
Effects of Cigarette Smoking on Influenza Virus/Host Interplay


Abstract
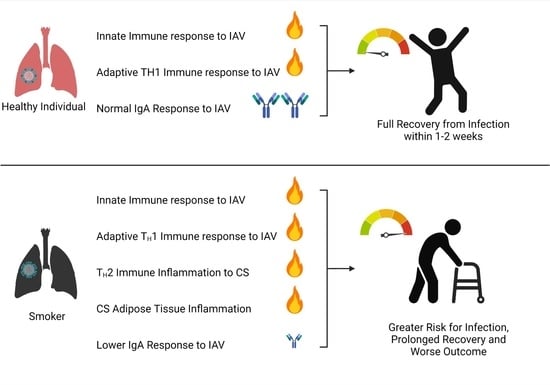
:
{kind=link}
{kind=link}
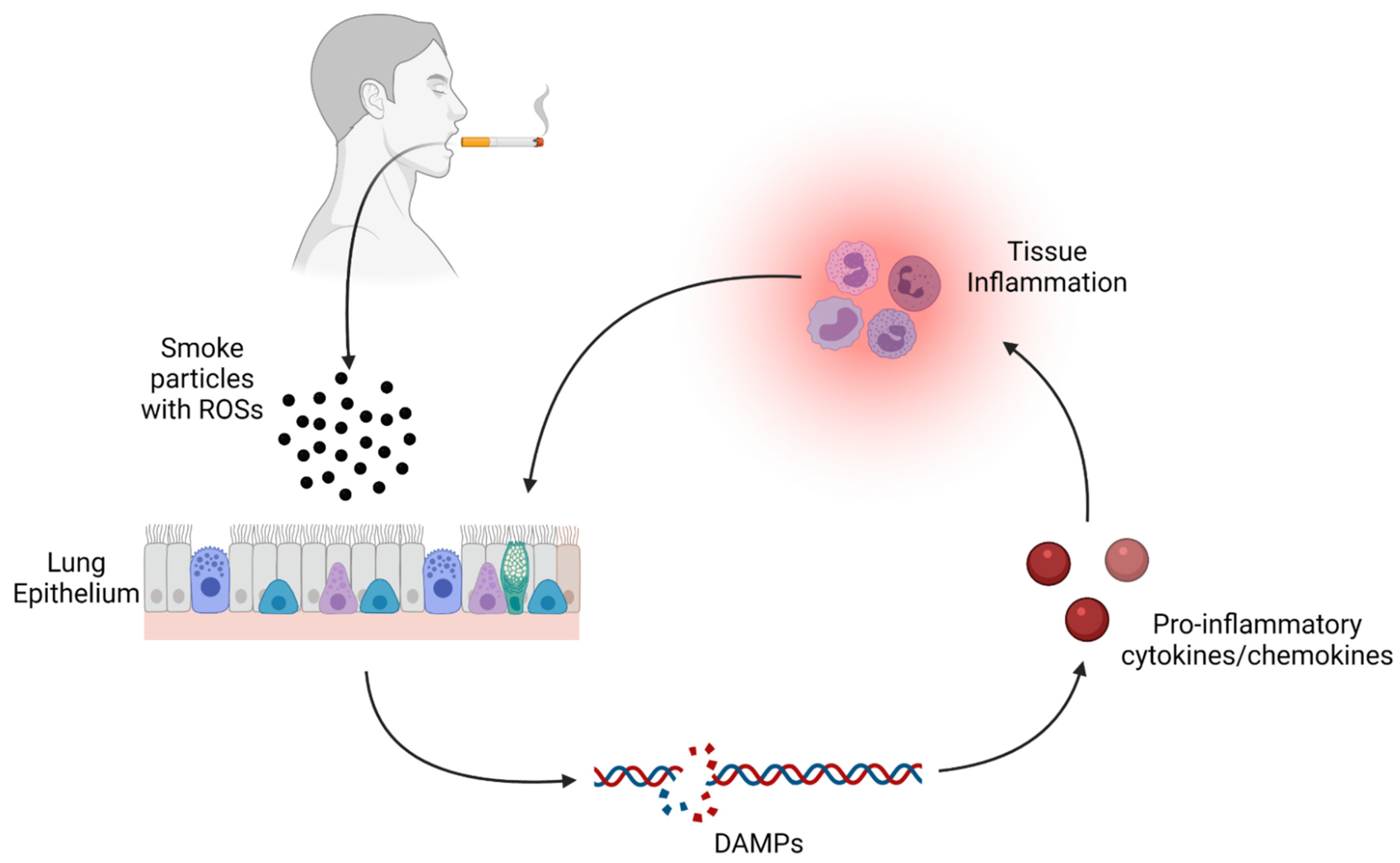{kind=link}
{kind=link}
1. Introduction
2. Influenza Virus and Response to Normal Infection
3. How Does CS Affect Immune Responses to IAV Pulmonary Insult?
4. Cigarette Smoke, Chronic Inflammation, and the Crossroads of Infection with Chronic Disease
5. Contribution of Adipose to Cardiovascular Disease and Chronic Inflammation
6. Adipose Tissue and IAV Infection
7. Host Factors Involved in CS Mediated Effects on IAV Infection
8. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- CDC. CDC Smoking & Tobacco Use Factsheet; CDC: Atlanta, GA, USA, 2020. [Google Scholar]
- Lawrence, H.; Hunter, A.; Murray, R.; Lim, W.S.; McKeever, T. Cigarette smoking and the occurrence of influenza—Systematic review. J. Infect. 2019, 79, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Arcavi, L.; Benowitz, N.L. Cigarette smoking and infection. Arch. Intern. Med. 2003, 164, 2206–2216. [Google Scholar] [CrossRef]
- Kark, J.D.; Lebiush, M.; Rannon, L. Cigarette-Smoking as a Risk Factor for Epidemic a(H1n1) Influenza in Young Men. N. Engl. J. Med. 1982, 307, 1042–1046. [Google Scholar] [CrossRef]
- Hanshaoworakul, W.; Simmerman, J.M.; Narueponjirakul, U.; Sanasuttipun, W.; Shinde, V.; Kaewchana, S.; Areechokechai, D.; Levy, J.; Ungchusak, K. Severe Human Influenza Infections in Thailand: Oseltamivir Treatment and Risk Factors for Fatal Outcome. PLoS ONE 2009, 4, e6015. [Google Scholar] [CrossRef] [Green Version]
- Ducatez, M.F.; Pelletier, C.; Meyer, G. Influenza D Virus in Cattle. France, 2011–2014. Emerg. Infect. Dis. 2015, 21, 368–371. [Google Scholar] [CrossRef] [PubMed]
- Hause, B.M.; Collin, E.A.; Liu, R.X.; Huang, B.; Sheng, Z.Z.; Lu, W.X.; Wang, D.; Nelson, E.A.; Li, F. Characterization of a Novel Influenza Virus in Cattle and Swine: Proposal for a New Genus in the Orthomyxoviridae Family. Mbio 2014, 5, e00031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, S.; Kamimoto, L.; Bramley, A.M.; Schmitz, A.M.; Benoit, S.R.; Louie, J.; Sugerman, D.E.; Druckenmiller, J.K.; Ritger, K.A.; Chugh, R.; et al. Hospitalized Patients with 2009 H1N1 Influenza in the United States, April–June 2009. N. Engl. J. Med. 2009, 361, 1935–1944. [Google Scholar] [CrossRef] [Green Version]
- Thompson, W.W.; Shay, D.K.; Weintraub, E.; Brammer, I.; Bridges, C.B.; Cox, N.J.; Fukuda, K. Influenza-associated hospitalizations in the United States. JAMA-J. Am. Med. Assoc. 2004, 292, 1333–1340. [Google Scholar] [CrossRef] [PubMed]
- Simonsen, L.; Fukuda, K.; Schonberger, L.B.; Cox, N.J. The impact of influenza epidemics on hospitalizations. J. Infect. Dis. 2000, 181, 831–837. [Google Scholar] [CrossRef] [Green Version]
- Morens, D.M.; Taubenberger, J.K.; Fauci, A.S. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: Implications for pandemic influenza preparedness. J. Infect. Dis. 2008, 198, 962–970. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.R.; Sheng, Z.M.; Ely, S.F.; Guinee, D.G.; Beasley, M.B.; Suh, J.; Deshpande, C.; Mollura, D.J.; Morens, D.M.; Bray, M.; et al. Pulmonary Pathologic Findings of Fatal 2009 Pandemic Influenza A/H1N1 Viral Infections. Arch. Pathol. Lab. Med. 2010, 134, 235–243. [Google Scholar] [CrossRef]
