Effect of the Process Parameters on the Energy Transfer during the Synthesis of the 2LiBH4-MgH2 Reactive Hydride Composite for Hydrogen Storage

 ,
,  ,
,  ,
, 
 ,
, 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Material Preparation
2.2. Characterizations
2.2.1. Sievert Apparatus
2.2.2. X-ray Diffraction
2.2.3. Physisorption Analyzer
2.2.4. Scanning Electron Microscopy
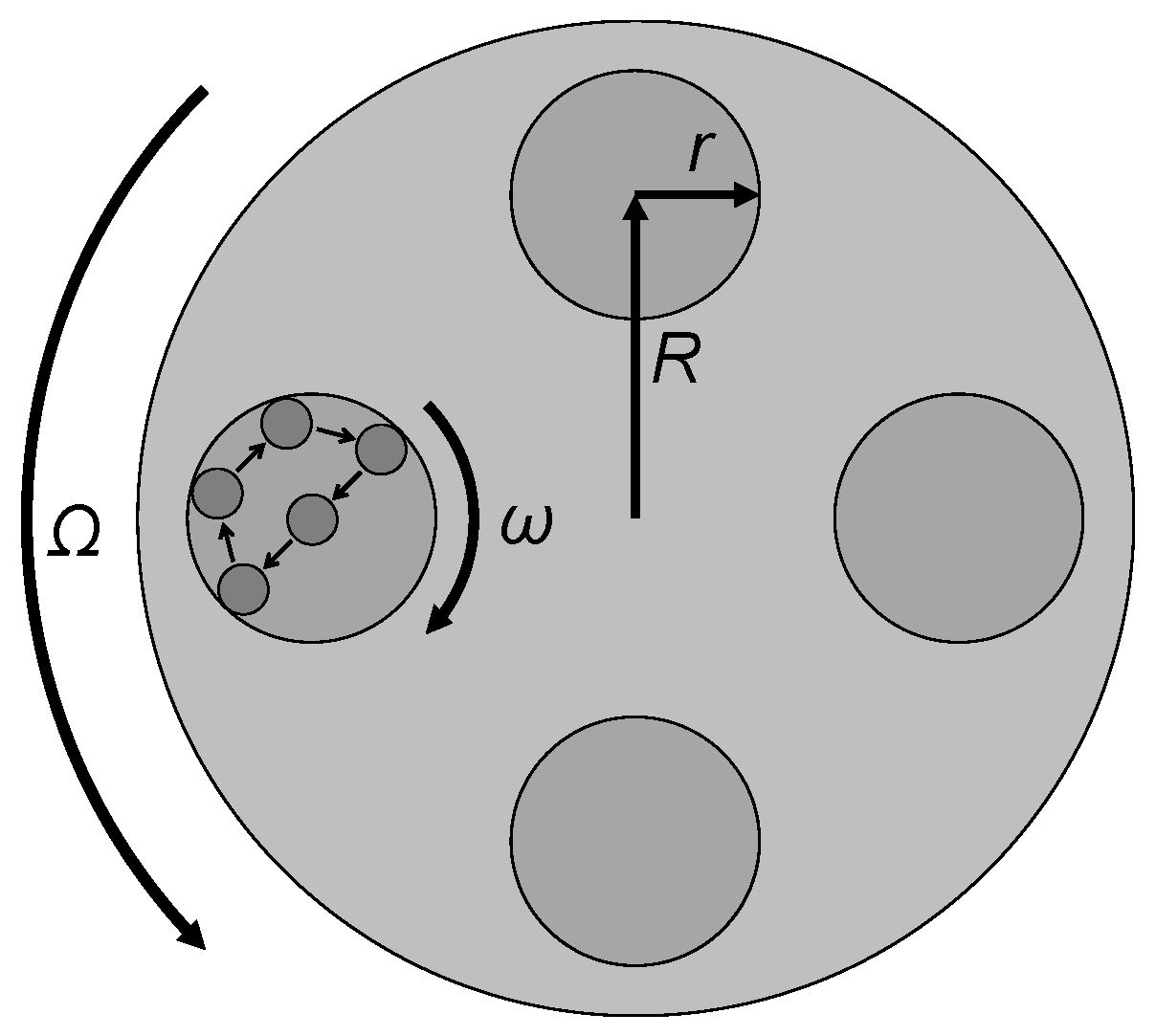
2.3. Modeling for Energy Transfer Quantification
- φb Milling balls interaction (yield coefficient);
- Ωp Plate absolute angular velocity;
- ωv Vial absolute angular velocity;
- db Ball diameter;
- mb Ball mass;
- Nb Ball number;
