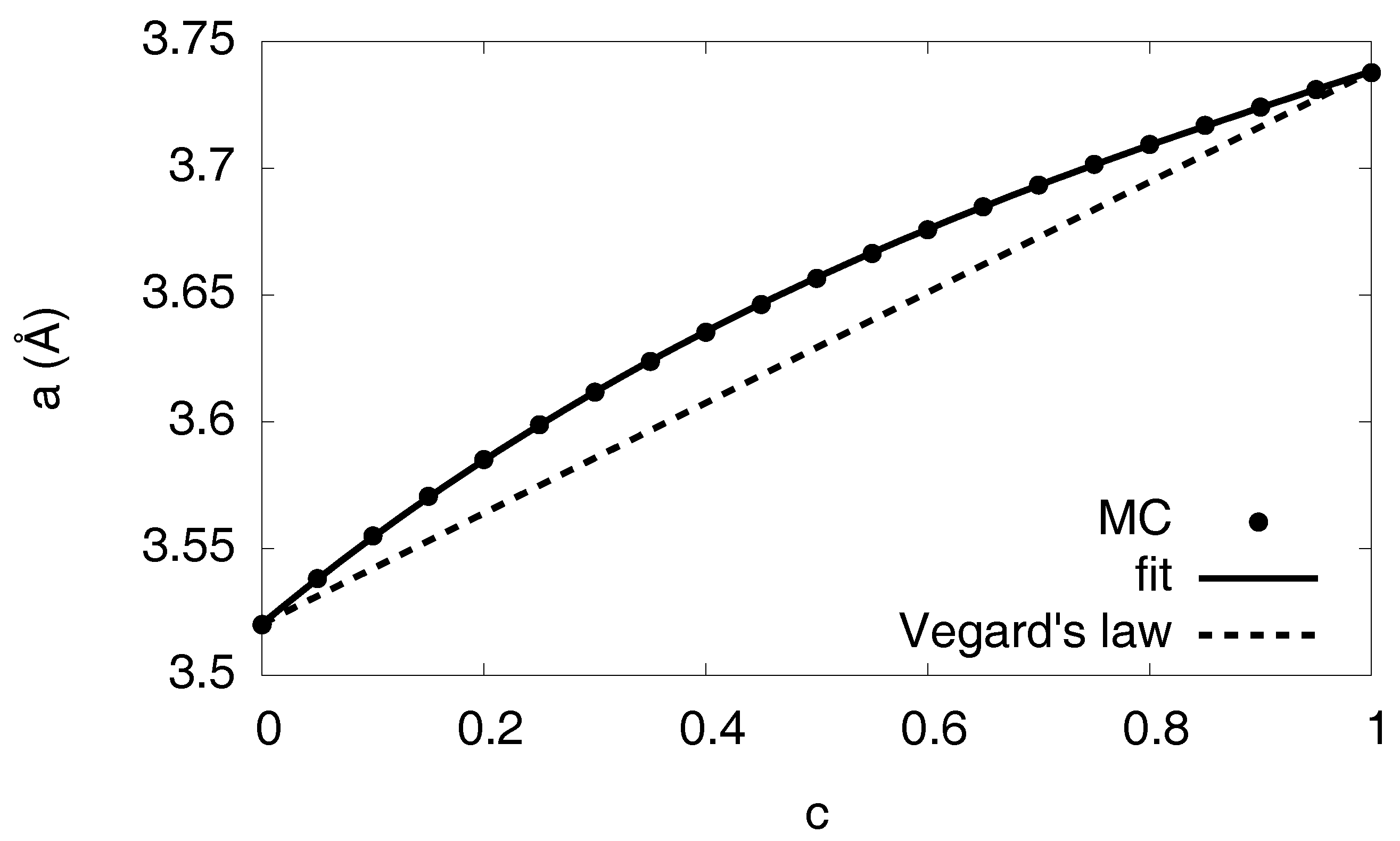
3.10. Volume Relaxation
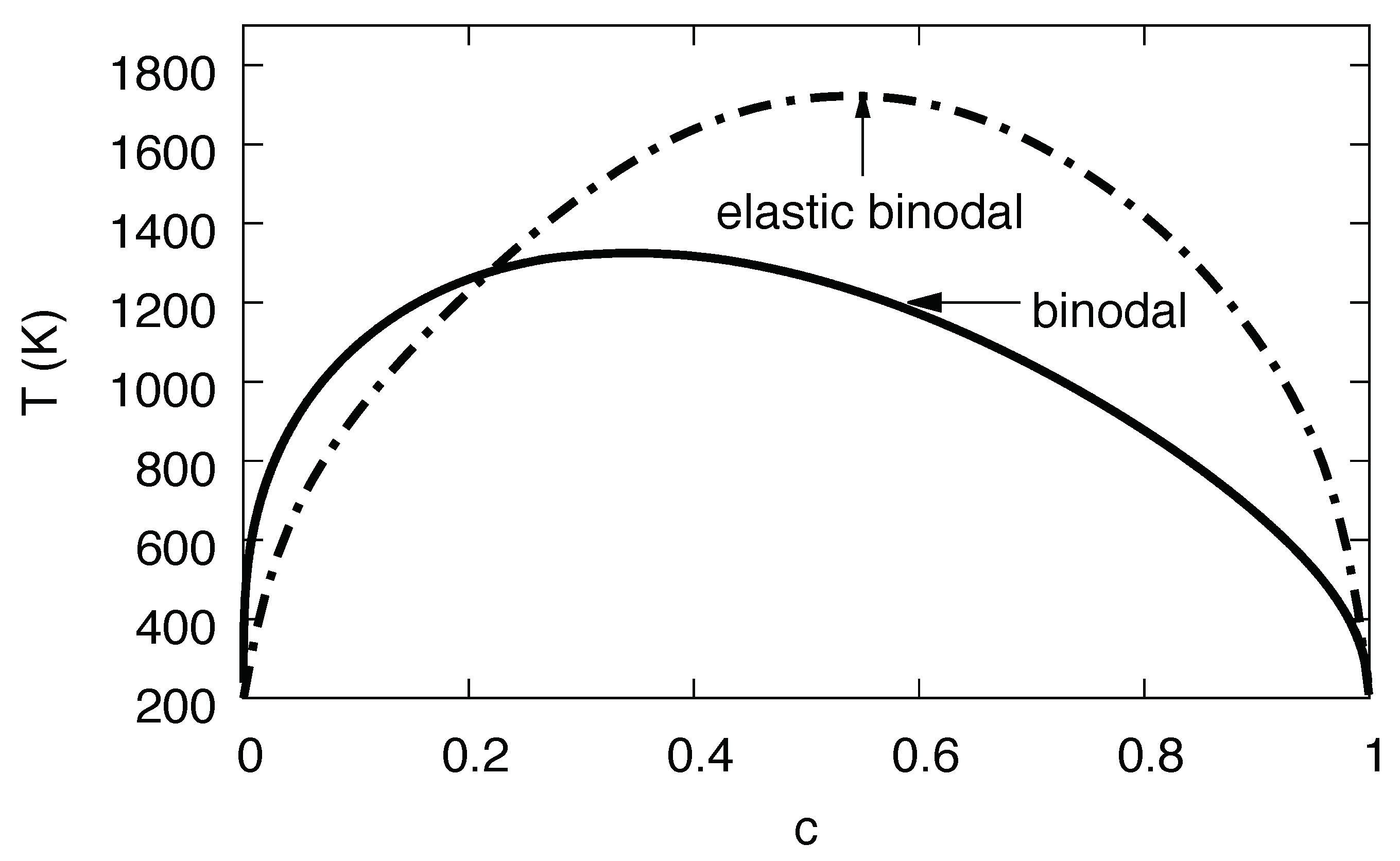
3.11. Phase Diagram
4. Discussion and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| MC | Monte Carlo |
Appendix A. Thermodynamic Framework
Appendix B. Reference State
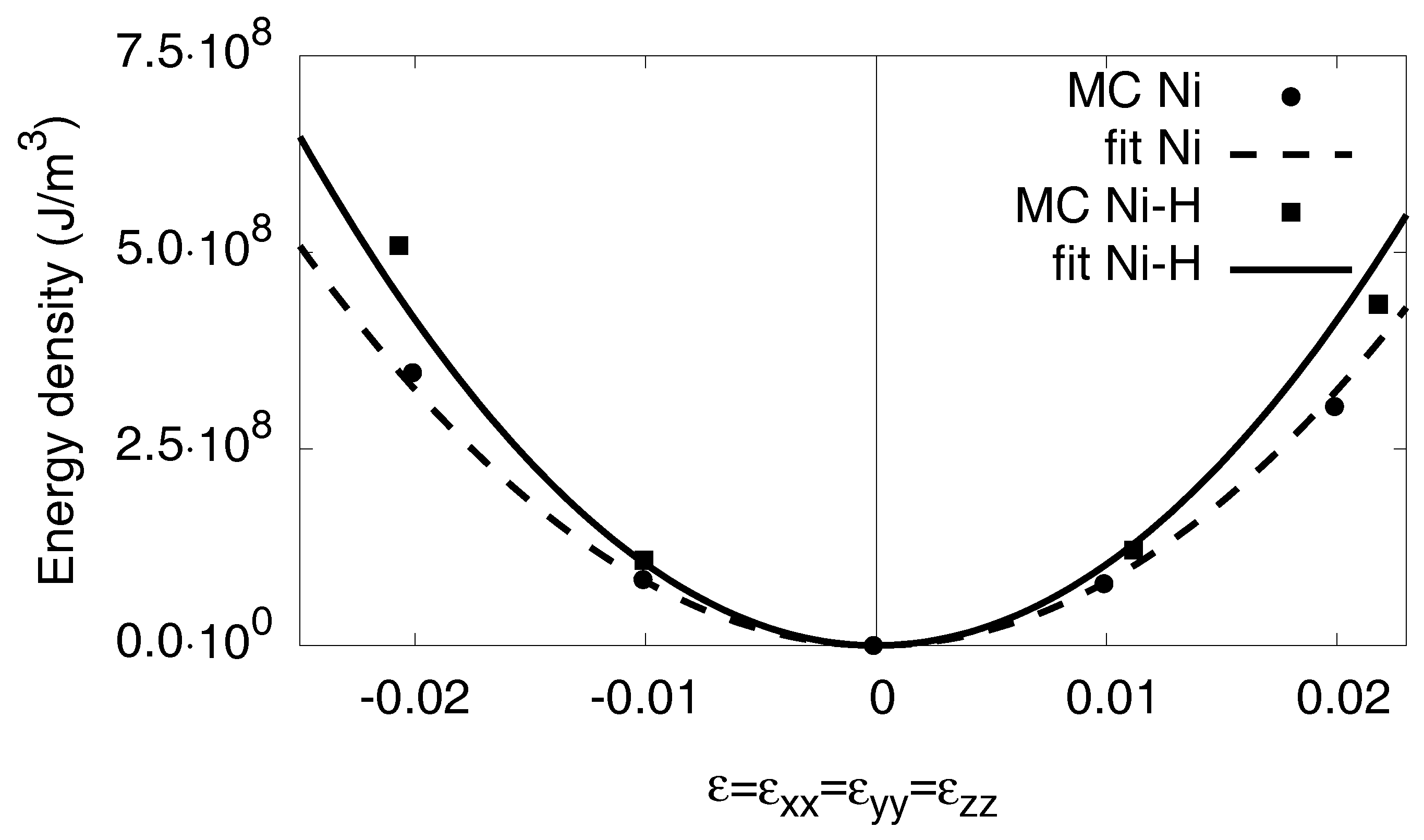
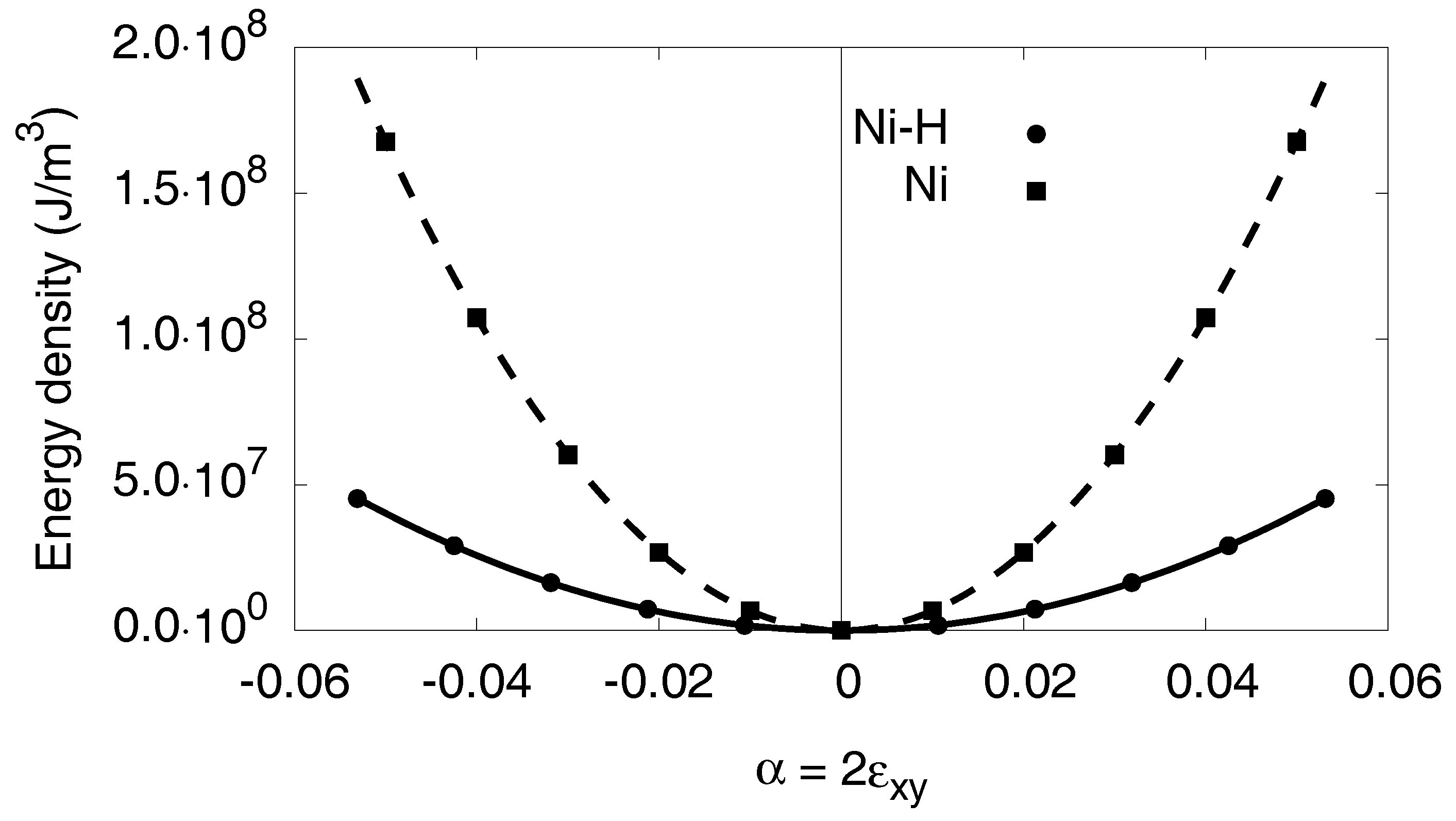
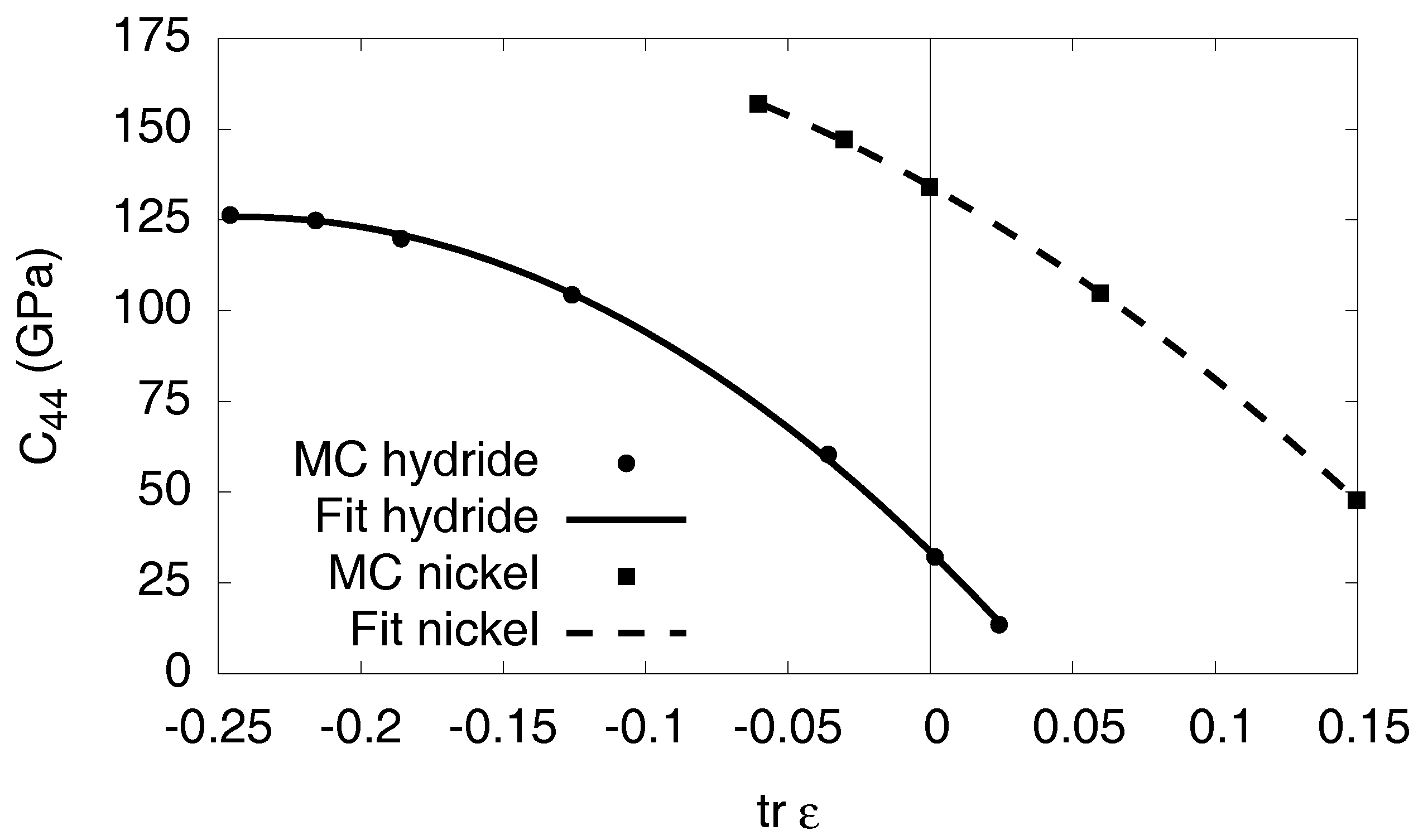
Appendix C. Elastic Constants
Appendix C.1. Bulk Modulus

Appendix C.2. Monoclinic Distortion

Appendix C.3. Orthorhombic Distortion

Appendix C.4. Nonlinear Elastic Coefficients




Appendix D. The Isotropic Model
Appendix E. Interfacial Effects