- Kalil, A.C.; Thomas, P.G. Influenza virus-related critical illness: Pathophysiology and epidemiology. Crit. Care 2019, 23, 258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. WHO 2018 Seasonal Influenza Factsheet; Organization WHO: Geneva, Switzerland, 2018. [Google Scholar]
- Putri, W.C.W.S.; Muscatello, D.J.; Stockwell, M.S.; Newall, A.T. Economic burden of seasonal influenza in the United States. Vaccine 2018, 36, 3960–3966. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.Q.; Lu, Y.; Raman, S.N.T.; Xu, F.; Wu, Q.; Li, Z.B.; Brownlie, R.; Liu, Q.; Zhou, Y. Nuclear-resident RIG-I senses viral replication inducing antiviral immunity. Nat. Commun. 2018, 9, 3199. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.Q.; Park, H.S.; Pyo, H.M.; Liu, Q.; Zhou, Y. Influenza A Virus Panhandle Structure Is Directly Involved in RIG-I Activation and Interferon Induction. J. Virol. 2015, 89, 6067–6079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, G.Q.; Zhou, Y. Cytoplasm and Beyond: Dynamic Innate Immune Sensing of Influenza A Virus by RIG-I. J. Virol. 2019, 93, e0299-18. [Google Scholar] [CrossRef] [Green Version]
- Allen, I.C.; Scull, M.A.; Moore, C.B.; Holl, E.K.; McElvania-TeKippe, E.; Taxman, D.J.; Guthrie, E.H.; Pickles, R.J.; Ting, J.P.Y. The NLRP3 Inflammasome Mediates In Vivo Innate Immunity to Influenza A Virus through Recognition of Viral RNA. Immunity 2009, 30, 556–565. [Google Scholar] [CrossRef] [Green Version]
- Thomas, P.G.; Dash, P.; Aldridge, J.R.; Ellebedy, A.H.; Reynolds, C.; Funk, A.J.; Martin, W.J.; Lamkanfi, M.; Webby, R.J.; Boyd, K.L.; et al. The Intracellular Sensor NLRP3 Mediates Key Innate and Healing Responses to Influenza A Virus via the Regulation of Caspase-1. Immunity 2009, 30, 566–575. [Google Scholar]
- Teijaro, J.R.; Walsh, K.B.; Cahalan, S.; Fremgen, D.M.; Roberts, E.; Scott, F.; Martinborough, E.; Peach, R.; Oldstone, M.B.A.; Rosen, H. Endothelial Cells Are Central Orchestrators of Cytokine Amplification during Influenza Virus Infection. Cell 2011, 146, 980–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, A.; Jones, D.; Bailey, M.; Beca, J.; Bellomo, R.; Blackwell, N.; Forrest, P.; Gattas, D.; Granger, E.; Herkes, R.; et al. Extracorporeal Membrane Oxygenation for 2009 Influenza A(H1N1) Acute Respiratory Distress Syndrome. JAMA-J. Am. Med. Assoc. 2009, 302, 1888–1895. [Google Scholar]
- To, K.K.W.; Hung, I.F.N.; Li, I.W.S.; Lee, K.L.; Koo, C.K.; Yan, W.W.; Liu, R.; Ho, K.Y.; Chu, K.H.; Watt, C.L.; et al. Delayed Clearance of Viral Load and Marked Cytokine Activation in Severe Cases of Pandemic H1N1 2009 Influenza Virus Infection. Clin. Infect. Dis. 2010, 50, 850–859. [Google Scholar] [CrossRef] [Green Version]
- Cheung, C.Y.; Poon, L.L.; Lau, A.S.; Luk, W.; Lau, Y.L.; Shortridge, K.F.; Gordon, S.; Guan, Y.; Peiris, J.S. Induction of proinflammatory cytokines in human macrophages by influenza A (H5N1) viruses: A mechanism for the unusual severity of human disease? Lancet 2002, 360, 1831–1837. [Google Scholar] [CrossRef]
- Gao, R.B.; Cao, B.; Hu, Y.W.; Feng, Z.J.; Wang, D.Y.; Hu, W.F.; Chen, J.; Jie, Z.J.; Qiu, H.B.; Xu, K.; et al. Human Infection with a Novel Avian-Origin Influenza A (H7N9) Virus. N. Engl. J. Med. 2013, 368, 1888–1897. [Google Scholar] [CrossRef] [Green Version]