- P* Total transferred power per unit mass (from the milling process to the material);
- PW Total weight of the powder;
- Rp Vector distance from the mill center to the vial center;
- rv Vector distance from the vial center to the vial wall;
- t Total time of milling.
3. Results
4. Discussion
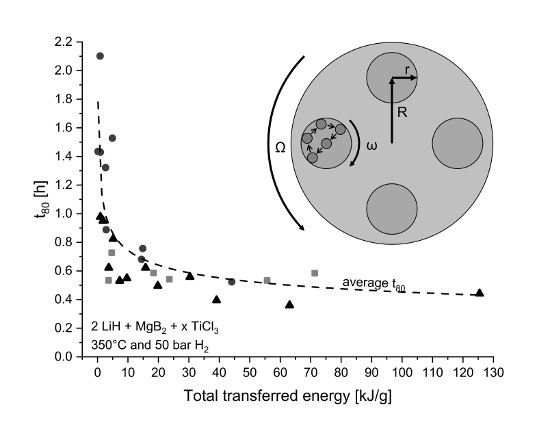
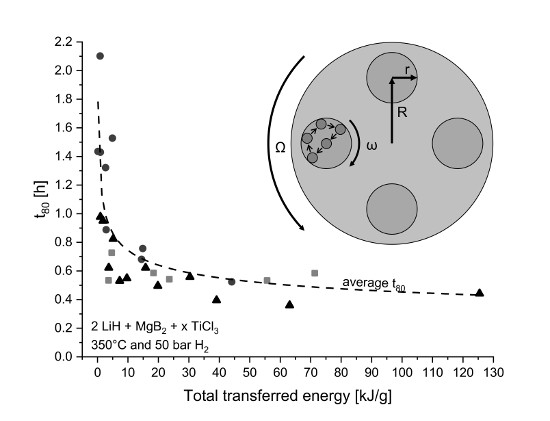
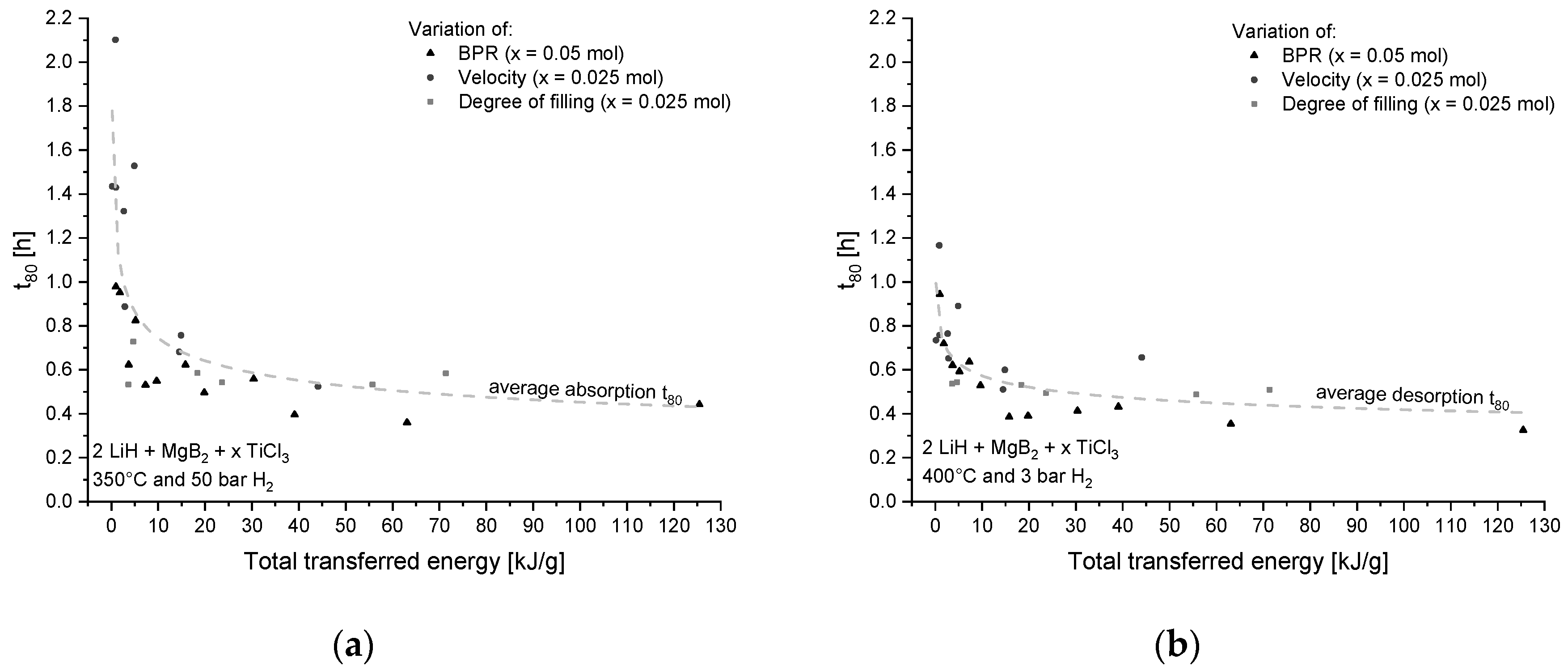
4.1. Transferred Energy During Milling
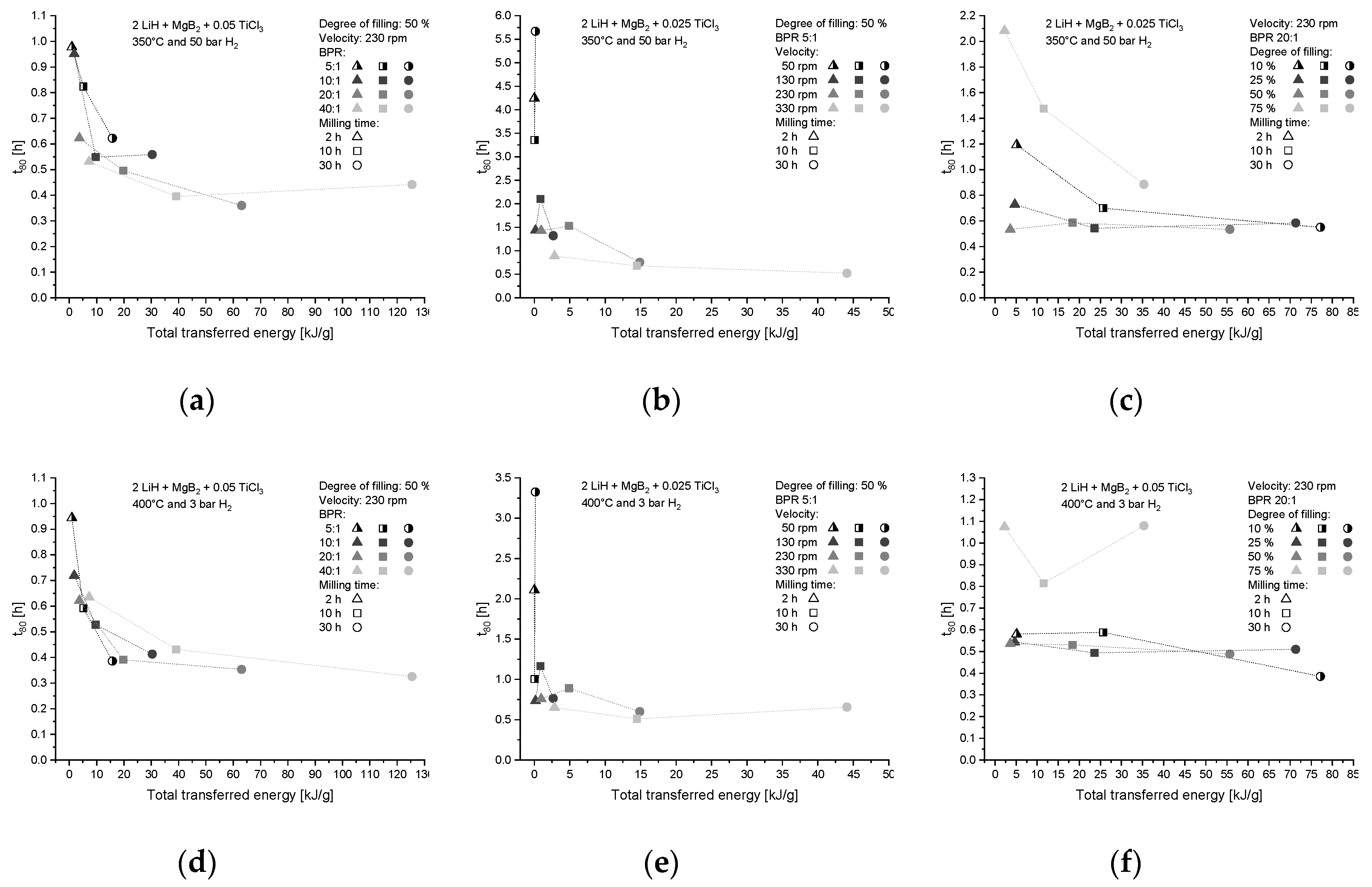
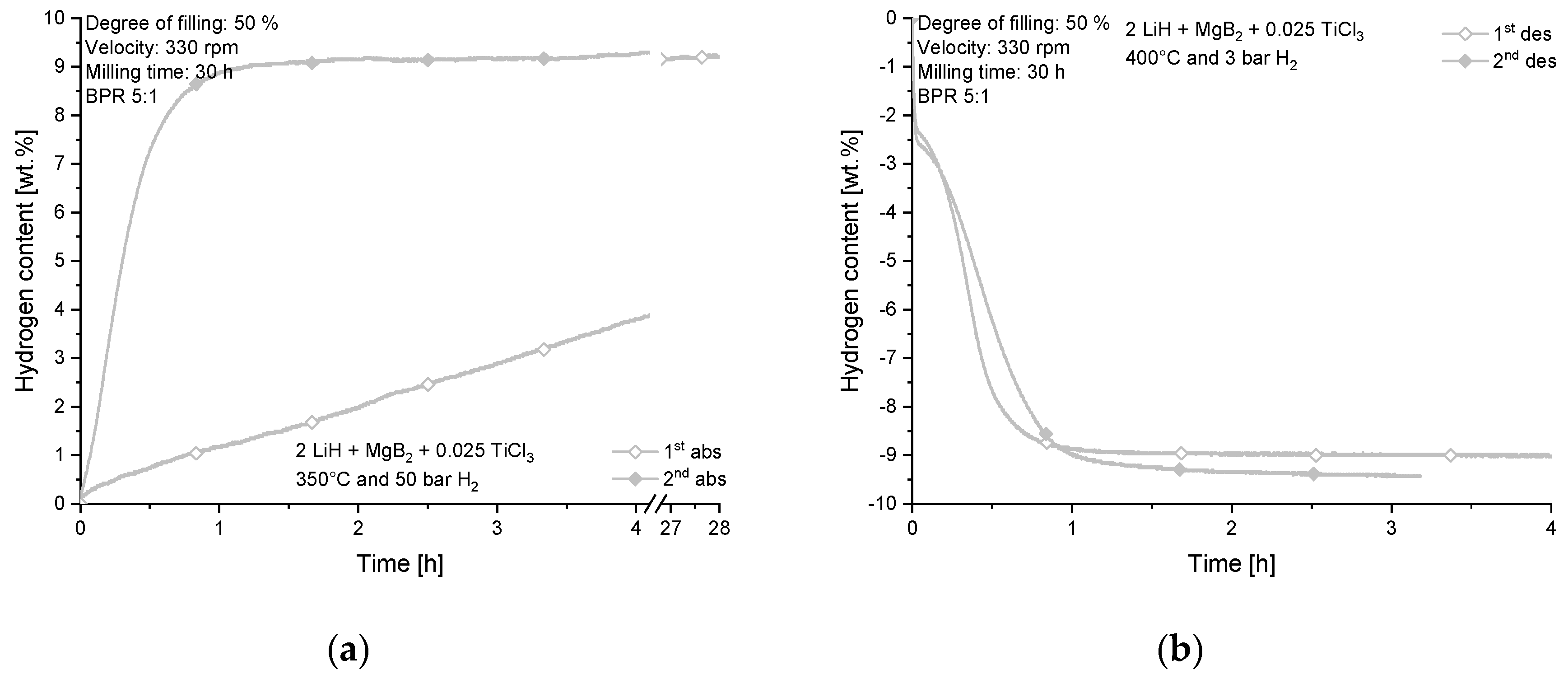
4.1.1. Influence on Kinetic Behavior
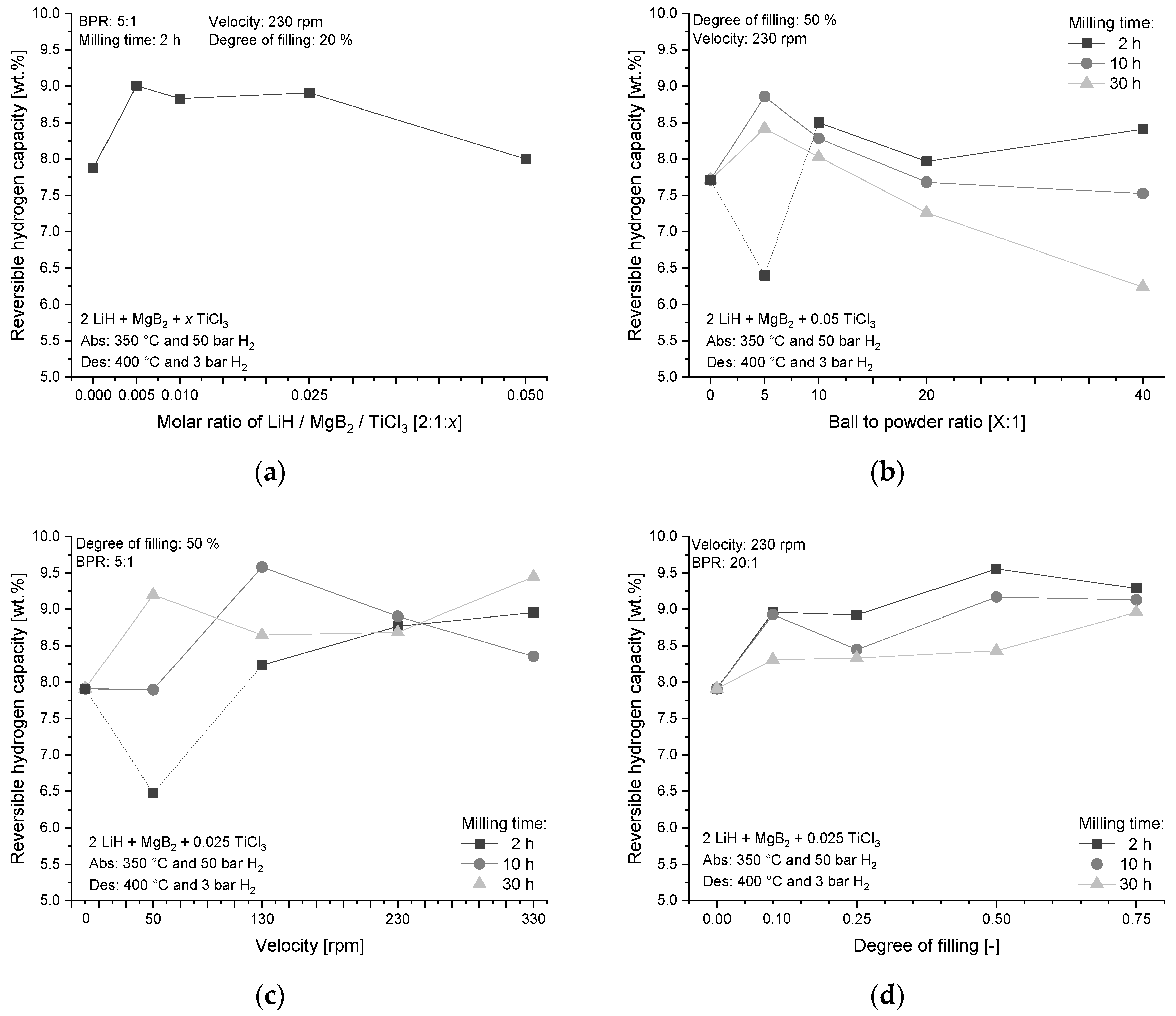
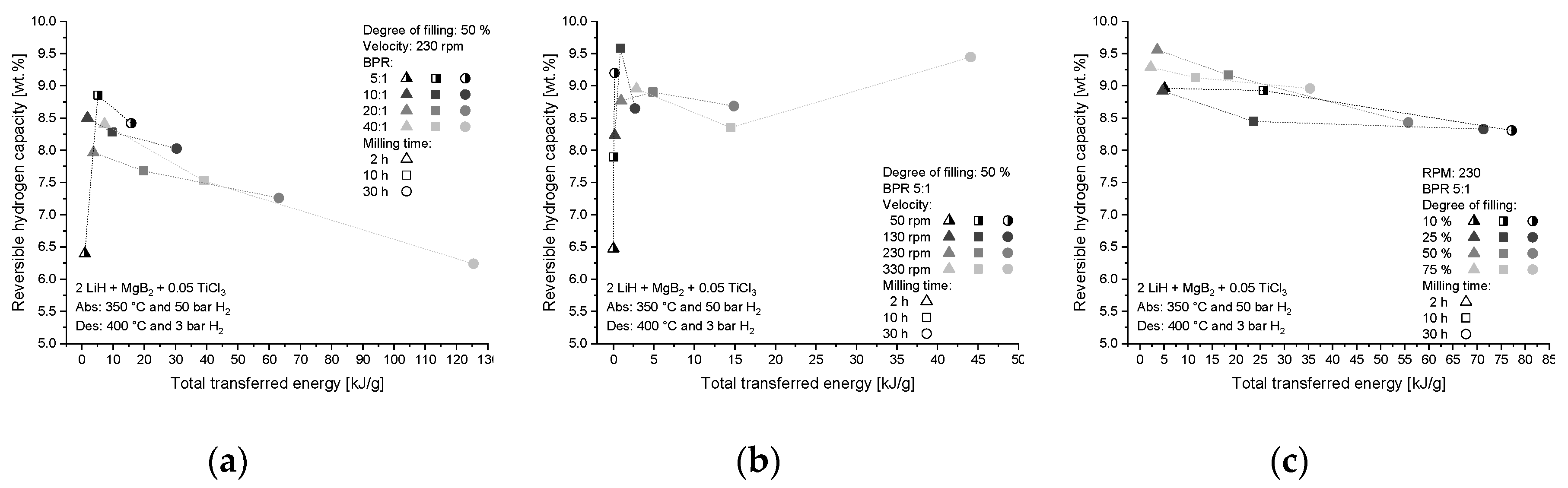
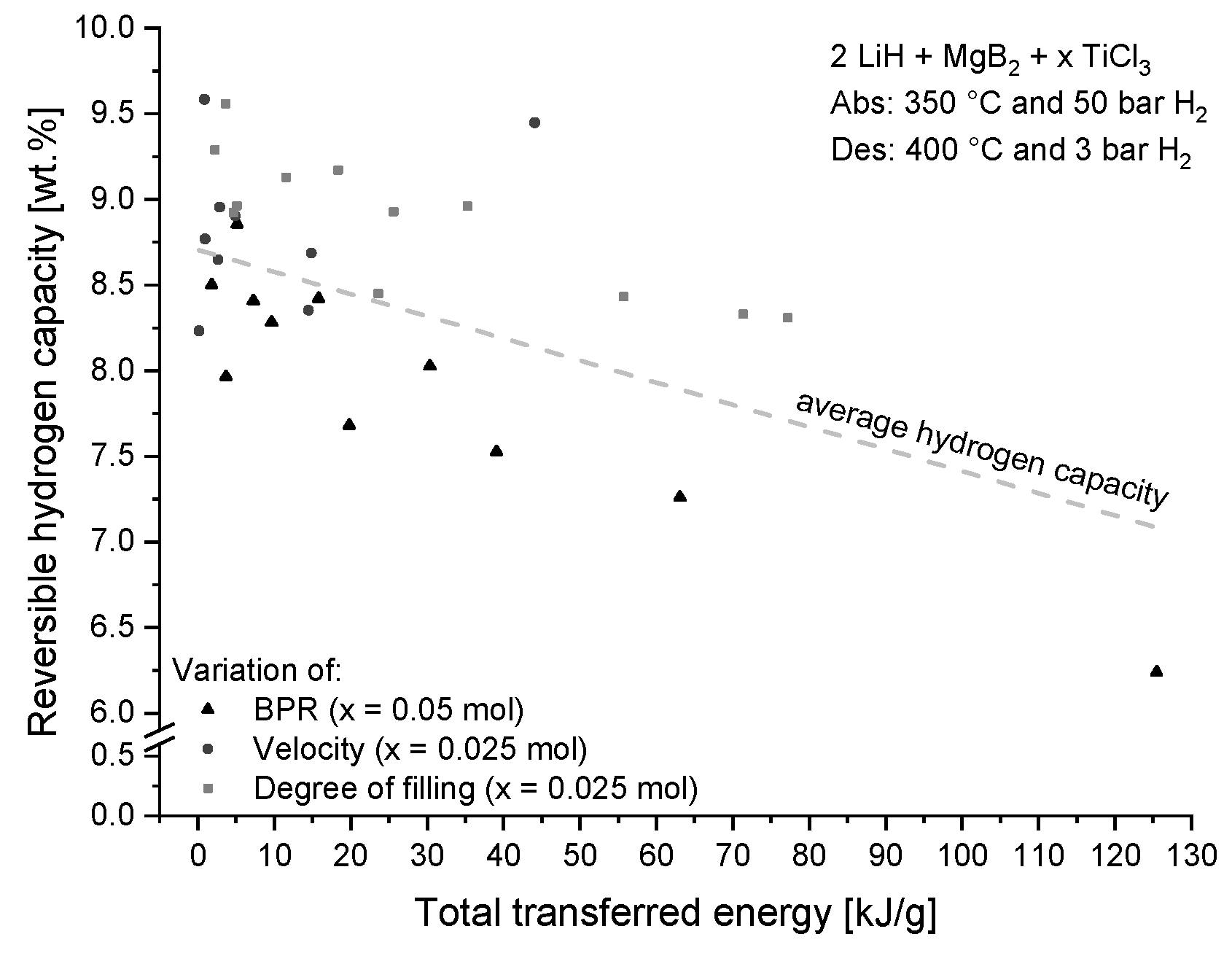
4.1.2. Influence on Hydrogen Storage Capacity
