A Heterothermic Kinetic Model of Hydrogen Absorption in Metals with Subsurface Transport



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
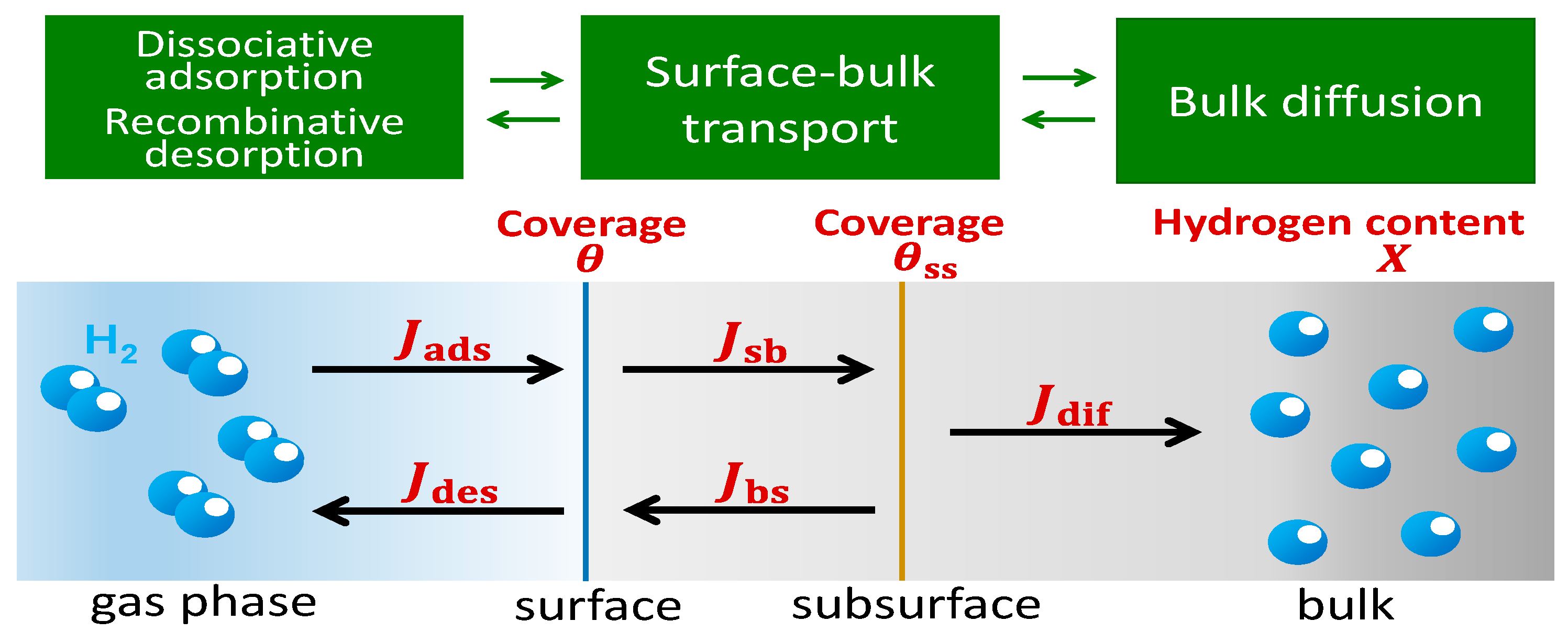
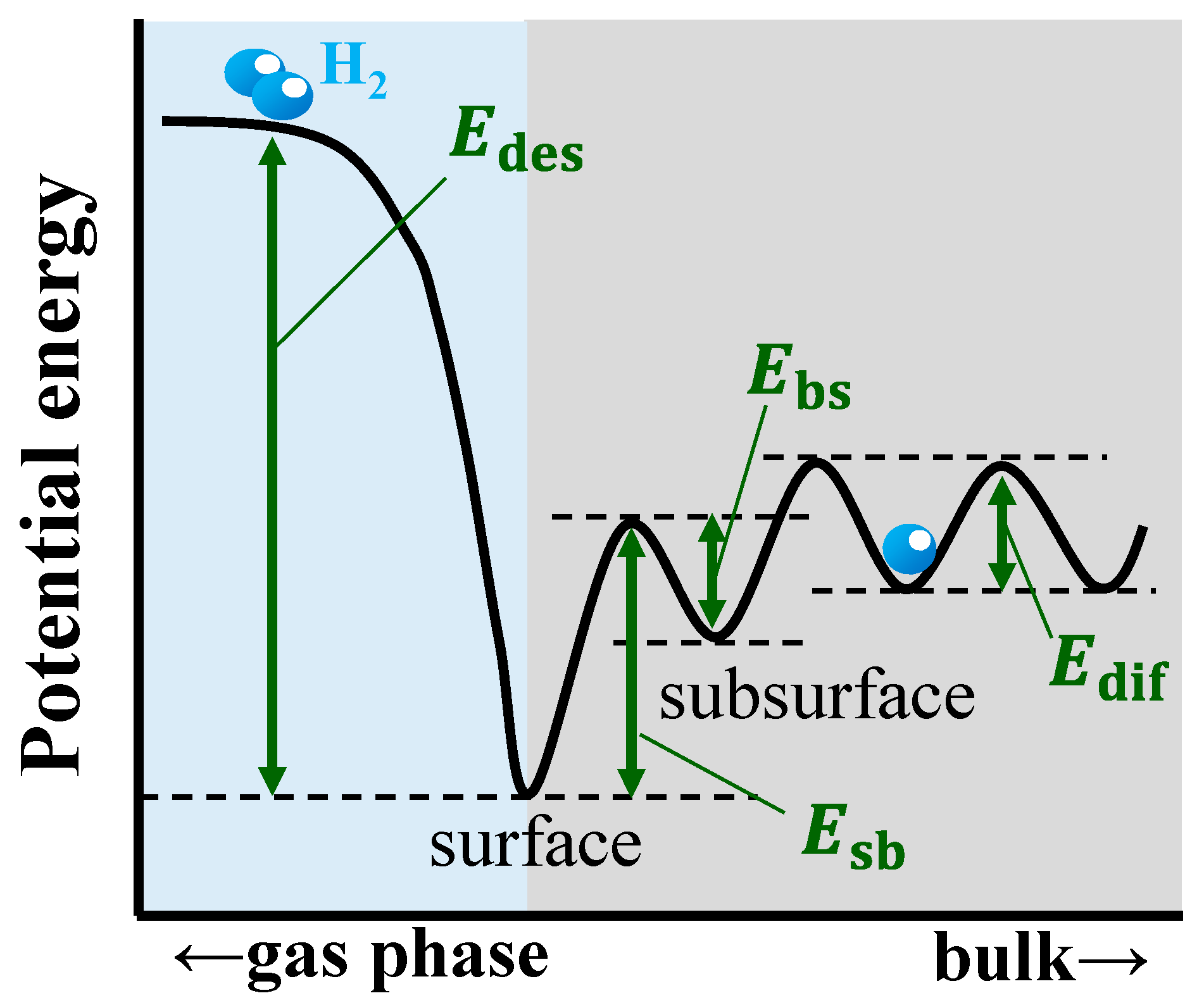
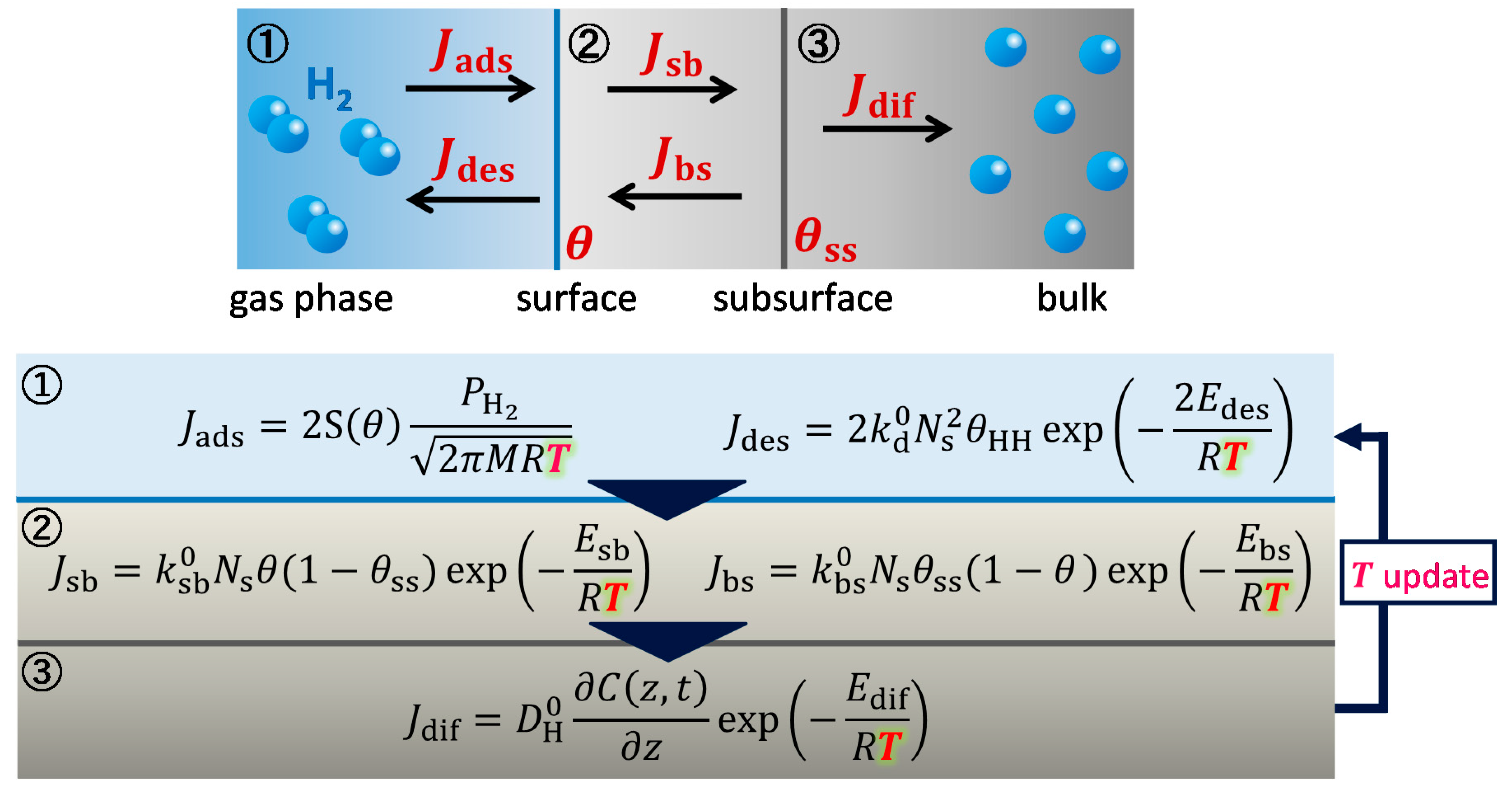
2. Theory and Calculation Methods
3. Experimental Methods
4. Results and Discussion
4.1. Hydrogen Absorption Rate at Room Temperature
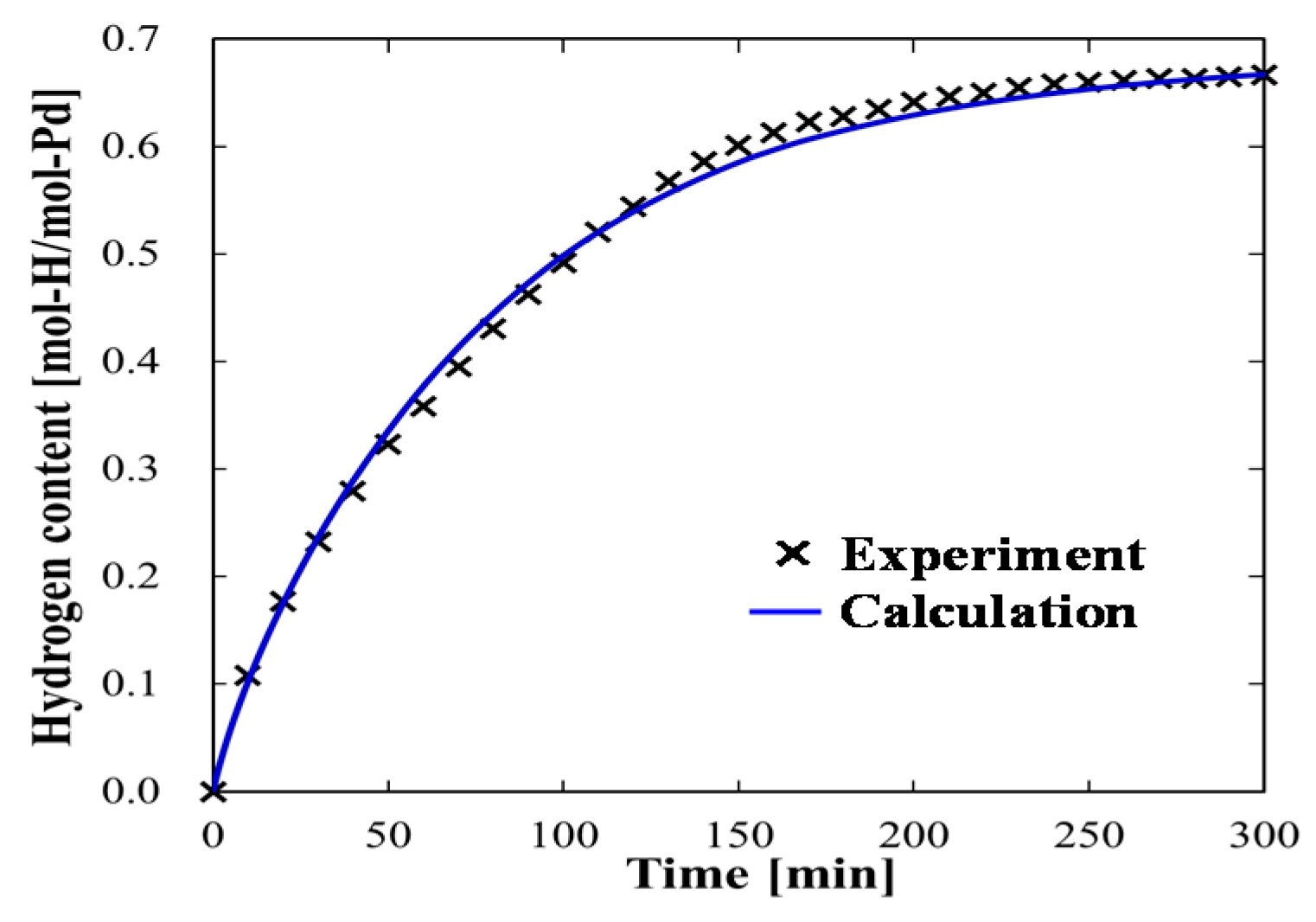
4.1.1. Experimental Data and Numerical Fitting
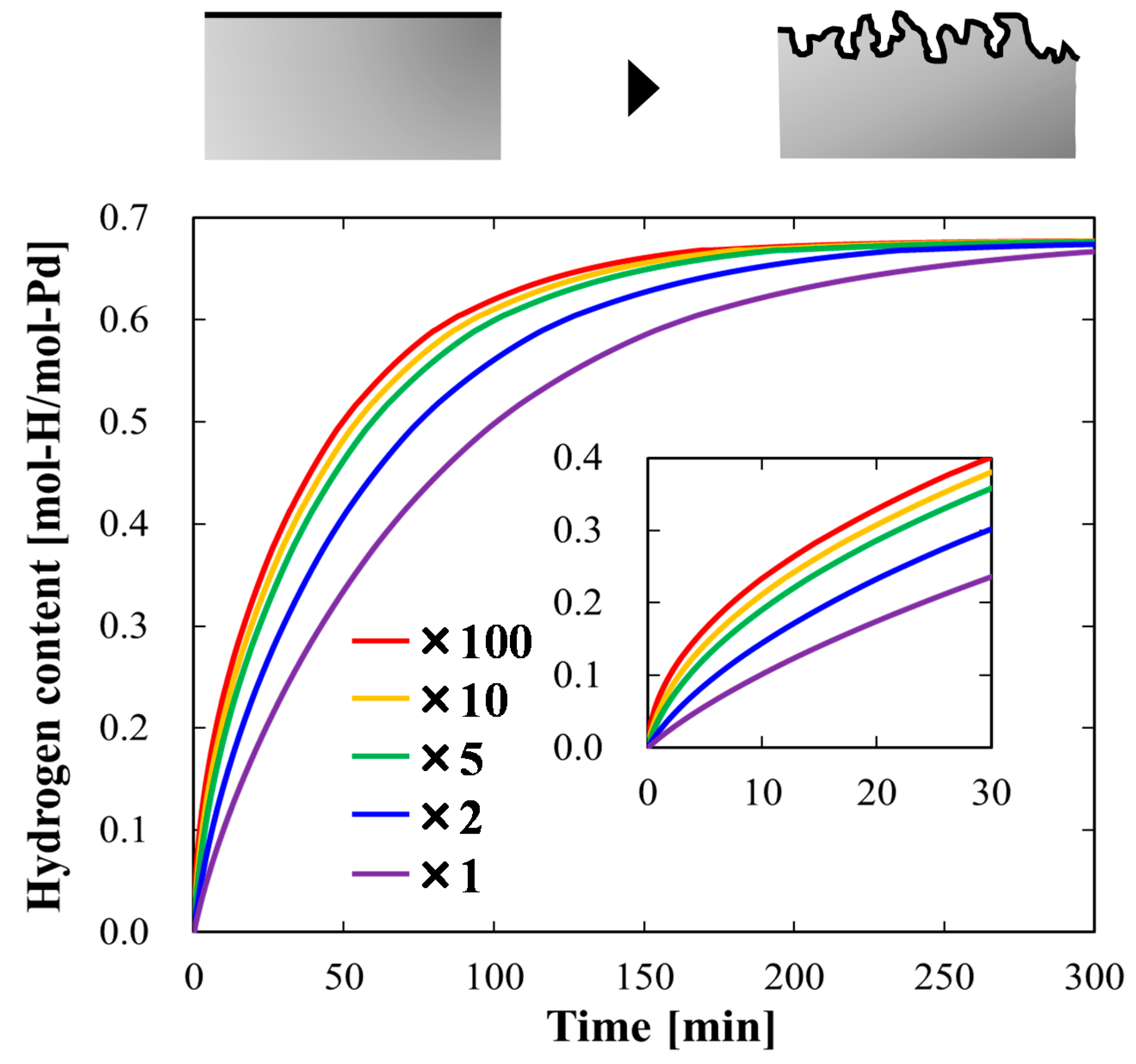
4.1.2. Case Study Simulation I: Effective Surface Area and Hydrogen Absorption Rate
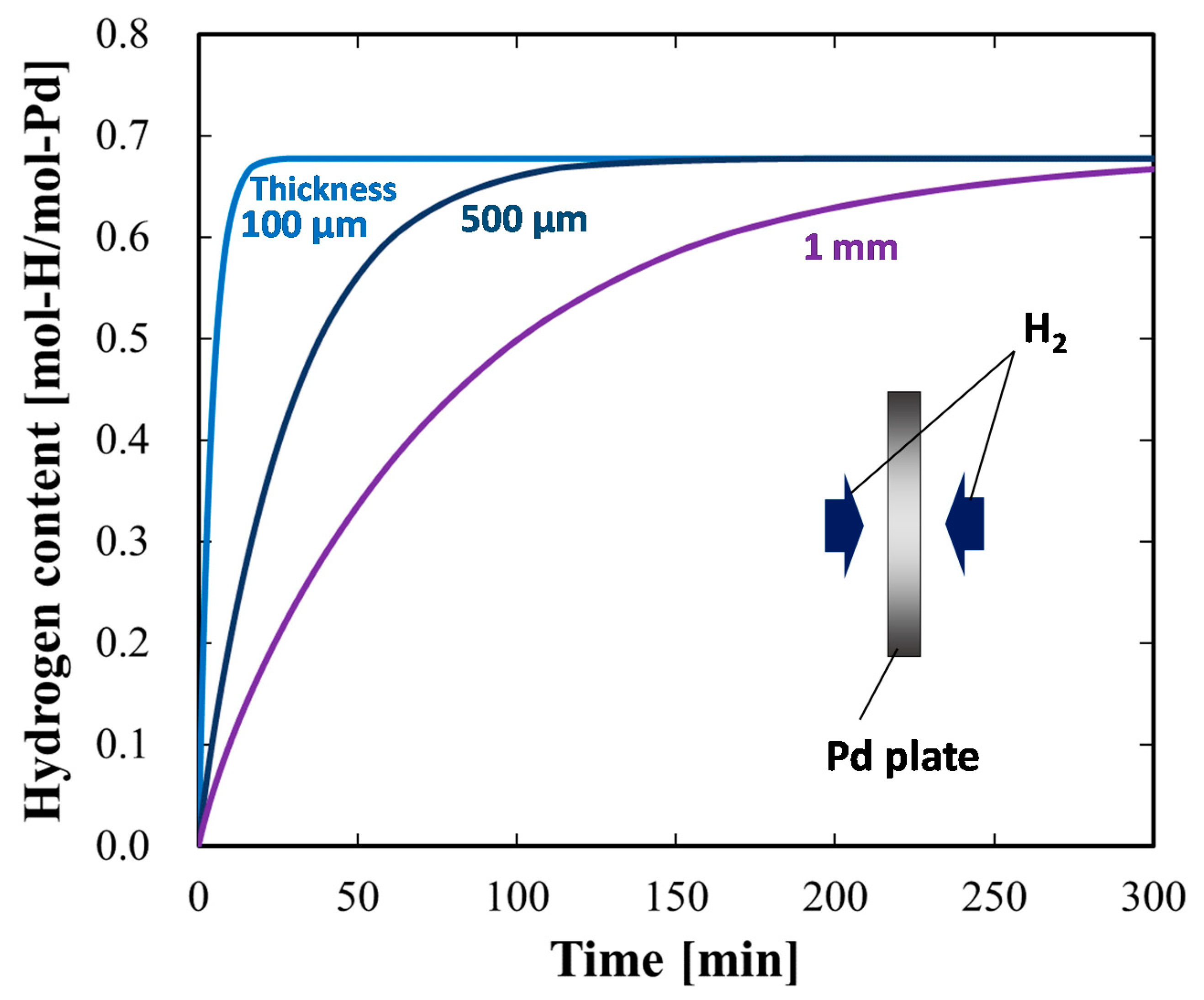
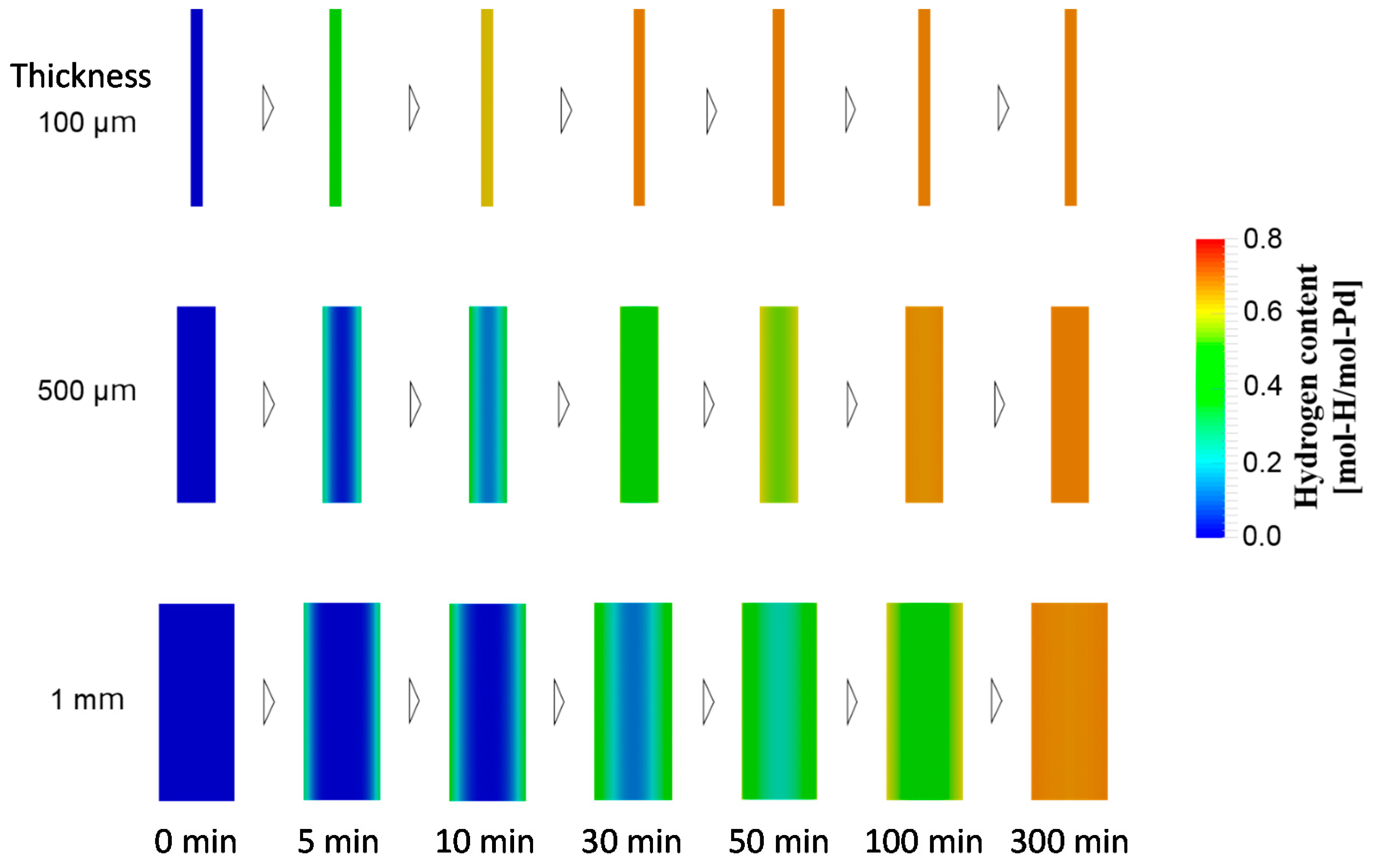
4.1.3. Case Study Simulation II: Plate Thickness and Hydrogen Absorption Rate
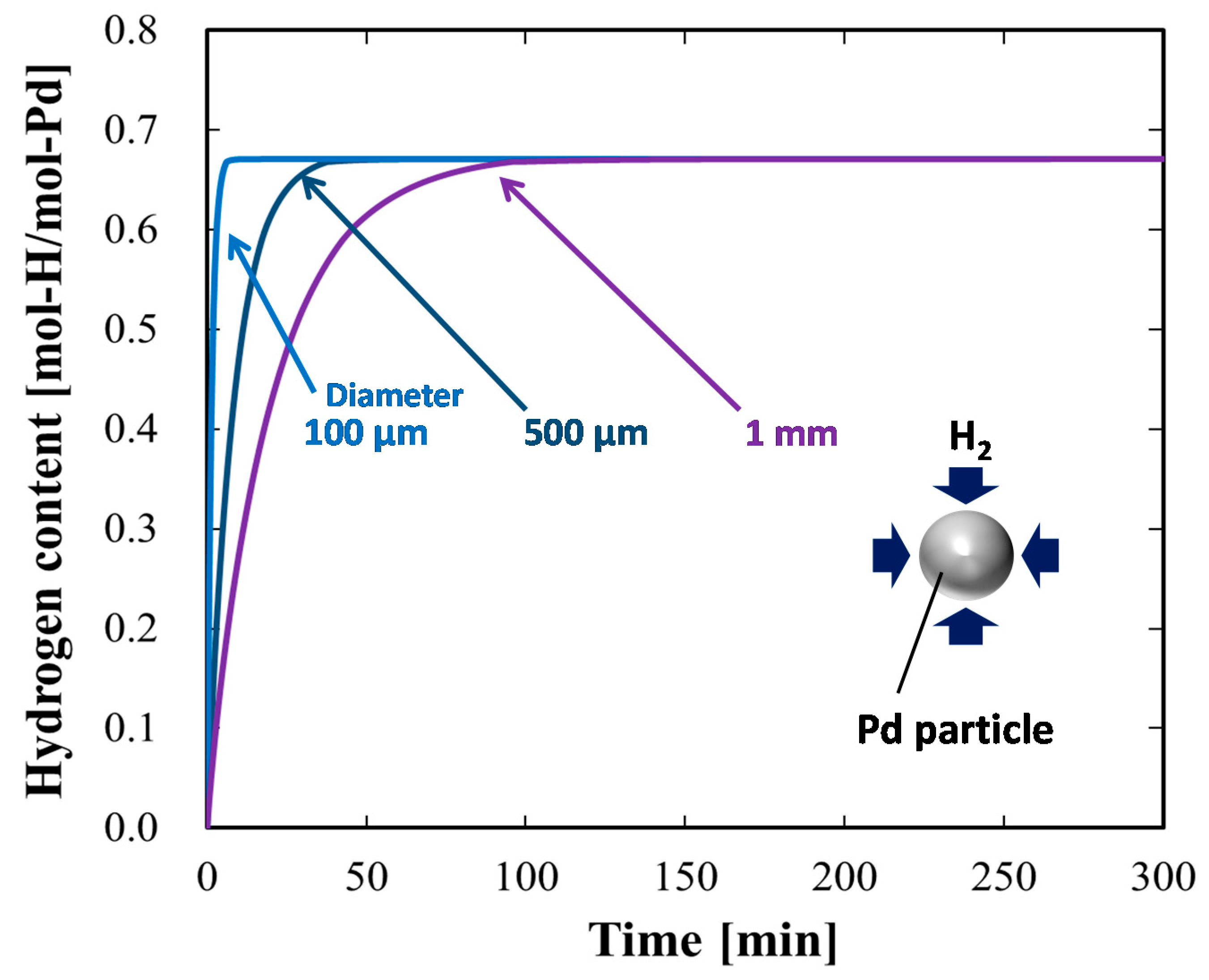
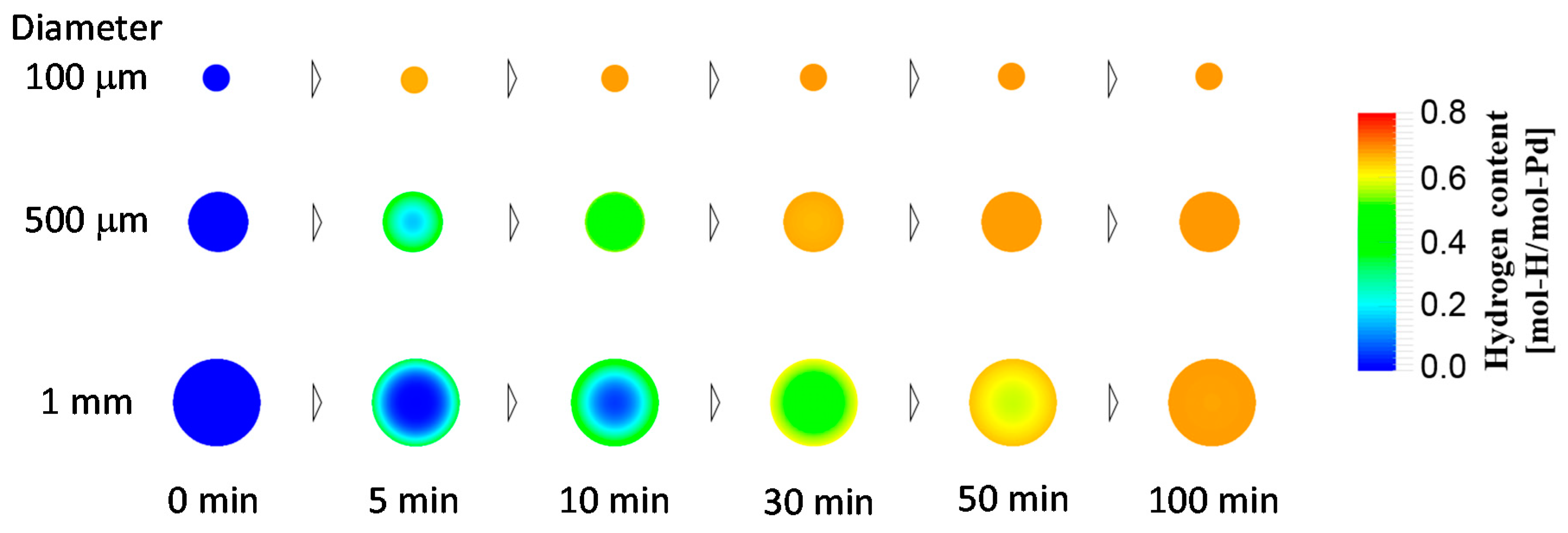
4.1.4. Case Study Simulation III: Particle Size and Hydrogen Absorption Rate