- Gao, H.N.; Lu, H.Z.; Cao, B.; Du, B.; Shang, H.; Gan, J.H.; Lu, S.H.; Yang, Y.D.; Fang, Q.; Shen, Y.Z.; et al. Clinical Findings in 111 Cases of Influenza A (H7N9) Virus Infection. N. Engl. J. Med. 2013, 368, 2277–2285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- To, K.F.; Chan, P.K.; Chan, K.F.; Lee, W.K.; Lam, W.Y.; Wong, K.F.; Tang, N.L.; Tsang, D.N.; Sung, R.Y.; Buckley, T.A.; et al. Pathology of fatal human infection associated with avian influenza A H5N1 virus. J. Med. Virol. 2001, 63, 242–246. [Google Scholar] [CrossRef]
- Le Goffic, R.; Arshad, M.I.; Rauch, M.; L’Helgoualc’h, A.; Delmas, B.; Piquet-Pellorce, C.; Samson, M. Infection with influenza virus induces IL-33 in murine lungs. Am. J. Respir. Cell Mol. Biol. 2011, 45, 1125–1132. [Google Scholar] [CrossRef]
- Cayrol, C.; Girard, J.P. IL-33: An alarmin cytokine with crucial roles in innate immunity, inflammation and allergy. Curr. Opin. Immunol. 2014, 31, 31–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuschner, W.G.; DAlessandro, A.; Wong, H.; Blanc, P.D. Dose-dependent cigarette smoking-related inflammatory responses in healthy adults. Eur. Respir. J. 1996, 9, 1989–1994. [Google Scholar] [CrossRef] [Green Version]
- Hunninghake, G.W.; Crystal, R.G. Cigarette-Smoking and Lung Destruction—Accumulation of Neutrophils in the Lungs of Cigarette Smokers. Am. Rev. Respir. Dis. 1983, 128, 833–838. [Google Scholar] [PubMed]
- Van Eeden, S.F.; Hogg, J.C. The response of human bone marrow to chronic cigarette smoking. Eur. Respir. J. 2000, 15, 915–921. [Google Scholar] [CrossRef] [Green Version]
- Ferrero, M.R.; Garcia, C.C.; Dutra de Almeida, M.; Torres Braz da Silva, J.; Bianchi Reis Insuela, D.; Teixeira Ferreira, T.P.; de Sa Coutinho, D.; Trindade de Azevedo, C.; Machado Rodrigues, E.S.P.; Martins, M.A. CCR5 Antagonist Maraviroc Inhibits Acute Exacerbation of Lung Inflammation Triggered by Influenza Virus in Cigarette Smoke-Exposed Mice. Pharmaceuticals 2021, 14, 620. [Google Scholar] [CrossRef]
- Gualano, R.C.; Hansen, M.J.; Vlahos, R.; Jones, J.E.; Park-Jones, R.A.; Deliyannis, G.; Turner, S.J.; Duca, K.A.; Anderson, G.P. Cigarette smoke worsens lung inflammation and impairs resolution of influenza infection in mice. Respir. Res. 2008, 9, 53. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Kong, Y.; Barnes, P.F.; Huang, F.F.; Klucar, P.; Wang, X.S.; Samten, B.; Sengupta, M.; Machona, B.; Donis, R.; et al. Exposure to Cigarette Smoke Inhibits the Pulmonary T-Cell Response to Influenza Virus and Mycobacterium tuberculosis. Infect. Immun. 2011, 79, 229–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.; Ling, M.T.; Mao, H.W.; Zheng, J.; Liu, M.; Lam, K.T.; Liu, Y.; Tu, W.W.; Lau, Y.L. Influenza Virus-Induced Lung Inflammation Was Modulated by Cigarette Smoke Exposure in Mice. PLoS ONE 2014, 9, e86166. [Google Scholar]
- Hong, M.J.; Gu, B.H.; Madison, M.C.; Landers, C.; Tung, H.Y.; Kim, M.; Yuan, X.; You, R.; Machado, A.A.; Gilbert, B.E.; et al. Protective role of gamma delta T cells in cigarette smoke and influenza infection. Mucosal Immunol. 2018, 11, 894–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.W.; Sharma, L.; Kang, Y.A.; Kim, S.H.; Chandrasekharan, S.; Losier, A.; Brady, V.; Bermejo, S.; Andrews, N.; Yoon, C.M.; et al. Impact of Cigarette Smoke Exposure on the Lung Fibroblastic Response after Influenza Pneumonia. Am. J. Respir. Cell Mol. Biol. 2018, 59, 770–781. [Google Scholar] [CrossRef] [PubMed]