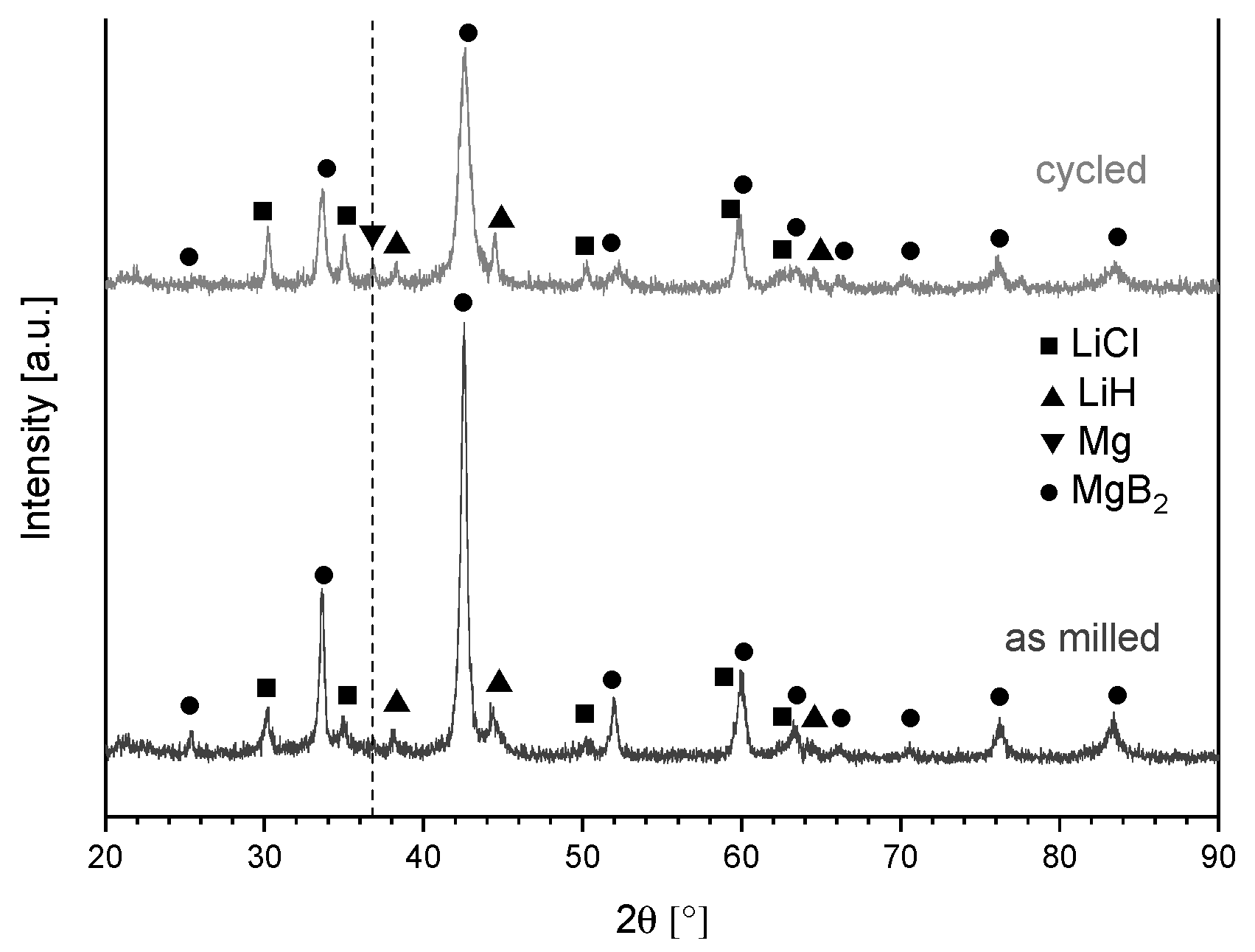
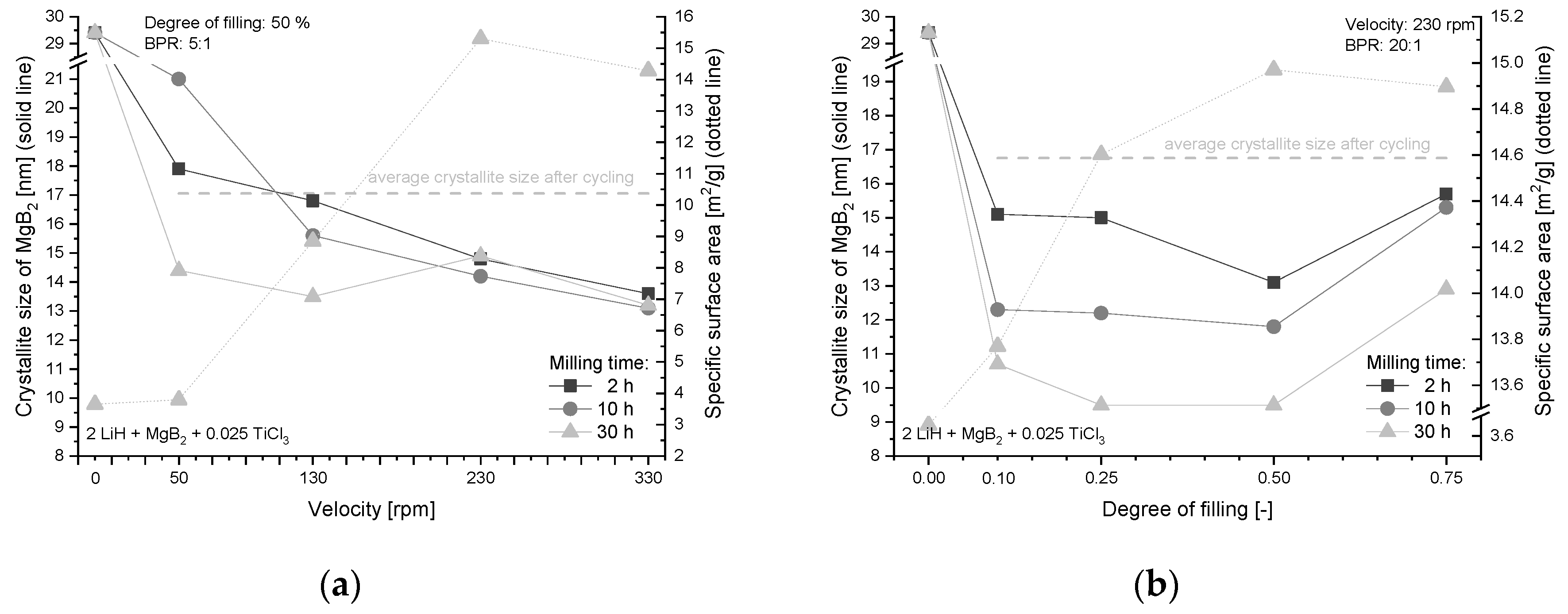
4.2. Effect of Additive and Milling
Nucleation Sites and Iron Contamination
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A




Appendix B

References
- Felderhoff, M.; Weidenthaler, C.; von Helmolt, R.; Eberle, U. Hydrogen storage: The remaining scientific and technological challenges. Phys. Chem. Chem. Phys. 2007, 9, 2643–2653. [Google Scholar] [CrossRef] [PubMed]
- Ross, D.K. Hydrogen storage: The major technological barrier to the development of hydrogen fuel cell cars. Vacuum 2006, 80, 1084–1089. [Google Scholar] [CrossRef]
- Bellosta von Colbe, J.; Ares, J.-R.; Barale, J.; Baricco, M.; Buckley, C.; Capurso, G.; Gallandat, N.; Grant, D.M.; Guzik, M.N.; Jacob, I.; et al. Application of hydrides in hydrogen storage and compression: Achievements, outlook and perspectives. Int. J. Hydrogen Energy 2019, in press. [Google Scholar] [CrossRef]
- Lide, D.R. Handbook of Chemistry and Physics; CRC Press: London, UK, 1994. [Google Scholar]
- Varin, R.A.; Czujko, T.; Wronski, Z.S. Nanomaterials for Solid State Hydrogen Storage; Springer: New York, NY, USA, 2009. [Google Scholar]
- Bossel, U.; Eliasson, B. Energy and the Hydrogen Economy, US DOE, EERE. Available online: http://www.afdc.energy.gov/pdfs/hyd_economy_bossel_eliasson.pdf (accessed on 15 February 2019).