4.2. Hydrogen Absorption Rate at Higher Temperature
4.2.1. Heterothermic Model Development
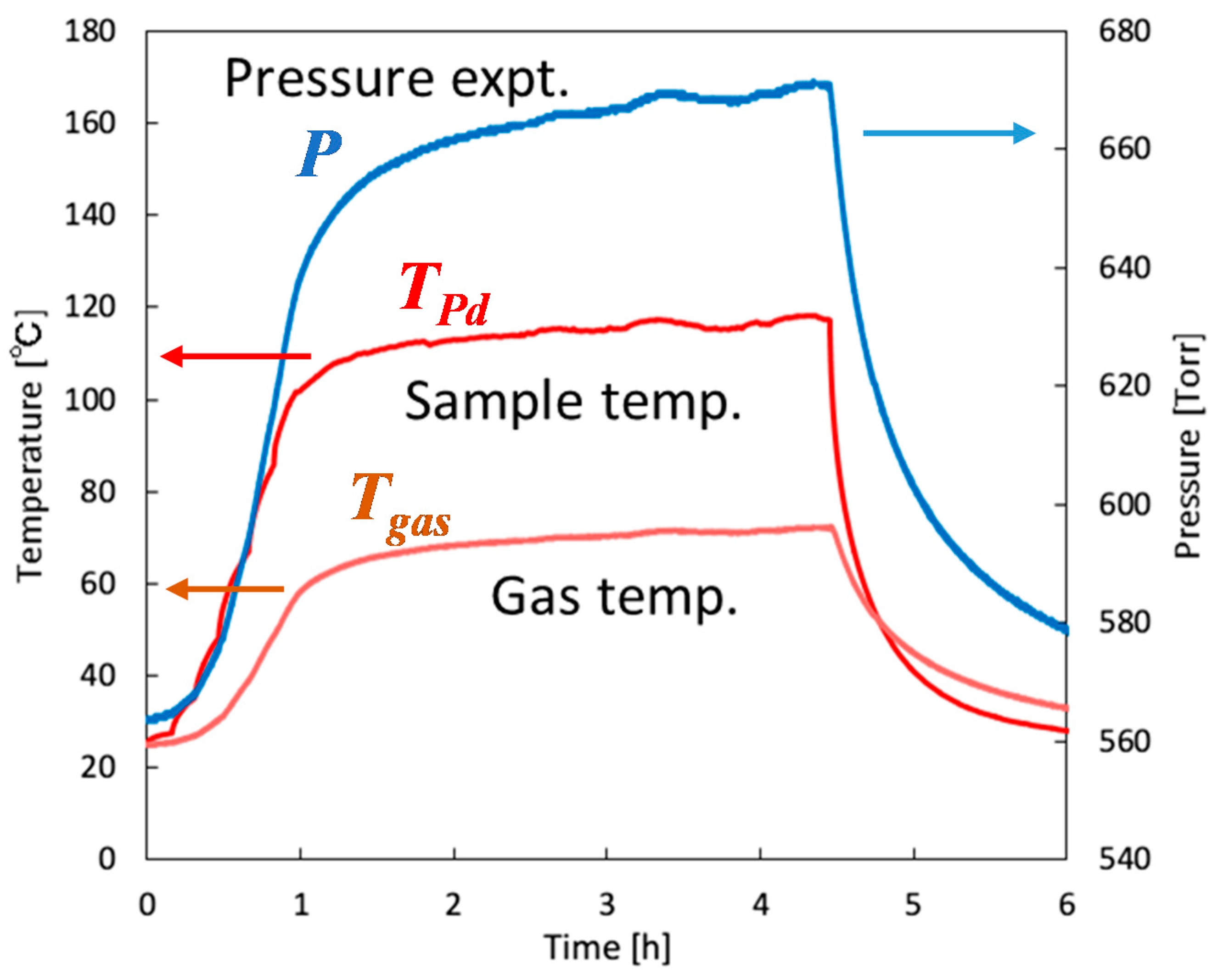
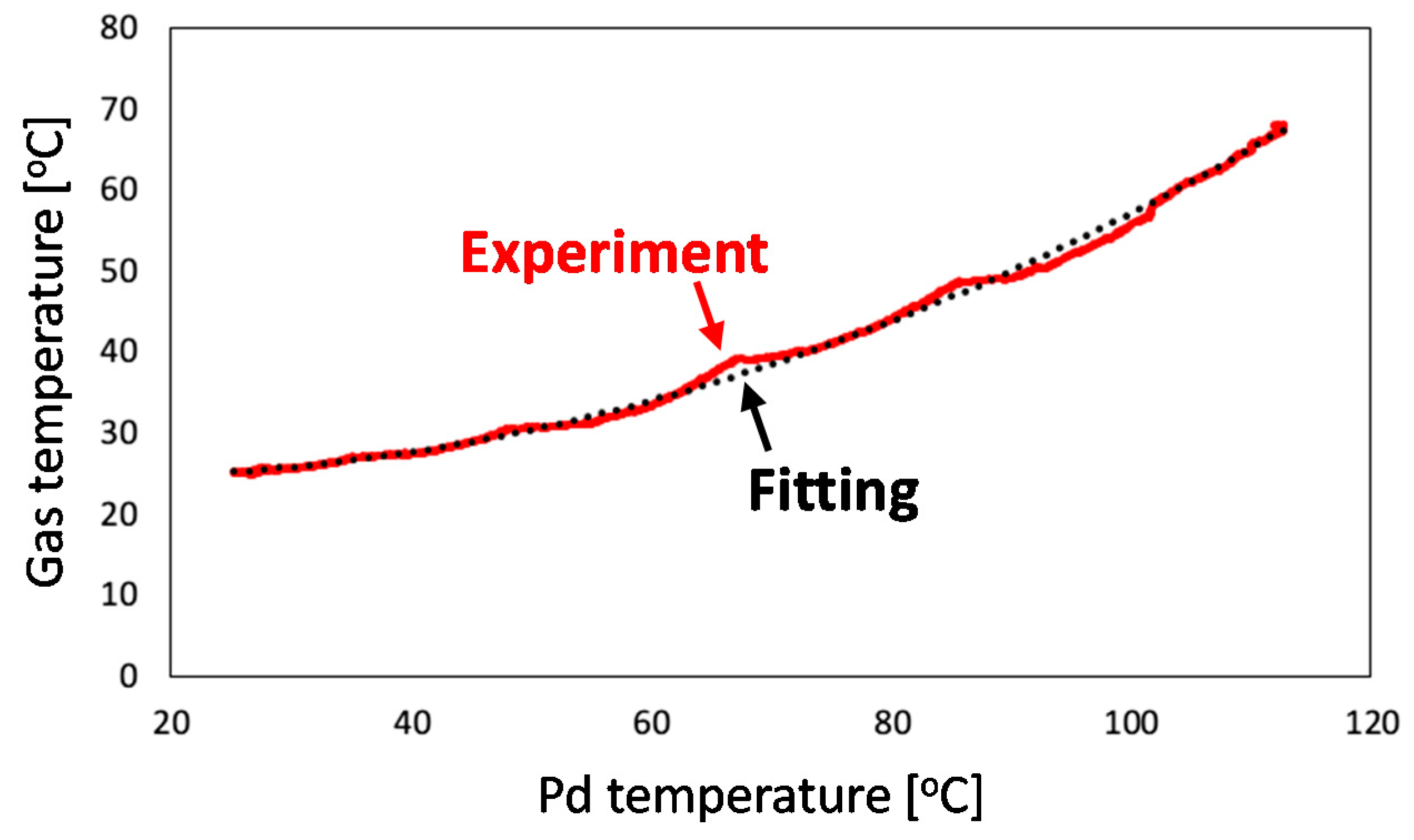
4.2.2. Calibration of Gas Phase Temperature
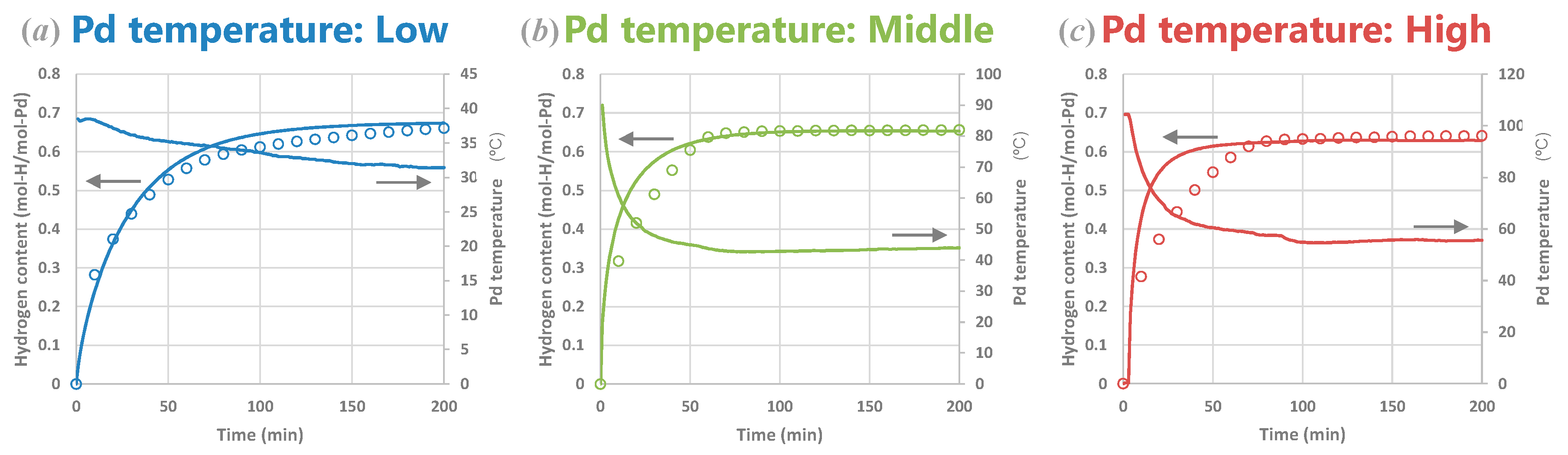
4.2.3. Experiments and Calculations at Various Temperatures
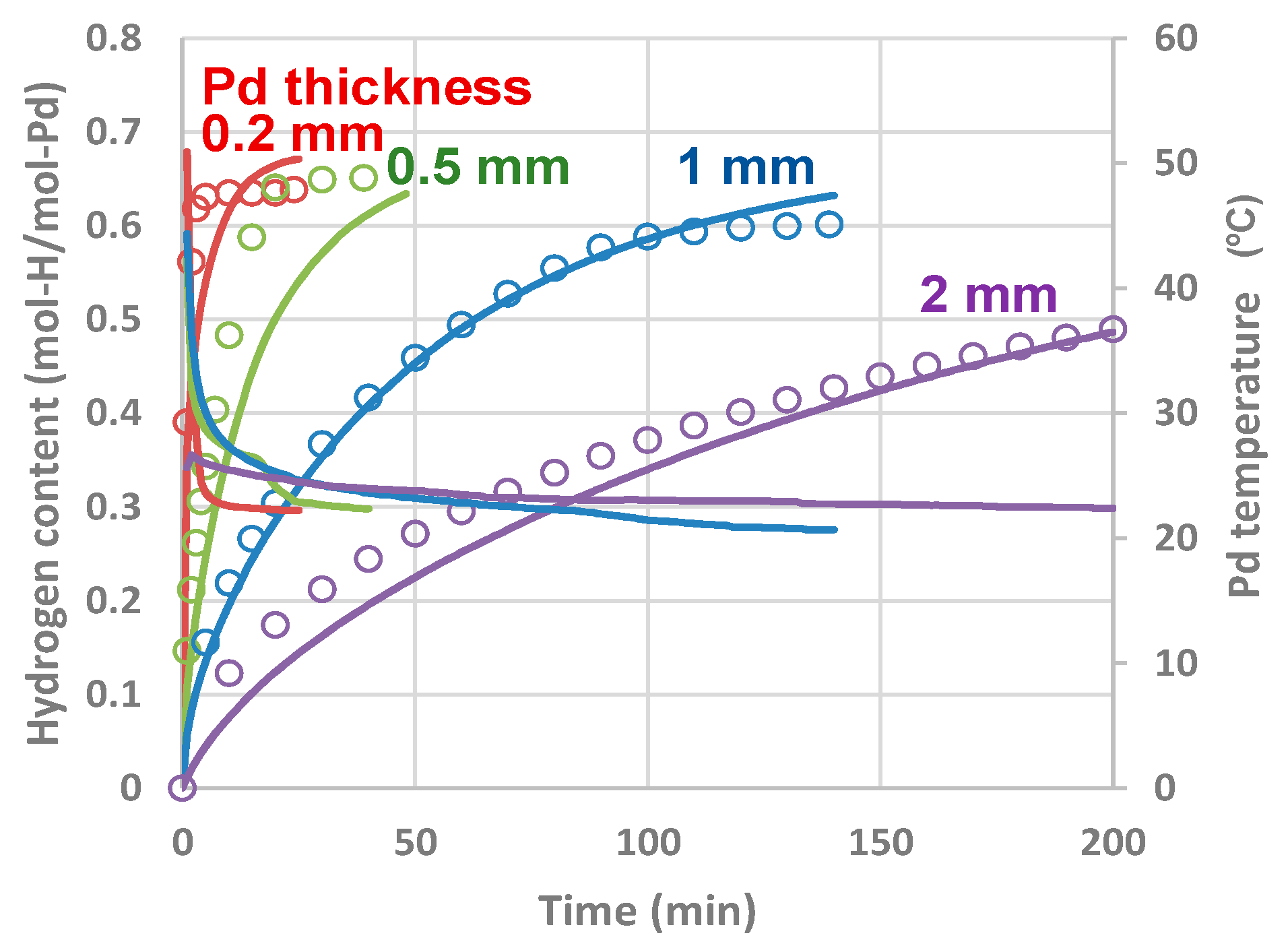
4.2.4. Experiments and Calculations for Various Metal Sizes
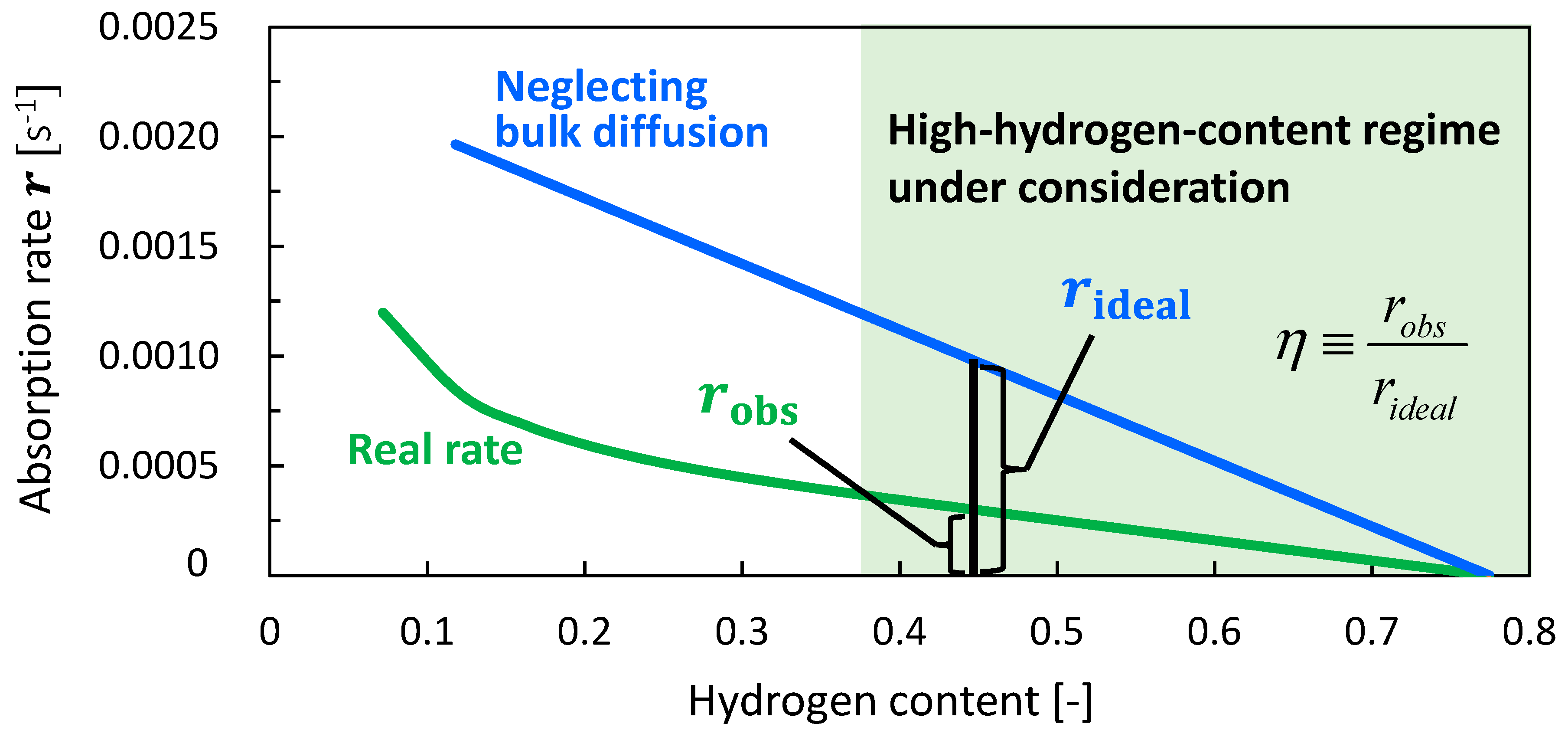
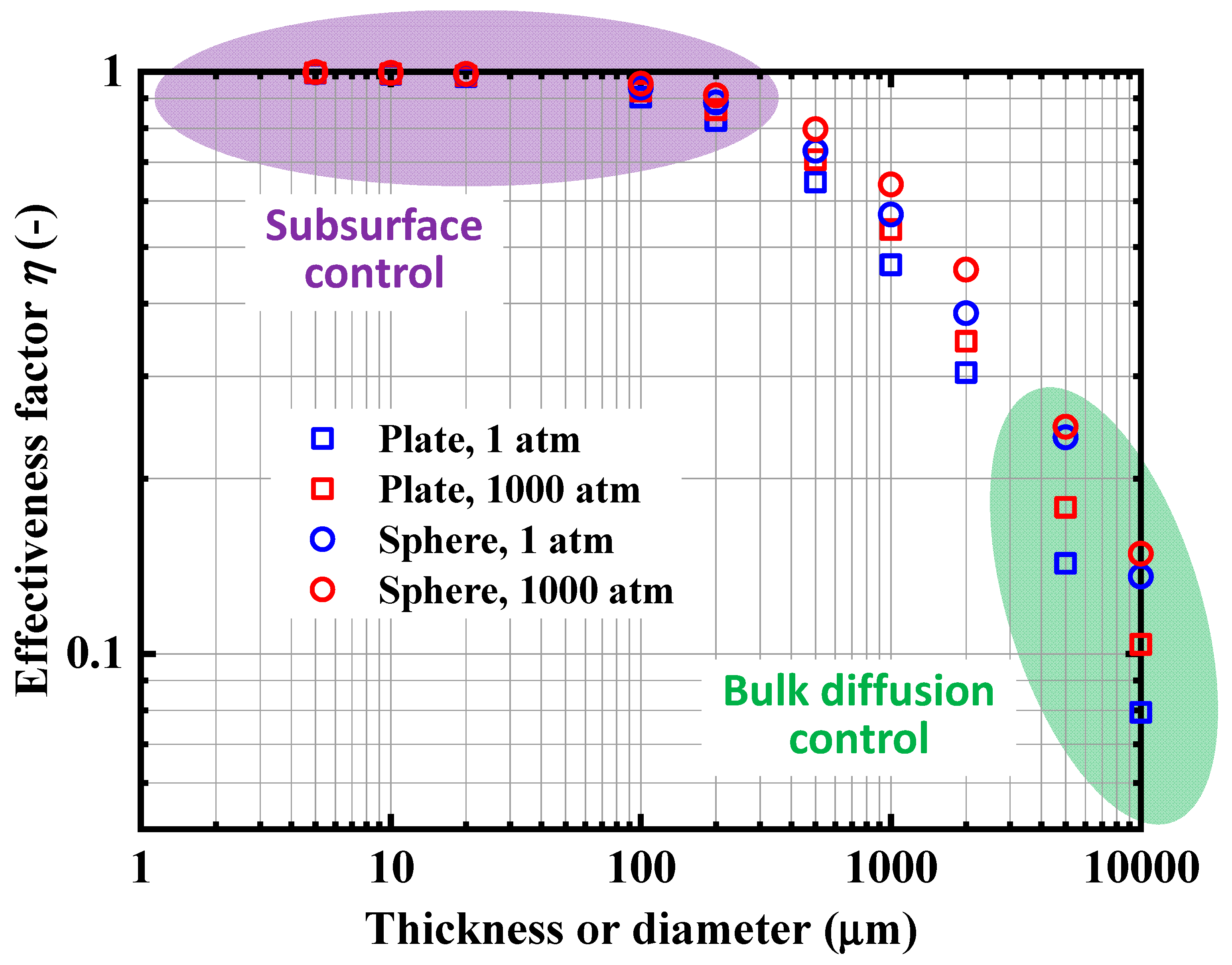
4.3. Analysis of Rate-Determining Steps
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schlapbach, L.; Zuttel, A. Hydrogen-storage materials for mobile applications. Nature 2001, 414, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Ley, M.B.; Jepsen, L.H.; Lee, Y.S.; Cho, Y.W.; von Colbe, J.M.B.; Dornheim, M.; Rokni, M.; Jensen, J.O.; Sloth, M.; Filinchuk, Y.; et al. Complex hydrides for hydrogen storage—New perspectives. Mater. Today 2014, 17, 122–128. [Google Scholar] [CrossRef]