References
- Williams, R.O. The calculation of coherent phase equilibria. Calphad 1984, 8, 1–14. [Google Scholar] [CrossRef]
- Cahn, J.W.; Larché, F. A simple model for coherent equilibrium. Acta Metall. 1984, 32, 1915–1923. [Google Scholar] [CrossRef]
- Johnson, W.C.; Vorhees, P.W. Phase equilibrium in two-phase coherent solids. Metall. Trans. A 1987, 18, 1213–1228. [Google Scholar] [CrossRef]
- Von Pezold, J.; Lymperakis, L.; Neugebauer, J. Hydrogen-enhanced local plasticity at dilute bulk H concentrations: The role of H–H interactions and the formation of local hydrides. Acta Mater. 2011, 59, 2969–2980. [Google Scholar] [CrossRef]
- Haftbaradaran, H.; Song, J.; Curtin, W.A.; Gao, H. Continuum and atomistic models of strongly coupled diffusion, stress, and solute concentration. J. Power Sour. 2011, 196, 361–370. [Google Scholar] [CrossRef]
- Hüter, C.; Fu, S.; Finsterbusch, M.; Figgemeier, E.; Wells, L.; Spatschek, R. Electrode-Electrolyte Interface Stability in Solid State Electrolyte Systems: Influence of Coating Thickness under Varying Residual Stresses. AIMS Mater. Sci. 2016, 4, 867. [Google Scholar] [CrossRef]
- Markus, I.M.; Prill, M.; Dang, S.; Markus, T.; Spatschek, R.; Singheiser, L. High temperature investigation of electrochemical lithium insertion into Li4Ti5O12. Phys. Chem. Chem. Phys. 2016, 18, 31640. [Google Scholar] [CrossRef] [PubMed]