- Von Helmolt, R.; Eberle, U. Fuel cell vehicles: Status 2007. J. Power Sources 2007, 165, 833–843. [Google Scholar] [CrossRef]
- Jepsen, J.; Bellosta von Colbe, J.M.; Klassen, T.; Dornheim, M. Economic potential of complex hydrides compared to conventional hydrogen storage systems. Int. J. Hydrogen Energy 2012, 37, 4204–4214. [Google Scholar] [CrossRef]
- Crivello, J.C.; Dam, B.; Denys, R.V.; Dornheim, M.; Grant, D.M.; Huot, J.; Jensen, T.R.; de Jongh, P.; Latroche, M.; Milanese, C.; et al. Review of magnesium hydride-based materials: Development and optimisation. Appl. Phys. A 2016, 122, 97. [Google Scholar] [CrossRef]
- Yartys, V.A.; Lototskyy, M.V.; Akiba, E.; Albert, R.; Antonov, V.E.; Ares, J.R.; Baricco, M.; Bourgeois, N.; Buckley, C.E.; Bellosta von Colbe, J.M.; et al. Magnesium based materials for hydrogen based energy storage: Past, present and future. Int. J. Hydrogen Energy 2019, in press. [Google Scholar] [CrossRef]
- Milanese, C.; Jensen, T.R.; Hauback, B.C.; Pistidda, C.; Dornheim, M.; Yang, H.; Lombardo, L.; Zuettel, A.; Filinchuk, Y.; Ngene, P.; et al. Complex hydrides for energy storage. Int. J. Hydrogen Energy 2019, 44, 7860–7874. [Google Scholar] [CrossRef]
- Puszkiel, J.; Garroni, S.; Milanese, C.; Gennari, F.; Klassen, T.; Dornheim, M.; Pistidda, C. Tetrahydroborates: Development and Potential as Hydrogen Storage Medium. Inorganics 2017, 5, 74. [Google Scholar] [CrossRef]
- Milanese, C.; Garroni, S.; Gennari, F.; Marini, A.; Klassen, T.; Dornheim, M.; Pistidda, C. Solid State Hydrogen Storage in Alanates and Alanate-Based Compounds: A Review. Metals 2018, 8, 567. [Google Scholar] [CrossRef]
- Garroni, S.; Santoru, A.; Cao, H.; Dornheim, M.; Klassen, T.; Milanese, C.; Gennari, F.; Pistidda, C. Recent Progress and New Perspectives on Metal Amide and Imide Systems for Solid-State Hydrogen Storage. Energies 2018, 11, 1027. [Google Scholar] [CrossRef]
- Barkhordarian, G.; Klassen, T.; Bormann, R. Fast hydrogen sorption kinetics of nanocrystalline Mg using Nb2O5 as catalyst. Scr. Mater. 2003, 49, 213–217. [Google Scholar] [CrossRef]
- Bösenberg, U.; Kim, J.W.; Gosslar, D.; Eigen, N.; Jensen, T.R.; von Colbe, J.M.B.; Zhou, Y.; Dahms, M.; Kim, D.H.; Günther, R.; et al. Role of additives in LiBH4-MgH2 reactive hydride composites for sorption kinetics. Acta Mater. 2010, 58, 3381–3389. [Google Scholar] [CrossRef]
- Pighin, S.A.; Capurso, G.; Lo Russo, S.; Peretti, H.A. Hydrogen sorption kinetics of magnesium hydride enhanced by the addition of Zr8Ni21 alloy. J. Alloys Compd. 2012, 530, 111–115. [Google Scholar] [CrossRef]
- Suryanarayana, C. Mechanical alloying and milling. Prog. Mater. Sci. 2001, 46, 1–184. [Google Scholar] [CrossRef]
- Garroni, S.; Pistidda, C.; Brunelli, M.; Vaughan, G.B.M.; Surinach, S.; Baro, M.D. Hydrogen desorption mechanism of 2NaBH4 + MgH2 composite prepared by high-energy ball milling. Scr. Mater. 2009, 60, 1129–1132. [Google Scholar] [CrossRef]
- Huot, J.; Ravnsbæk, D.B.; Zhang, J.; Cuevas, F.; Latroche, M.; Jensen, T.R. Mechanochemical synthesis of hydrogen storage materials. Prog. Mater. Sci. 2013, 58, 30–75. [Google Scholar] [CrossRef]
- Gosalawit–Utke, R.; Thiangviriya, S.; Javadian, P.; Laipple, D.; Pistidda, C.; Bergemann, N.; Horstmann, C.; Jensen, T.R.; Klassen, T.; Dornheim, M. Effective nanoconfinement of 2LiBH4–MgH2 via simply MgH2 premilling for reversible hydrogen storages. Int. J. Hydrogen Energy 2014, 39, 15614–15626. [Google Scholar] [CrossRef]