- Eddleston, J.; Lee, R.U.; Doerner, A.M.; Herschbach, J.; Zuraw, B.L. Cigarette Smoke Decreases Innate Responses of Epithelial Cells to Rhinovirus Infection. Am. J. Respir. Cell Mol. Biol. 2011, 44, 118–126. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.J.; Lee, C.G.; Lee, J.Y.; Dela Cruz, C.S.; Chen, Z.J.; Enelow, R.; Elias, J.A. Cigarette smoke selectively enhances viral PAMP- and virus-induced pulmonary innate immune and remodeling responses in mice. J. Clin. Investig. 2008, 118, 2771–2784. [Google Scholar] [CrossRef] [Green Version]
- Robbins, C.S.; Bauer, C.M.T.; Vujicic, N.; Gaschler, G.J.; Lichty, B.D.; Brown, E.G.; Stampfli, M.R. Cigarette smoke impacts immune inflammatory responses to influenza in mice. Am. J. Respir. Crit. Care Med. 2006, 174, 1342–1351. [Google Scholar] [CrossRef] [Green Version]
- Finklea, J.F.; Sandifer, S.H.; Smith, D.D. Cigarette Smoking and Epidemic Influenza. Am. J. Epidemiol. 1969, 90, 390–399. [Google Scholar] [CrossRef]
- Aronson, M.D.; Weiss, S.T.; Ben, R.L.; Komaroff, A.L. Association between cigarette smoking and acute respiratory tract illness in young adults. JAMA 1982, 248, 181–183. [Google Scholar] [CrossRef] [PubMed]
- Godoy, P.; Castilla, J.; Soldevila, N.; Mayoral, J.M.; Toledo, D.; Martin, V.; Astray, J.; Egurrola, M.; Morales-Suarez-Varela, M.; Dominguez, A.; et al. Smoking may increase the risk of influenza hospitalization and reduce influenza vaccine effectiveness in the elderly. Eur. J. Public Health 2018, 28, 150–155. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, K.G.; Kent, J.; Hammersley, V. Influenza A among community-dwelling elderly persons in Leicestershire during winter 1993-4; cigarette smoking as a risk factor and the efficacy of influenza vaccination. Epidemiol. Infect. 1999, 123, 103–108. [Google Scholar] [CrossRef]
- Thomson, N.C.; Chaudhuri, R.; Livingston, E. Asthma and cigarette smoking. Eur. Respir. J. 2004, 24, 822–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, H.F.; Dyer, K.D.; Foster, P.S. Eosinophils: Changing perspectives in health and disease. Nat. Rev. Immunol. 2013, 13, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Minty, A.; Chalon, P.; Derocq, J.M.; Dumont, X.; Guillemot, J.C.; Kaghad, M.; Labit, C.; Leplatois, P.; Liauzun, P.; Miloux, B.; et al. Interleukin-13 is a new human lymphokine regulating inflammatory and immune-responses. Nature 1993, 362, 248–250. [Google Scholar] [CrossRef]
- McKenzie, A.N.J.; Culpepper, J.A.; Malefyt, R.D.; Briere, F.; Punnonen, J.; Aversa, G.; Sato, A.; Dang, W.; Cocks, B.G.; Menon, S.; et al. Interleukin-13, a T-cell-derived cytokine that regulates human monocyte and B-cell function. Proc. Natl. Acad. Sci. USA 1993, 90, 3735–3739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Punnonen, J.; Aversa, G.; Cocks, B.G.; McKenzie, A.N.J.; Menon, S.; Zurawski, G.; Malefyt, R.D.; Devries, J.E. Interleukin-13 induces interleukin-4-independent IGG4 and IGE synthesis and CD23 expression by human B-cells. Proc. Natl. Acad. Sci. USA 1993, 90, 3730–3734. [Google Scholar] [CrossRef] [Green Version]
- Zheng, T.; Zhu, Z.; Wang, Z.D.; Homer, R.J.; Ma, B.; Riese, R.J.; Chapman, H.A.; Shapiro, S.D.; Elias, J.A. Inducible targeting of IL-13 to the adult lung causes matrix metalloproteinase-and cathepsin-dependent emphysema. J. Clin. Investig. 2000, 106, 1081–1093. [Google Scholar] [CrossRef] [Green Version]