- Mohtadi, R.; Orimo, S. The renaissance of hydrides as energy materials. Nat. Rev. Mater. 2016, 2, 16091. [Google Scholar] [CrossRef]
- Behm, R.J.; Penka, V.; Cattania, M.G.; Christmann, K.; Ertl, G. Evidence for “subsurface” hydrogen on Pd(100): An intermediate between chemisorbed and dissolved species. J. Chem. Phys. 1983, 78, 7486–7490. [Google Scholar] [CrossRef]
- Wilde, M.; Fukutani, K. Penetration mechanisms of surface-adsorbed hydrogen atoms into bulk metals: Experiment and model. Phys. Rev. B 2008, 78, 115411. [Google Scholar] [CrossRef]
- Greeley, J.; Mavrikakis, M. Surface and subsurface hydrogen: Adsorption properties on transition metals and near-surface alloys. J. Phys. Chem. B 2005, 109, 3460–3471. [Google Scholar] [CrossRef]
- Christmann, K. Interaction of hydrogen with solid surfaces. Surf. Sci. Rep. 1988, 9, 1–163. [Google Scholar] [CrossRef]
- Dino, W.A.; Kasai, H.; Okiji, A. Orientational effects in dissociative adsorption/associative desorption dynamics of H2 (D2) on Cu and Pd. Prog. Surf. Sci. 2000, 63, 63–134. [Google Scholar] [CrossRef]
- Pundt, A.; Kirchheim, R. Hydrogen in metals: Microstructural aspects. Annu. Rev. Mater. Res. 2006, 36, 555–608. [Google Scholar] [CrossRef]
- Du, A.J.; Smith, S.C.; Yao, X.D.; Lu, G.Q. Hydrogen spillover mechanism on a Pd-doped Mg surface as revealed by ab initio density functional calculation. J. Am. Chem. Soc. 2007, 129, 10201–10204. [Google Scholar] [CrossRef]
- Firmino, T.; Marquardt, R.; Gatti, F.; Dong, W. Diffusion rates for hydrogen on Pd(111) from molecular quantum dynamics calculations. J. Phys. Chem. Lett. 2014, 5, 4270–4274. [Google Scholar] [CrossRef] [PubMed]