- Capurso, G.; Agresti, F.; Crociani, L.; Rossetto, G.; Schiavo, B.; Maddalena, A.; Lo Russo, S.; Principi, G. Nanoconfined mixed Li and Mg borohydrides as materials for solid state hydrogen storage. Int. J. Hydrogen Energy 2012, 37, 10768–10773. [Google Scholar] [CrossRef]
- Comănescu, C.; Capurso, G.; Maddalena, A. Nanoconfinement in activated mesoporous carbon of calcium borohydride for improved reversible hydrogen storage. Nanotechnology 2012, 23, 385401. [Google Scholar] [CrossRef] [PubMed]
- Dornheim, M.; Doppiu, S.; Barkhordarian, G.; Boesenberg, U.; Klassen, T.; Gutfleisch, O.; Bormann, R. Hydrogen storage in magnesium-based hydrides and hydride composites. Scr. Mater. 2007, 56, 841–846. [Google Scholar] [CrossRef]
- Westerwaal, R.J.; Haije, W.G. Evaluation solid-state hydrogen storage systems. ECN Hydrog. Clean Foss. Fuels 2008, 3, 19–21. [Google Scholar]
- Barkhordarian, G.; Klassen, T.; Dornheim, M.; Bormann, R. Unexpected kinetic effect of MgB2 in reactive hydride composites containing complex borohydrides. J. Alloys Compd. 2007, 440, L18–L21. [Google Scholar] [CrossRef]
- Vajo, J.J.; Skeith, S.L.; Mertens, F. Reversible Storage of Hydrogen in Destabilized LiBH4. J. Phys. Chem. B 2005, 109, 3719–3722. [Google Scholar] [CrossRef] [PubMed]
- Züttel, A.; Wenger, P.; Rentsch, S.; Sudan, P.; Mauron, P.; Emmenegger, C. LiBH4 a new hydrogen storage material. J. Power Sources 2003, 118, 1–7. [Google Scholar] [CrossRef]
- Jepsen, J.; Milanese, C.; Puszkiel, J.; Girella, A.; Schiavo, B.; Lozano, A.G.; Capurso, G.; Bellosta von Colbe, J.M.; Marini, A.; Kabelac, S.; et al. Fundamental Material Properties of the 2LiBH4-MgH2 Reactive Hydride Composite for Hydrogen Storage: (I) Thermodynamic and Heat Transfer Properties. Energies 2018, 11, 1081. [Google Scholar] [CrossRef]
- Jepsen, J.; Milanese, C.; Puszkiel, J.; Girella, A.; Schiavo, B.; Lozano, A.G.; Capurso, G.; Bellosta von Colbe, J.M.; Marini, A.; Kabelac, S.; et al. Fundamental Material Properties of the 2LiBH4-MgH2 Reactive Hydride Composite for Hydrogen Storage: (II) Kinetic Properties. Energies 2018, 11, 1170. [Google Scholar] [CrossRef]
- Puszkiel, J.A.; Castro Riglos, M.V.; Karimi, F.; Santoru, A.; Pistidda, C.; Klassen, T.; Bellosta von Colbe, J.M.; Dornheim, M. Changing the dehydrogenation pathway of LiBH4–MgH2 via nanosized lithiated TiO2. Phys. Chem. Chem. Phys. 2017, 19, 7455–7460. [Google Scholar] [CrossRef] [PubMed]
- Karimi, F.; Riglos, M.V.C.; Santoru, A.; Hoell, A.; Raghuwanshi, V.S.; Milanese, C.; Bergemann, N.; Pistidda, C.; Nolis, P.; Baro, M.D.; et al. In Situ Formation of TiB2 Nanoparticles for Enhanced Dehydrogenation/Hydrogenation Reaction Kinetics of LiBH4–MgH2 as a Reversible Solid-State Hydrogen Storage Composite System. J. Phys. Chem. C 2018, 122, 11671–11681. [Google Scholar] [CrossRef]
- Le, T.-T.; Pistidda, C.; Puszkiel, J.; Castro Riglos, M.V.; Karimi, F.; Skibsted, J.; GharibDoust, S.P.; Richter, B.; Emmler, T.; Milanese, C.; et al. Design of a Nanometric AlTi Additive for MgB2-Based Reactive Hydride Composites with Superior Kinetic Properties. J. Phys. Chem. C 2018, 122, 7642–7655. [Google Scholar] [CrossRef]