- Gern, J.E.; Lemanske, R.F., Jr.; Busse, W.W. Early life origins of asthma. J. Clin. Investig. 1999, 104, 837–843. [Google Scholar] [CrossRef] [Green Version]
- Moran, T.M.; Isobe, H.; Fernandez-Sesma, A.; Schulman, J.L. Interleukin-4 causes delayed virus clearance in influenza virus-infected mice. J. Virol. 1996, 70, 5230–5235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.M.; Li, Q.H.; Xie, J.G.; Xu, Y.J. Cigarette smoke inhibits BAFF expression and mucosal immunoglobulin A responses in the lung during influenza virus infection. Respir. Res. 2015, 16, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Duffney, P.F.; Embong, A.K.; McGuire, C.C.; Thatcher, T.H.; Phipps, R.P.; Sime, P.J. Cigarette smoke increases susceptibility to infection in lung epithelial cells by upregulating caveolin-dependent endocytosis. PLoS ONE 2020, 15, e0232102. [Google Scholar] [CrossRef] [PubMed]
- Danov, O.; Wolff, M.; Bartel, S.; Bohlen, S.; Obernolte, H.; Wronski, S.; Jonigk, D.; Hammer, B.; Kovacevic, D.; Reuter, S.; et al. Cigarette Smoke Affects Dendritic Cell Populations, Epithelial Barrier Function, and the Immune Response to Viral Infection With H1N1. Front. Med. 2020, 7, 571003. [Google Scholar] [CrossRef] [PubMed]
- Jaspers, I.; Horvath, K.M.; Zhang, W.L.; Brighton, L.E.; Carson, J.L.; Noah, T.L. Reduced Expression of IRF7 in Nasal Epithelial Cells from Smokers after Infection with Influenza. Am. J. Respir. Cell Mol. Biol. 2010, 43, 368–375. [Google Scholar] [CrossRef] [Green Version]
- Bauer, C.M.T.; Zavitz, C.C.J.; Botelho, F.M.; Lambert, K.N.; Brown, E.G.; Mossman, K.L.; Taylor, J.D.; Stampfli, M.R. Treating Viral Exacerbations of Chronic Obstructive Pulmonary Disease: Insights from a Mouse Model of Cigarette Smoke and H1N1 Influenza Infection. PLoS ONE 2010, 5, e3251. [Google Scholar] [CrossRef] [Green Version]
- Boehme, S.A.; Franz-Bacon, K.; Ludka, J.; DiTirro, D.N.; Ly, T.W.; Bacon, K.B. MAP3K19 Is Overexpressed in COPD and Is a Central Mediator of Cigarette Smoke-Induced Pulmonary Inflammation and Lower Airway Destruction. PLoS ONE 2016, 11, e0167169. [Google Scholar] [CrossRef]
- Mebratu, Y.A.; Smith, K.R.; Agga, G.E.; Tesfaigzi, Y. Inflammation and emphysema in cigarette smoke-exposed mice when instilled with poly (I:C) or infected with influenza A or respiratory syncytial viruses. Respir. Res. 2016, 17, 75. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.X.; Zhang, W.; More, S.; Booth, J.L.; Duggan, E.S.; Liu, L.; Zhao, Y.D.; Metcalf, J.P. Cigarette smoke attenuates the RIG-I-initiated innate antiviral response to influenza infection in two murine models. Am. J. Physiol. -Lung Cell. Mol. Physiol. 2014, 307, L848–L858. [Google Scholar] [CrossRef] [Green Version]
- Duffney, P.F.; McCarthy, C.E.; Nogales, A.; Thatcher, T.H.; Martinez-Sobrido, L.; Phipps, R.P.; Sime, P.J. Cigarette smoke dampens antiviral signaling in small airway epithelial cells by disrupting TLR3 cleavage. Am. J. Physiol. -Lung Cell. Mol. Physiol. 2018, 314, L505–L513. [Google Scholar] [CrossRef] [Green Version]
- Hsu, A.C.Y.; Starkey, M.R.; Hanish, I.; Parsons, K.; Haw, T.J.; Howland, L.J.; Barr, I.; Mahony, J.B.; Foster, P.S.; Knight, D.A.; et al. Targeting PI3K-p110 alpha Suppresses Influenza Virus Infection in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2015, 191, 1012–1023. [Google Scholar] [CrossRef] [PubMed]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef] [PubMed]
