
Distribution and Genetic Diversity of Hepatitis E Virus in Wild and Domestic Rabbits in Australia

 , and
, and
Abstract
:1. Introduction
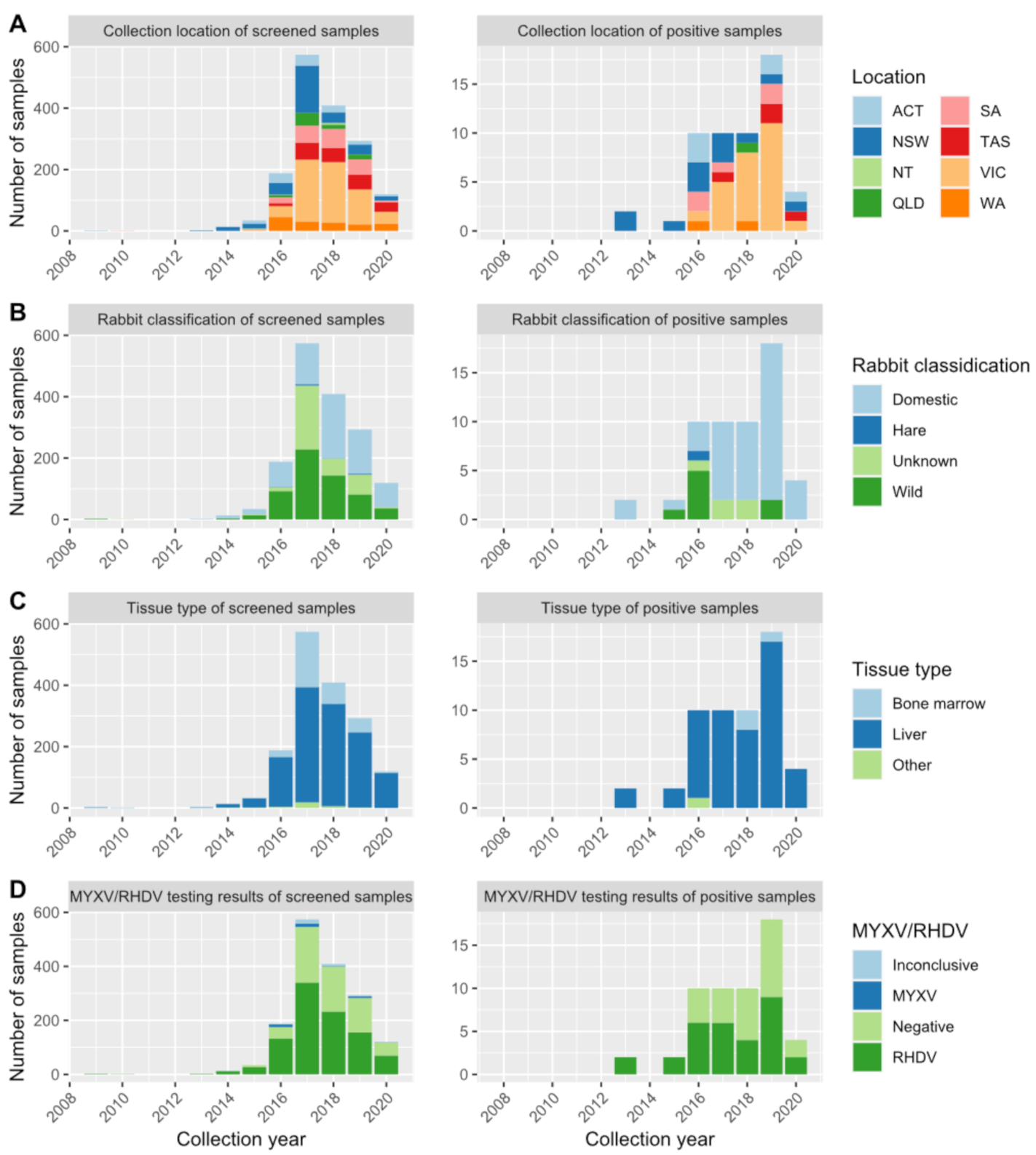
2. Results
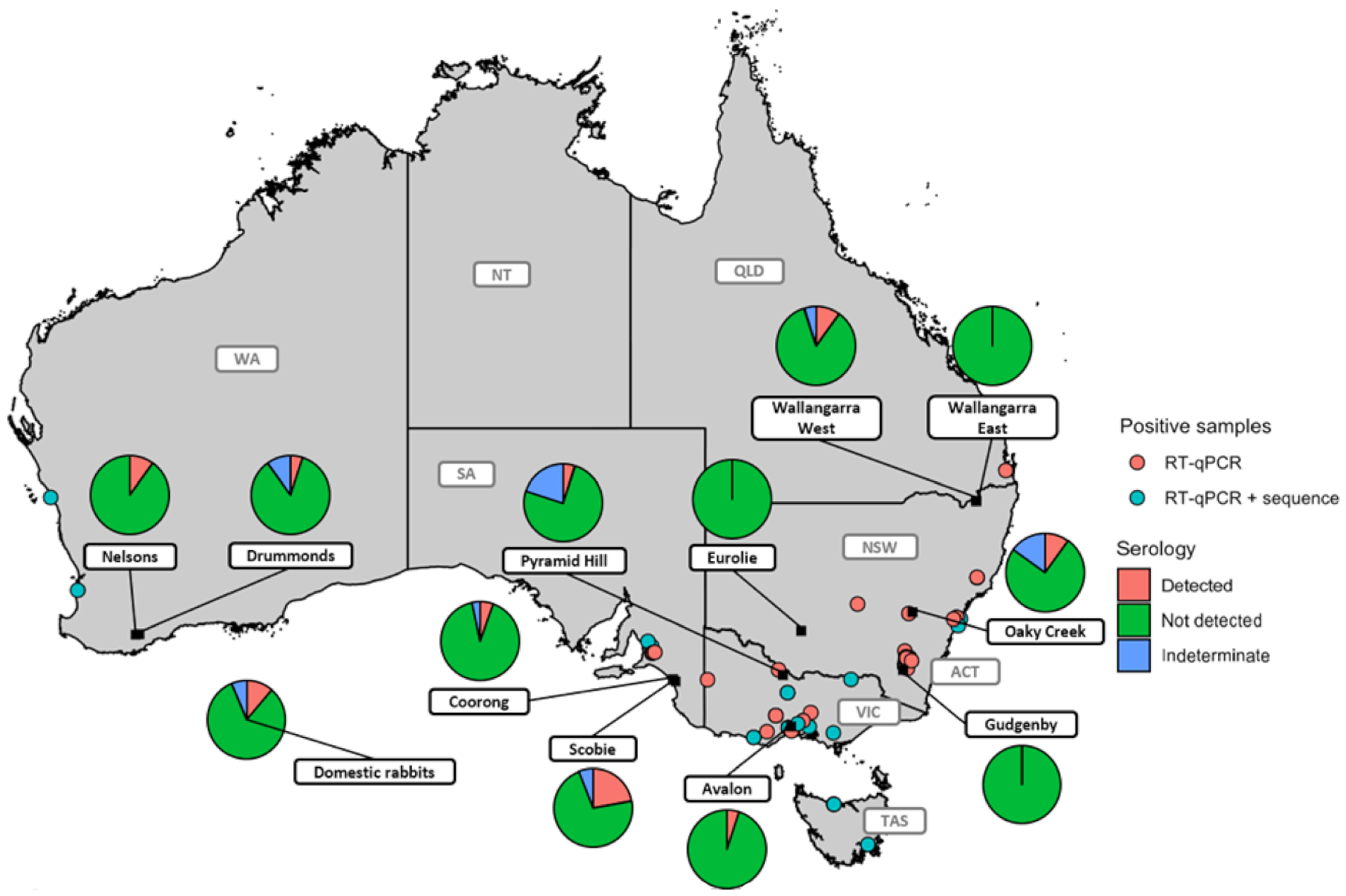
2.1. RT-qPCR Results and Seroepidemiology Show That Hepatitis E Virus is Widely Distributed in Australian Rabbit Populations
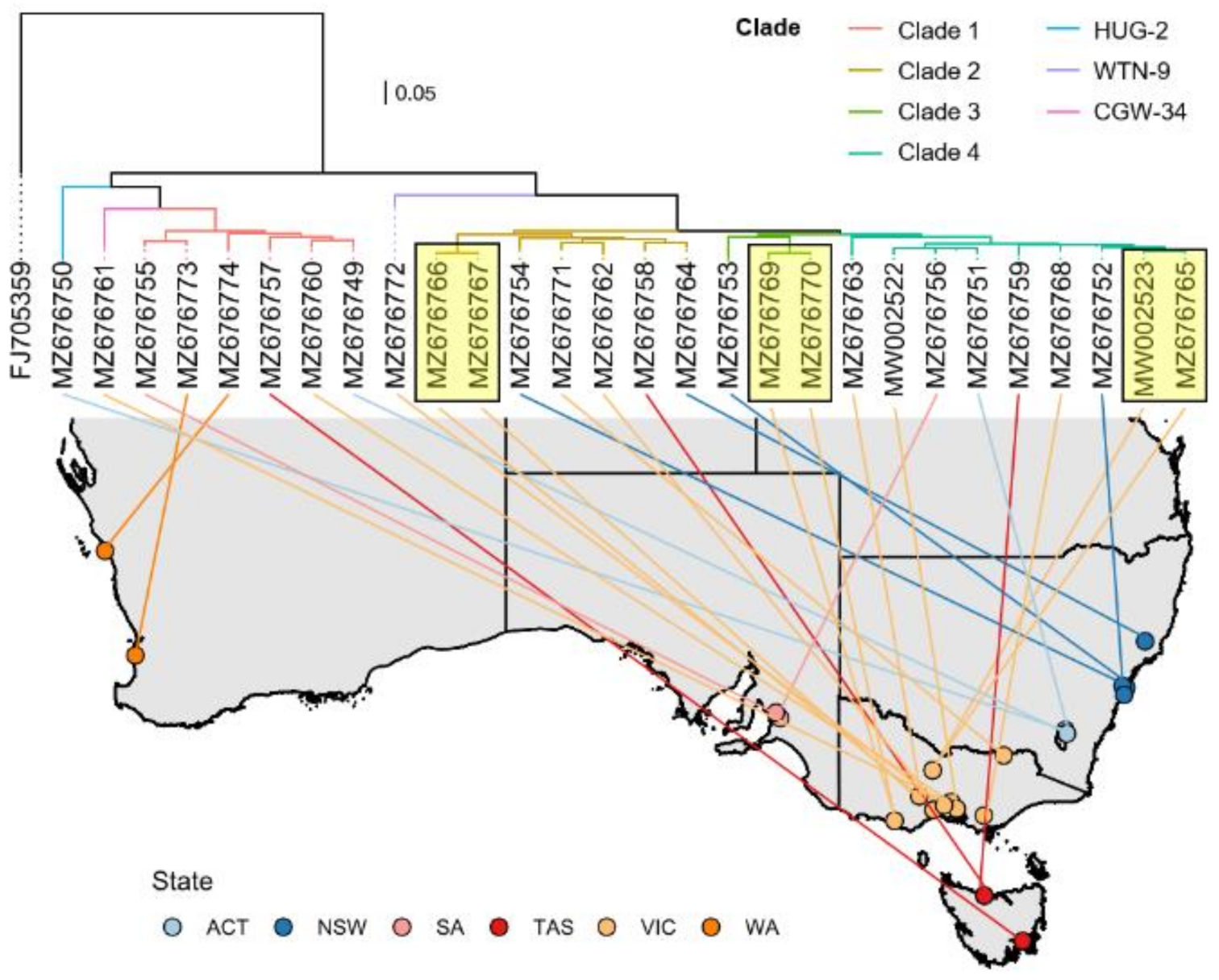
2.2. Australian HEV Ssequence Analysis Shows High Genetic Diversity with Minimal Phylogeographic Structure
2.3. Time-Structured Phylogenetic Analysis Indicates Multiple Independent Introductions of HEV into Australia
3. Discussion
4. Materials and Methods
4.1. Sample Collection
4.2. RT-qPCR
4.3. Sequencing
4.4. Sequence Data Analysis
4.5. Phylogeographic Analysis
4.6. Bayesian Evolutionary Analyses Analysis
4.7. Serology