- Spatschek, R.; Gobbi, G.; Hüter, C.; Chakrabarty, A.; Aydin, U.; Brinckmann, S.; Neugebauer, J. Scale bridging description of coherent phase equilibria in the presence of surfaces and interfaces. Phys. Rev. B 2016, 94, 134106. [Google Scholar] [CrossRef]
- Shizuku, Y.; Yamamoto, S.; Fukai, Y. Phase diagram of the Ni-H system at high hydrogen pressures. J. Alloys Comp. 2002, 336, 159. [Google Scholar] [CrossRef]
- Fukai, Y.; Yamatomo, S.; Harada, S.; Kanazawa, M. The phase diagram of the Ni-H system revisited. J. Alloys Comp. 2004, 372, L4–L5. [Google Scholar] [CrossRef]
- Hüter, C.; Friák, M.; Weikamp, W.; Neugebauer, J.; Goldenfeld, N.; Svendsen, B.; Spatschek, R. Nonlinear elastic effects in phase field crystal and amplitude equations: Comparison to ab initio simulations of bcc metals and graphene. Phys. Rev. B 2016, 93, 214105. [Google Scholar] [CrossRef]
- Sadigh, B.; Erhart, P. Calculation of excess free energies of precipitates via direct thermodynamic integration across phase boundaries. Phys. Rev. B 2012, 86, 134204. [Google Scholar] [CrossRef]
- Laks, D.B.; Ferreira, L.G.; Froyen, S.; Zunger, A. Efficient cluster expansion for substitutional systems. Phys. Rev. B 1992, 46, 12587. [Google Scholar] [CrossRef]
- Kurta, R.P.; Bugaev, V.N.; Ortiz, A.D. Long-Wavelength Elastic Interactions in Complex Crystals. Phys. Rev. Lett. 2010, 104, 085502. [Google Scholar] [CrossRef] [PubMed]
- Metropolis, N.; Rosenbluth, A.W.; Rosenbluth, M.N.; Teller, A.H. Equation of State Calculations by Fast Computing Machines. J. Chem. Phys 1953, 21, 1087–1092. [Google Scholar] [CrossRef]
- Plimpton, S.J. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comp. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- LAMMPS Molecular Dynamics Simulator. Available online: http://lammps.sandia.gov (accessed on 17 April 2018).