- De Torres, J.P.; Cordoba-Lanus, E.; Lopez-Aguilar, C.; Muros de Fuentes, M.; Montejo de Garcini, A.; Aguirre-Jaime, A.; Celli, B.R.; Casanova, C. C-reactive protein levels and clinically important predictive outcomes in stable COPD patients. Eur. Respir. J. 2006, 27, 902–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto-Plata, V.M.; Mullerova, H.; Toso, J.F.; Feudjo-Tepie, M.; Soriano, J.B.; Vessey, R.S.; Celli, B.R. C-reactive protein in patients with COPD, control smokers and non-smokers. Thorax 2006, 61, 23–28. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, T.; Church, D.F.; Pryor, W.A. Quantitative-Analysis of the Hydrogen-Peroxide Formed in Aqueous Cigarette Tar Extracts. Free. Radic. Biol. Med. 1989, 7, 9–15. [Google Scholar] [CrossRef]
- Pryor, W.A.; Prier, D.G.; Church, D.F. Electron-Spin Resonance Study of Mainstream and Sidestream Cigarette-Smoke—Nature of the Free-Radicals in Gas-Phase Smoke and in Cigarette Tar. Environ. Health Perspect. 1983, 47, 345–355. [Google Scholar] [CrossRef]
- Pryor, W.A.; Stone, K.; Zang, L.Y.; Bermudez, E. Fractionation of aqueous cigarette tar extracts: Fractions that contain the tar radical cause DNA damage. Chem. Res. Toxicol. 1998, 11, 441–448. [Google Scholar] [CrossRef]
- Zang, L.Y.; Stone, K.; Pryor, W.A. Detection of Free-Radicals in Aqueous Extracts of Cigarette Tar by Electron-Spin-Resonance. Free. Radic. Biol. Med. 1995, 19, 161–167. [Google Scholar] [CrossRef]
- Cross, C.E.; Traber, M.; Eiserich, J.; van der Vliet, A. Micronutrient antioxidants and smoking. Br. Med. Bull. 1999, 55, 691–704. [Google Scholar] [CrossRef] [Green Version]
- Pryor, W.A.; Terauchi, K.; Davis, W.H., Jr. Electron spin resonance (ESR) study of cigarette smoke by use of spin trapping techniques. Environ. Health Perspect. 1976, 16, 161–176. [Google Scholar] [CrossRef]
- Valavanidis, A.; Vlachogianni, T.; Fiotakis, K. Tobacco smoke: Involvement of reactive oxygen species and stable free radicals in mechanisms of oxidative damage, carcinogenesis and synergistic effects with other respirable particles. Int. J. Environ. Res. Public Health 2009, 6, 445–462. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Taneja, V.; Vassallo, R. Cigarette smoking and inflammation: Cellular and molecular mechanisms. J. Dent. Res. 2012, 91, 142–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zieglerheitbrock, H.W.L.; Wedel, A.; Schraut, W.; Strobel, M.; Wendelgass, P.; Sternsdorf, T.; Bauerle, P.A.; Haas, J.G.; Riethmuller, G. Tolerance to Lipopolysaccharide Involves Mobilization of Nuclear Factor Kappa-B with Predominance of P50 Homodimers. J. Biol. Chem. 1994, 269, 17001–17004. [Google Scholar] [CrossRef]
- World Health Organization. WHO Cardiovascular Diseases Factsheet 2020; Organization WHO: Geneva, Switzerland, 2020. [Google Scholar]
- Bouabdallaoui, N.; Messas, N.; Greenlaw, N.; Ferrari, R.; Ford, I.; Fox, K.M.; Tendera, M.; Datshana, P.N.; Hassager, C.; Gabriel Steg, P.; et al. Impact of smoking on cardiovascular outcomes in patients with stable coronary artery disease. Eur. J. Prev. Cardiol. 2020, 28, 1460–1466. [Google Scholar] [CrossRef] [PubMed]
- Wronska, A.; Kmiec, Z. Structural and biochemical characteristics of various white adipose tissue depots. Acta Physiol. 2012, 205, 194–208. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.M. Subcutaneous and visceral adipose tissue: Structural and functional differences. Obes. Rev. 2010, 11, 11–18. [Google Scholar] [CrossRef]