- Fukutani, K.; Wilde, M.; Ogura, S. Nuclear dynamics and electronic effects of hydrogen on solid surfaces. Chem. Rec. 2017, 17, 233–249. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.F.; Flanagan, T.B. An investigation of the dynamic equilibrium between chemisorbed and absorbed hydrogen in the palladium/hydrogen system. J. Phys. Chem. 1973, 77, 2628–2634. [Google Scholar] [CrossRef]
- Conrad, H.; Ertl, G.; Latta, E.E. Adsorption of hydrogen on palladium single crystal surfaces. Surf. Sci. 1974, 41, 435–446. [Google Scholar] [CrossRef]
- Wagner, S.; Pundt, A. Quasi-thermodynamic model on hydride formation in palladium-hydrogen thin films: Impact of elastic and microstructural constraints. Int. J. Hydrogen Energy 2016, 41, 2727–2738. [Google Scholar] [CrossRef]
- Huot, J.; Liang, G.; Boily, S.; Van Neste, A.; Schulz, R. Structural study and hydrogen sorption kinetics of ball-milled magnesium hydride. J. Alloys Compd. 1999, 293, 495–500. [Google Scholar] [CrossRef]
- Pozzo, M.; Alfe, D. Hydrogen dissociation and diffusion on transition metal (= Ti, Zr, V, Fe, Ru, Co, Rh, Ni, Pd, Cu, Ag)-doped Mg(0001) surfaces. Int. J. Hydrogen Energy 2009, 34, 1922–1930. [Google Scholar] [CrossRef]
- Evard, E.; Gabis, I.; Yartys, V.A. Kinetics of hydrogen evolution from MgH2: Experimental studies, mechanism and modelling. Int. J. Hydrogen Energy 2010, 35, 9060–9069. [Google Scholar] [CrossRef]
- Ward, T.L.; Dao, T. Model of hydrogen permeation behavior in palladium membranes. J. Membr. Sci. 1999, 153, 211–231. [Google Scholar] [CrossRef]
- Bhargav, A.; Jackson, G.S.; Ciora, R.J.; Liu, P.T.K. Model development and validation of hydrogen transport through supported palladium membranes. J. Membr. Sci. 2010, 356, 123–132. [Google Scholar] [CrossRef]
- Behm, R.J.; Christmann, K.; Ertl, G. Adsorption of hydrogen on Pd(100). Surf. Sci. 1980, 99, 320–340. [Google Scholar] [CrossRef]