- Angelo, J.E.; Moody, N.R.; Baskes, M.I. Trapping of hydrogen to lattice-defects in nickel. Model. Simul. Mater. Sci. Engr. 1995, 3, 289. [Google Scholar] [CrossRef]
- Baskes, M.I.; Sha, X.W.; Angelo, J.E.; Moody, N.R. Trapping of hydrogen to lattice defects in nickel. Model. Simul. Mater. Sci. Eng. 1997, 5, 651–652. [Google Scholar] [CrossRef]
- Haasen, P. Physical Metallurgy; Cambridge University Press: Cambridge, UK, 2003. [Google Scholar]
- Landau, L.D.; Lifshitz, E.M. Theory of Elasticity; Pergamon Press: Oxford, UK, 1987. [Google Scholar]
- Mura, T. Micromechanics of Defects in Solids; Springer: Berlin/Heidelberg, Germany, 1987. [Google Scholar]
- Marchenko, V.I. Theory of the equilibrium shape of crystals. Zh. Eksp. Teor. Fiz. (USSR) 1981, 81, 1141. [Google Scholar]
- Fischer, F.D.; Waitz, T.; Vollath, D.; Simha, N.K. On the role of surface energy and surface stress in phase-transforming nanoparticles. Progress Mater. Sci. 2008, 53, 481–527. [Google Scholar] [CrossRef]
- Cahn, J.W.; Larché, F. Surface Stress and Chemical Equilibrium of Small Crystals. II. Solid. Particles Embedded in a Solid Matrix. Acta Metall. 1982, 30, 51–56. [Google Scholar] [CrossRef]
- Armbruster, M.H. The Solubility of Hydrogen at Low Pressure in Iron, Nickel and Certain Steels at 400 to 600∘. J. Am. Chem. Soc. 1943, 65, 1043–1054. [Google Scholar] [CrossRef]
- Brener, E.A.; Marchenko, V.I.; Spatschek, R. Influence of strain on the kinetics of phase transitions in solids. Phys. Rev. E 2007, 75, 041604. [Google Scholar] [CrossRef] [PubMed]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ni | Ni-H | |
|---|---|---|
| 250.7 GPa | 295.3 GPa | |
| 145.5 GPa | 197.3 GPa | |
| 134.3 GPa | 33.5 GPa |
| Elastic Constant | Material | |||||
|---|---|---|---|---|---|---|
| Ni | −3.25 | −7.04 | – | – | – | |
| Ni | −2.01 | −12.51 | – | – | – | |
| K | Ni | −1.00 | 0.53 | −4.49 | 2.57 | 50.03 |
| Ni-H | −22.80 | −47.20 | – | – | – | |
| Ni-H | −9.77 | −14.69 | – | – | – | |
| K | Ni | −2.25 | 3.37 | 11.51 | 3.55 | −51.00 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korbmacher, D.; Von Pezold, J.; Brinckmann, S.; Neugebauer, J.; Hüter, C.; Spatschek, R. Modeling of Phase Equilibria in Ni-H: Bridging the Atomistic with the Continuum Scale. Metals 2018, 8, 280. https://doi.org/10.3390/met8040280
Korbmacher D, Von Pezold J, Brinckmann S, Neugebauer J, Hüter C, Spatschek R. Modeling of Phase Equilibria in Ni-H: Bridging the Atomistic with the Continuum Scale. Metals. 2018; 8(4):280. https://doi.org/10.3390/met8040280
Chicago/Turabian StyleKorbmacher, Dominique, Johann Von Pezold, Steffen Brinckmann, Jörg Neugebauer, Claas Hüter, and Robert Spatschek. 2018. "Modeling of Phase Equilibria in Ni-H: Bridging the Atomistic with the Continuum Scale" Metals 8, no. 4: 280. https://doi.org/10.3390/met8040280
APA StyleKorbmacher, D., Von Pezold, J., Brinckmann, S., Neugebauer, J., Hüter, C., & Spatschek, R. (2018). Modeling of Phase Equilibria in Ni-H: Bridging the Atomistic with the Continuum Scale. Metals, 8(4), 280. https://doi.org/10.3390/met8040280