- Lear, S.A.; Chockalingam, A.; Kohli, S.; Richardson, C.G.; Humphries, K.H. Elevation in cardiovascular disease risk in South Asians is mediated by differences in visceral adipose tissue. Obesity 2012, 20, 1293–1300. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Shim, K.W.; Yoon, Y.S.; Lee, S.Y.; Kim, S.S.; Oh, S.W. Cigarette Smoking Increases Abdominal and Visceral Obesity but Not Overall Fatness: An Observational Study. PLoS ONE 2012, 7, e45815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakanishi, K.; Nishida, M.; Ohama, T.; Moriyama, T.; Yamauchi-Takihara, K. Smoking Associates with Visceral Fat Accumulation Especially in Women. Circ. J. 2014, 78, 1259–1263. [Google Scholar] [CrossRef] [Green Version]
- Terry, J.G.; Hartley, K.G.; Steffen, L.M.; Nair, S.; Alman, A.C.; Wellons, M.F.; Jacobs, D.R.; Tindle, H.A.; Carr, J.J. Association of smoking with abdominal adipose deposition and muscle composition in Coronary Artery Risk Development in Young Adults (CARDIA) participants at mid-life: A population-based cohort study. PLoS Med. 2020, 17, e1003223. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef] [PubMed]
- Nerlekar, N.; Brown, A.J.; Muthalaly, R.G.; Talman, A.; Hettige, T.; Cameron, J.D.; Wong, D.T.L. Association of Epicardial Adipose Tissue and High-Risk Plaque Characteristics: A Systematic Review and Meta-Analysis. J. Am. Heart. Assoc. 2017, 6, e006379. [Google Scholar] [CrossRef]
- Hajsadeghi, F.; Nabavi, V.; Bhandari, A.; Choi, A.; Vincent, H.; Flores, F.; Budoff, M.; Ahmadi, N. Increased epicardial adipose tissue is associated with coronary artery disease and major adverse cardiovascular events. Atherosclerosis 2014, 237, 486–489. [Google Scholar] [CrossRef]
- De Vos, A.M.; Prokop, M.; Roos, C.J.; Meijs, M.F.L.; van der Schouw, Y.T.; Rutten, A.; Gorter, P.M.; Cramer, M.J.; Doevendans, P.A.; Rensing, B.J.; et al. Peri-coronary epicardial adipose tissue is related to cardiovascular risk factors and coronary artery calcification in post-menopausal women. Eur. Heart J. 2008, 29, 777–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.H.; Chung, J.H.; Kwon, B.J.; Song, S.W.; Choi, W.S. The Associations of Epicardial Adipose Tissue with Coronary Artery Disease and Coronary Atherosclerosis. Int. Heart J. 2014, 55, 197–203. [Google Scholar] [CrossRef] [Green Version]
- Ormseth, M.J.; Lipson, A.; Alexopoulos, N.; Hartlage, G.R.; Oeser, A.M.; Bian, A.; Gebretsadik, T.; Shintani, A.; Raggi, P.; Stein, C.M. Association of epicardial adipose tissue with cardiometabolic risk and metabolic syndrome in patients with rheumatoid arthritis. Arthritis Care Res. 2013, 65, 1410–1415. [Google Scholar] [CrossRef] [PubMed]
- Mach, L.; Bedanova, H.; Soucek, M.; Karpisek, M.; Nemec, P.; Orban, M. Tobacco smoking and cytokine levels in human epicardial adipose tissue: Impact of smoking cessation. Atherosclerosis 2016, 255, 37–42. [Google Scholar] [CrossRef]
- Itoh, M.; Tsuji, T.; Nakamura, H.; Yamaguchi, K.; Fuchikami, J.; Takahashi, M.; Morozumi, Y.; Aoshiba, K. Systemic effects of acute cigarette smoke exposure in mice. Inhal. Toxicol. 2014, 26, 464–473. [Google Scholar] [CrossRef] [PubMed]
- Saltiel, A.R.; Olefsky, J.M. Inflammatory mechanisms linking obesity and metabolic disease. J. Clin. Investig. 2017, 127, 1–4. [Google Scholar] [CrossRef]