- Ali-Khan, I.; Dietz, K.J.; Waelbroeck, F.G.; Wienhold, P. The rate of hydrogen release out of clean metallic surfaces. J. Nucl. Mater. 1978, 76, 337–343. [Google Scholar] [CrossRef]
- Kitagawa, Y.; Tanabe, K. Development of a kinetic model of hydrogen absorption and desorption in magnesium and analysis of the rate-determining step. Chem. Phys. Lett. 2018, 699, 132–138. [Google Scholar] [CrossRef]
- Tanabe, K. Modeling of hydrogen/deuterium dynamics and heat generation on palladium nanoparticles for hydrogen storage and solid-state nuclear fusion. Heliyon 2016, 2, e00057. [Google Scholar] [CrossRef]
- Frieske, H.; Wicke, E. Magnetic susceptibility and equilibrium diagram of PdHn. Ber. Der Bunsen-Ges. Fur Phys. Chem. 1973, 77, 48–52. [Google Scholar] [CrossRef]















© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ono, S.; Uchikoshi, T.; Hayashi, Y.; Kitagawa, Y.; Yeh, G.; Yamaguchi, E.; Tanabe, K. A Heterothermic Kinetic Model of Hydrogen Absorption in Metals with Subsurface Transport. Metals 2019, 9, 1131. https://doi.org/10.3390/met9101131
Ono S, Uchikoshi T, Hayashi Y, Kitagawa Y, Yeh G, Yamaguchi E, Tanabe K. A Heterothermic Kinetic Model of Hydrogen Absorption in Metals with Subsurface Transport. Metals. 2019; 9(10):1131. https://doi.org/10.3390/met9101131
Chicago/Turabian StyleOno, Shunsuke, Takeru Uchikoshi, Yusuke Hayashi, Yuta Kitagawa, George Yeh, Eiichi Yamaguchi, and Katsuaki Tanabe. 2019. "A Heterothermic Kinetic Model of Hydrogen Absorption in Metals with Subsurface Transport" Metals 9, no. 10: 1131. https://doi.org/10.3390/met9101131
APA StyleOno, S., Uchikoshi, T., Hayashi, Y., Kitagawa, Y., Yeh, G., Yamaguchi, E., & Tanabe, K. (2019). A Heterothermic Kinetic Model of Hydrogen Absorption in Metals with Subsurface Transport. Metals, 9(10), 1131. https://doi.org/10.3390/met9101131