- Santos, F.A.; Ramos, A.S.; Santos, C. d.; Suyuki, P.A.; Rodrigues Júnior, D. Efficiency Evaluation of ZrB2 Incorporation in the MgB2 Matrix Phase Using High-Energy Milling. Mater. Sci. Forum 2010, 660–661, 82–87. [Google Scholar] [CrossRef]
- Pitt, M.P.; Paskevicius, M.; Webb, C.J.; Sheppard, D.A.; Buckley, C.E.; Gray, E.M. The synthesis of nanoscopic Ti based alloys and their effects on the MgH2 system compared with the MgH2 + 0.01Nb2O5 benchmark. Int. J. Hydrogen Energy 2012, 37, 4227–4237. [Google Scholar] [CrossRef]
- Burgio, N.; Iasonna, A.; Magini, M.; Martelli, S.; Padella, F. Mechanical alloying of the Fe−Zr system. Correlation between input energy and end products. Il Nuovo Cimento D 1991, 13, 459–476. [Google Scholar] [CrossRef]
- Busch, N. Optimierung des Herstellungsprozesses und der Zusammensetzung von Wasserstoffspeichermaterialien; Fachhochschule Lübeck: Lübeck, Germany, 2012. [Google Scholar]
- Werner, T. Verbesserung der Wasserstoffspeichereigenschaften Eines Reaktiven Hydridkomposits durch Optimierung des Herstellungsprozesses; Hochschule für Angewandte Wissenschaften Hamburg: Hamburg, Germany, 2013. [Google Scholar]
- Pinkerton, F.E.; Meyer, M.S.; Meisner, G.P.; Balogh, M.P.; Vajo, J.J. Phase Boundaries and Reversibility of LiBH4/MgH2 Hydrogen Storage Material. J. Phys. Chem. C 2007, 111, 12881–12885. [Google Scholar] [CrossRef]
- Bösenberg, U. LiBH4-MgH2 Composites for Hydrogen Storage; Technische Universität Hamburg-Harburg: Hamburg, Germany, 2009. [Google Scholar]
- Scherrer, P. Bestimmung der Größe und der inneren Struktur von Kolloidteilchen mittels Röntgenstrahlen. Nachrichten von der Gesellschaft der Wissenschaften zu Göttingen, Mathematisch-Physikalische Klasse 1918, 1918, 98–100. [Google Scholar] [CrossRef]
- Pretzel, F.E.; Rupert, G.N.; Mader, C.L.; Storms, E.K.; Gritton, G.V.; Rushing, C.C. Properties of lithium hydride I. Single crystals. J. Phys. Chem. Solids 1960, 16, 10–20. [Google Scholar] [CrossRef]
- MatWeb. Available online: http://www.matweb.com (accessed on 15 September 2018).
- Puszkiel, J.A.; Gennari, F.C.; Larochette, P.A.; Ramallo-López, J.M.; Vainio, U.; Karimi, F.; Pranzas, P.K.; Troiani, H.; Pistidda, C.; Jepsen, J.; et al. Effect of Fe additive on the hydrogenation-dehydrogenation properties of 2LiH + MgB2/2LiBH4 + MgH2 system. J. Power Sources 2015, 284, 606–616. [Google Scholar] [CrossRef]
- Busch, N.; Jepsen, J.; Pistidda, C.; Puszkiel, J.A.; Karimi, F.; Milanese, C.; Tolkiehn, M.; Chaudhary, A.-L.; Klassen, T.; Dornheim, M. Influence of milling parameters on the sorption properties of the LiH–MgB2 system doped with TiCl3. J. Alloys Compd. 2015, 645, S299–S303. [Google Scholar] [CrossRef]
- Deprez, E.; MunÞoz-Maìrquez, M.A.; Roldaìn, M.A.; Prestipino, C.; Palomares, F.J.; Minella, C.B.; Bösenberg, U.; Dornheim, M.; Bormann, R.; Fernaìndez, A. Oxidation State and Local Structure of Ti-Based Additives in the Reactive Hydride Composite 2LiBH4 + MgH2. J. Phys. Chem. C 2010, 114, 3309–3317. [Google Scholar] [CrossRef]
- Deprez, E.; Justo, A.; Rojas, T.C.; López-Cartés, C.; Bonatto Minella, C.; Bösenberg, U.; Dornheim, M.; Bormann, R.; Fernández, A. Microstructural study of the LiBH4-MgH2 reactive hydride composite with and without Ti-isopropoxide additive. Acta Mater. 2010, 58, 5683–5694. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, H.Y.; Yang, Y.F.; Jiang, Q.C. Solidification behavior of cast TiB2 particulate reinforced Mg composites. Mater. Sci. Eng. A 2008, 478, 9–15. [Google Scholar] [CrossRef]