- Papathanassoglou, E.; El-Haschimi, K.; Li, X.C.; Matarese, G.; Strom, T.; Mantzoros, C. Leptin receptor expression and signaling in lymphocytes: Kinetics during lymphocyte activation, role in lymphocyte survival, and response to high fat diet in mice. J. Immunol. 2006, 176, 7745–7752. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Mizuarai, S.; Araki, H.; Mashiko, S.; Ishihara, A.; Kanatani, A.; Itadani, H.; Kotani, H. Adiposity elevates plasma MCP-1 levels leading to the increased CD11b-positive monocytes in mice. J. Biol. Chem. 2003, 278, 46654–46660. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.H.; Tian, J.; Tian, X.Y.; Tang, X.Y.; Rui, K.; Tong, J.; Lu, L.W.; Xu, H.X.; Wang, S.J. Adipose Tissue Dendritic Cells Enhances Inflammation by Prompting the Generation of Th17 Cells. PLoS ONE 2014, 9, e92450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.G.; Lee, L.A.; Wu, Y.C.; Hsiao, M.J.; Horng, J.T.; Kuo, R.L.; Huang, C.H.; Lin, Y.C.; Tsao, K.C.; Chen, M.C.; et al. A pilot study on primary cultures of human respiratory tract epithelial cells to predict patients’ responses to H7N9 infection. Oncotarget 2018, 9, 14492–14508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.G.; Sheridan, P.A.; Harp, J.B.; Beck, M.A. Diet-induced obese mice have increased mortality and altered immune responses when infected with influenza virus. J. Nutr. 2007, 137, 1236–1243. [Google Scholar] [CrossRef] [Green Version]
- Bennett, L.D.; Fox, J.M.; Signoret, N. Mechanisms regulating chemokine receptor activity. Immunology 2011, 134, 246–256. [Google Scholar] [CrossRef]
- Chu, H.X.; Arumugam, T.V.; Gelderblom, M.; Magnus, T.; Drunnnnond, G.R.; Sobey, C.G. Role of CCR2 in inflammatory conditions of the central nervous system. J. Cereb. Blood Flow Metab. 2014, 34, 1425–1429. [Google Scholar] [CrossRef] [Green Version]
- Dawson, T.C.; Beck, M.A.; Kuziel, W.A.; Henderson, F.; Maeda, N. Contrasting effects of CCR5 and CCR2 deficiency in the pulmonary inflammatory response to influenza A virus. Am. J. Pathol. 2000, 156, 1951–1959. [Google Scholar] [CrossRef] [Green Version]
- Lin, K.L.; Suzuki, Y.; Nakano, H.; Ramsburg, E.; Gunn, M.D. CCR2+ monocyte-derived dendritic cells and exudate macrophages produce influenza-induced pulmonary immune pathology and mortality. J. Immunol. 2008, 180, 2562–2572. [Google Scholar] [CrossRef] [Green Version]
- Doyle, I.; Ratcliffe, M.; Walding, A.; Vanden Bon, E.; Dymond, M.; Tomlinson, W.; Tilley, D.; Shelton, P.; Dougall, I. Differential gene expression analysis in human monocyte-derived macrophages: Impact of cigarette smoke on host defence. Mol. Immunol. 2010, 47, 1058–1065. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chavez, J.; Hai, R. Effects of Cigarette Smoking on Influenza Virus/Host Interplay. Pathogens 2021, 10, 1636. https://doi.org/10.3390/pathogens10121636
Chavez J, Hai R. Effects of Cigarette Smoking on Influenza Virus/Host Interplay. Pathogens. 2021; 10(12):1636. https://doi.org/10.3390/pathogens10121636
Chicago/Turabian StyleChavez, Jerald, and Rong Hai. 2021. "Effects of Cigarette Smoking on Influenza Virus/Host Interplay" Pathogens 10, no. 12: 1636. https://doi.org/10.3390/pathogens10121636
APA StyleChavez, J., & Hai, R. (2021). Effects of Cigarette Smoking on Influenza Virus/Host Interplay. Pathogens, 10(12), 1636. https://doi.org/10.3390/pathogens10121636