- Qiu, D.; Zhang, M.X.; Fu, H.M.; Kelly, P.M.; Taylor, J.A. Crystallography of recently developed grain refiners for Mg–Al alloys. Philos. Mag. Lett. 2007, 87, 505–514. [Google Scholar] [CrossRef]
- Jepsen, J.; Milanese, C.; Girella, A.; Lozano, G.A.; Pistidda, C.; Bellosta von Colbe, J.M.; Marini, A.; Klassen, T.; Dornheim, M. Compaction pressure influence on material properties and sorption behaviour of LiBH4–MgH2 composite. Int. J. Hydrogen Energy 2013, 38, 8357–8366. [Google Scholar] [CrossRef]
- De Rango, P.; Chaise, A.; Charbonnier, J.; Fruchart, D.; Jehan, M.; Marty, P.; Miraglia, S.; Rivoirard, S.; Skryabina, N. Nanostructured magnesium hydride for pilot tank development. J. Alloys Compd. 2007, 446–447, 52–57. [Google Scholar] [CrossRef]
- Çakmak, G.; Öztürk, T. Milling of magnesium powders without additives. Powder Technol. 2013, 237, 484–488. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
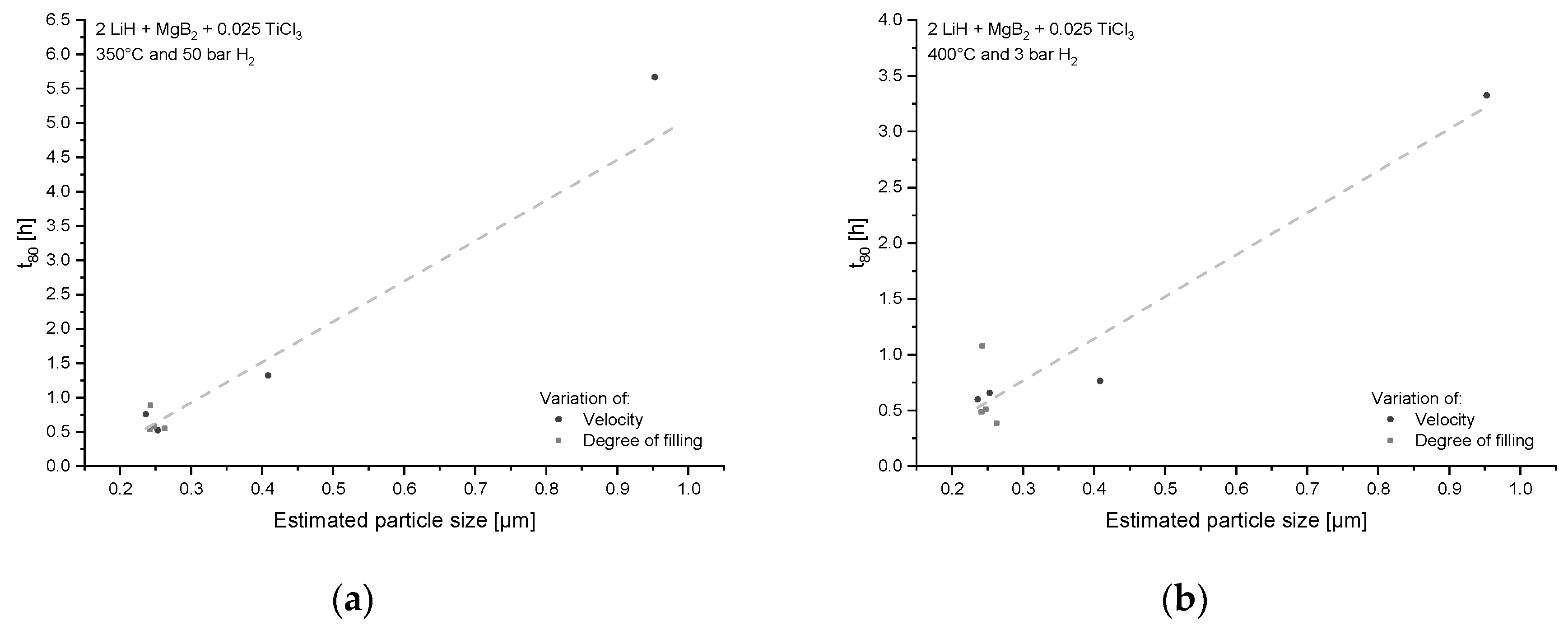{kind=link}
{kind=link}
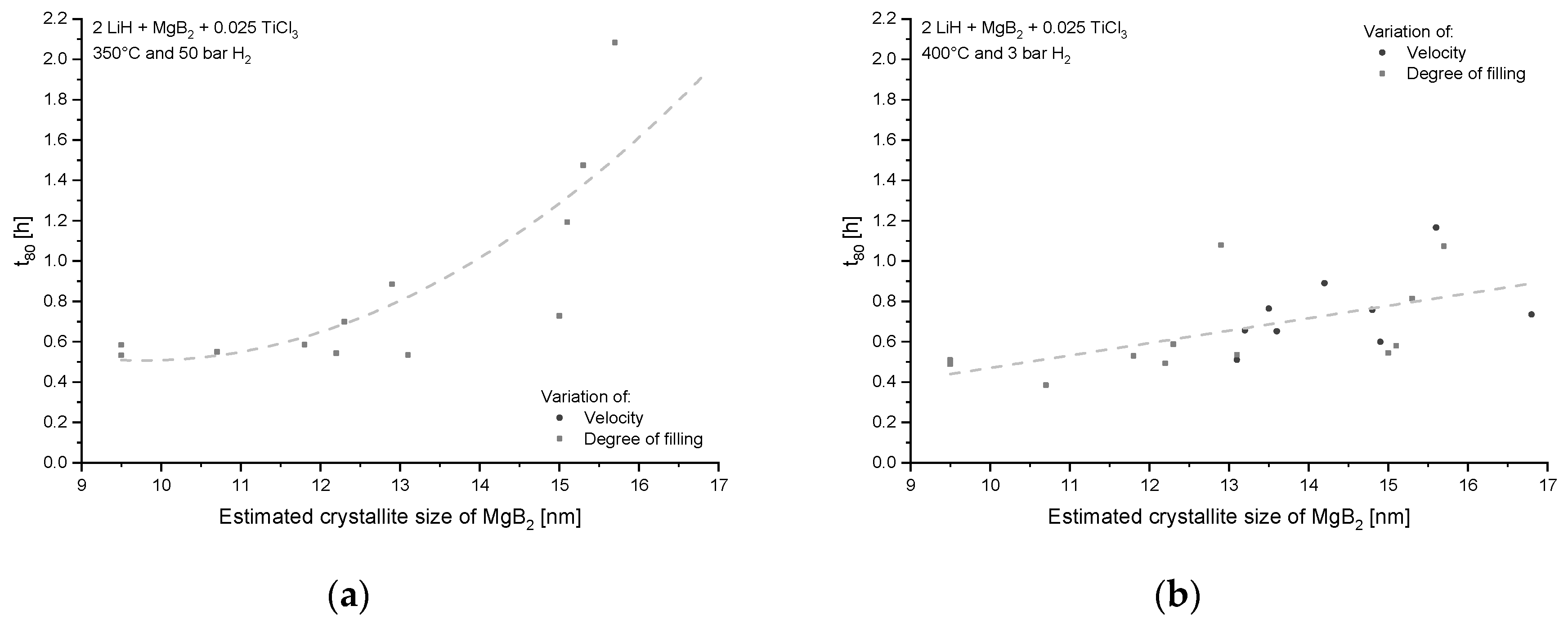{kind=link}
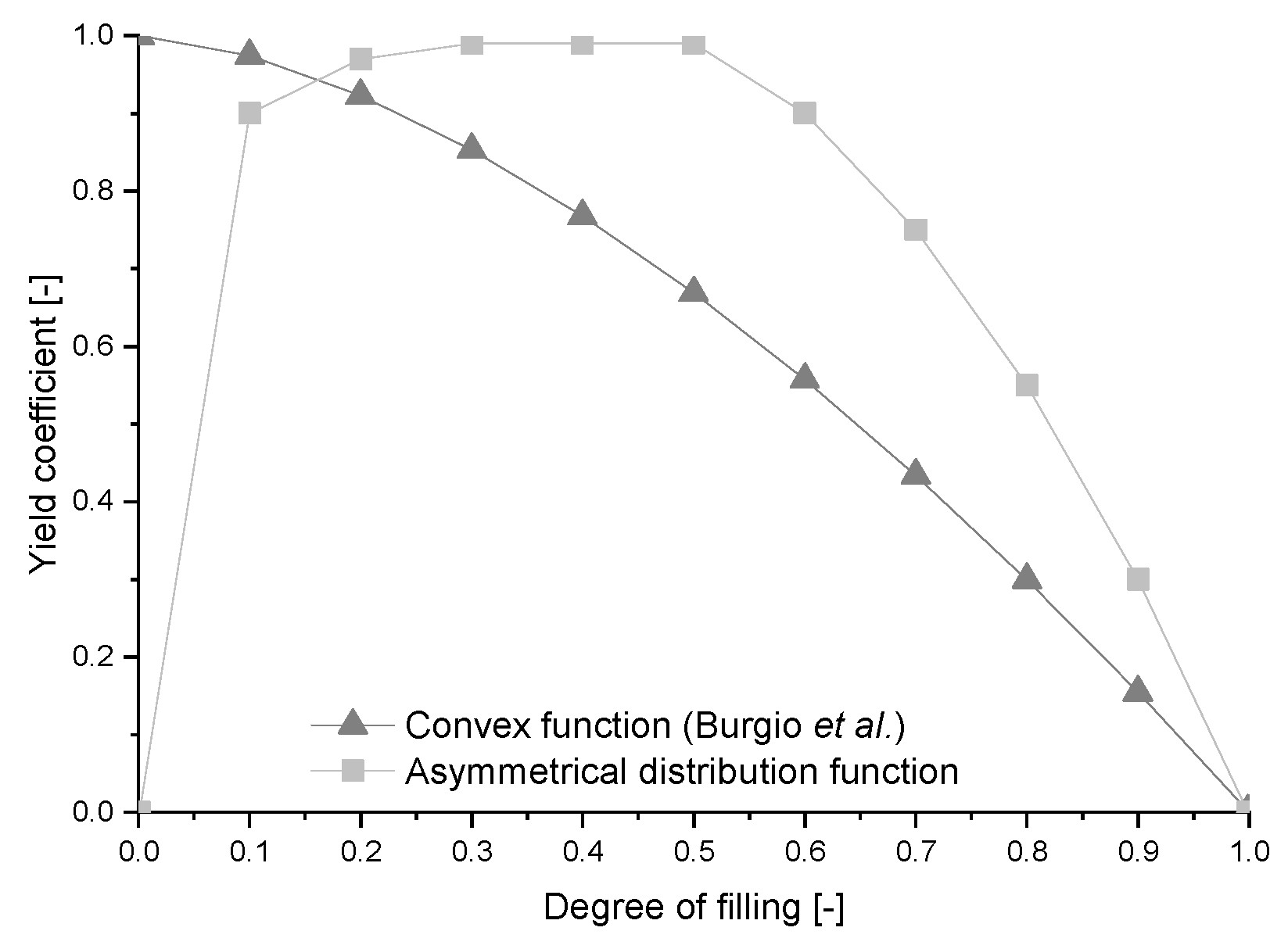{kind=link}
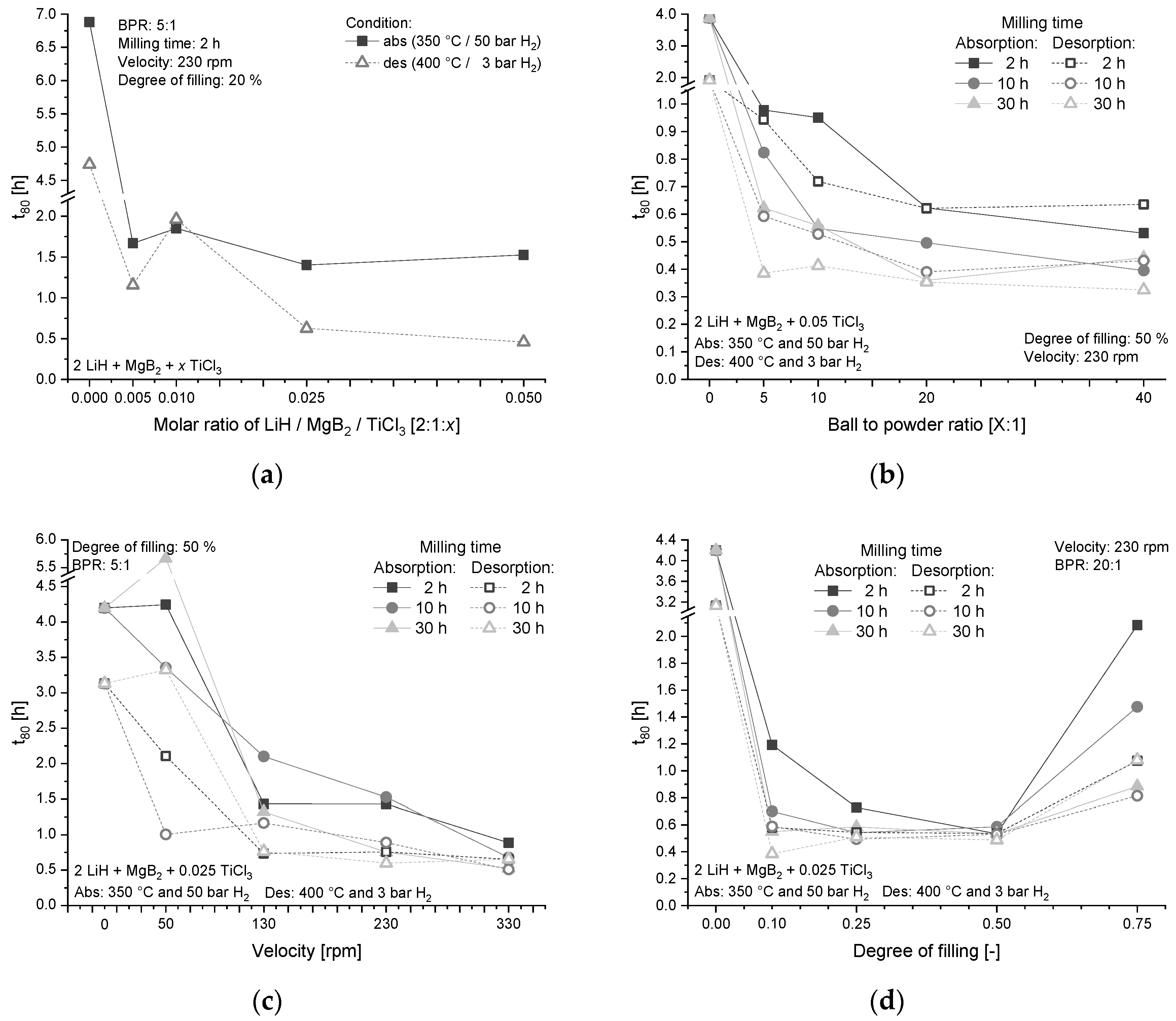
| Label | Milling Parameter | Variation |
|---|---|---|
| (a) | Milling time | 2 h, 10 h, 30 h |
| (b) | BPR–Ball-to-powder ratio | 5:1, 10:1, 20:1, 40:1 |
| (c) | Velocity | 50 rpm, 130 rpm, 230 rpm, 330 rpm |
| (d) | Degree of filling | 10%, 25%, 50%, 75% |
| (e) | Milling vials/balls material | Tempered steel |
| (f) | Milling balls diameter | 1.0 cm |
| (g) | Milling vials diameter | 7.6 cm |
| (h) | Milling vials height | 6.6 cm |
| (i) | Type/model of mill | Planetary ball mill (Pulverisette 5, Fritsch) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jepsen, J.; Capurso, G.; Puszkiel, J.; Busch, N.; Werner, T.; Milanese, C.; Girella, A.; Bellosta von Colbe, J.; Dornheim, M.; Klassen, T. Effect of the Process Parameters on the Energy Transfer during the Synthesis of the 2LiBH4-MgH2 Reactive Hydride Composite for Hydrogen Storage. Metals 2019, 9, 349. https://doi.org/10.3390/met9030349
Jepsen J, Capurso G, Puszkiel J, Busch N, Werner T, Milanese C, Girella A, Bellosta von Colbe J, Dornheim M, Klassen T. Effect of the Process Parameters on the Energy Transfer during the Synthesis of the 2LiBH4-MgH2 Reactive Hydride Composite for Hydrogen Storage. Metals. 2019; 9(3):349. https://doi.org/10.3390/met9030349
Chicago/Turabian StyleJepsen, Julian, Giovanni Capurso, Julián Puszkiel, Nina Busch, Tobias Werner, Chiara Milanese, Alessandro Girella, José Bellosta von Colbe, Martin Dornheim, and Thomas Klassen. 2019. "Effect of the Process Parameters on the Energy Transfer during the Synthesis of the 2LiBH4-MgH2 Reactive Hydride Composite for Hydrogen Storage" Metals 9, no. 3: 349. https://doi.org/10.3390/met9030349
APA StyleJepsen, J., Capurso, G., Puszkiel, J., Busch, N., Werner, T., Milanese, C., Girella, A., Bellosta von Colbe, J., Dornheim, M., & Klassen, T. (2019). Effect of the Process Parameters on the Energy Transfer during the Synthesis of the 2LiBH4-MgH2 Reactive Hydride Composite for Hydrogen Storage. Metals, 9(3), 349. https://doi.org/10.3390/met9030349