4.8. Visualisation and Atatistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Purdy, M.A.; Harrison, T.J.; Jameel, S.; Meng, X.J.; Okamoto, H.; Van der Poel, W.H.M.; Smith, D.B.; ICTV Report Consortium. Ictv Virus Taxonomy Profile: Hepeviridae. J. Gen. Virol. 2017, 98, 2645–2646. [Google Scholar] [CrossRef]
- Smith, D.B.; Izopet, J.; Nicot, F.; Simmonds, P.; Jameel, S.; Meng, X.J.; Norder, H.; Okamoto, H.; van der Poel, W.H.M.; Reuter, G.; et al. Update: Proposed reference sequences for subtypes of hepatitis E virus (species Orthohepevirus A). J. Gen. Virol. 2020, 101, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.H.; Tan, B.H.; Teo, E.C.; Lim, S.G.; Dan, Y.Y.; Wee, A.; Aw, P.P.; Zhu, Y.; Hibberd, M.L.; Tan, C.K.; et al. Chronic Infection with Camelid Hepatitis E Virus in a Liver Transplant Recipient Who Regularly Consumes Camel Meat and Milk. Gastroenterology 2016, 150, 355–357. [Google Scholar] [CrossRef] [Green Version]
- Park, W.J.; Park, B.J.; Ahn, H.S.; Lee, J.B.; Park, S.Y.; Song, C.S.; Lee, S.W.; Yoo, H.S.; Choi, I.S. Hepatitis E virus as an emerging zoonotic pathogen. J. Vet. Sci. 2016, 17, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Navaneethan, U.; Al Mohajer, M.; Shata, M.T. Hepatitis E and pregnancy: Understanding the pathogenesis. Liver Int. 2008, 28, 1190–1199. [Google Scholar] [CrossRef] [Green Version]
- Dalton, H.R.; Saunders, M.; Woolson, K.L. Hepatitis E virus in developed countries: One of the most successful zoonotic viral diseases in human history? J. Virus Erad. 2015, 1, 23–29. [Google Scholar] [CrossRef]
- Abravanel, F.; Lhomme, S.; El Costa, H.; Schvartz, B.; Peron, J.M.; Kamar, N.; Izopet, J. Rabbit Hepatitis E Virus Infections in Humans, France. Emerg. Infect. Dis. 2017, 23, 1191–1193. [Google Scholar] [CrossRef] [Green Version]
- Izopet, J.; Dubois, M.; Bertagnoli, S.; Lhomme, S.; Marchandeau, S.; Boucher, S.; Kamar, N.; Abravanel, F.; Guerin, J.L. Hepatitis E virus strains in rabbits and evidence of a closely related strain in humans, France. Emerg. Infect. Dis. 2012, 18, 1274–1281. [Google Scholar] [CrossRef] [PubMed]
- Jenckel, M.; Hall, R.N.; Strive, T. First description of hepatitis E virus in Australian rabbits. Aust. Vet. J. 2021, 99, 356–358. [Google Scholar] [CrossRef]
- Quick, J.; Grubaugh, N.D.; Pullan, S.T.; Claro, I.M.; Smith, A.D.; Gangavarapu, K.; Oliveira, G.; Robles-Sikisaka, R.; Rogers, T.F.; Beutler, N.A.; et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat. Protoc. 2017, 12, 1261–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Liu, L.; Wang, L. An overview: Rabbit hepatitis E virus (HEV) and rabbit providing an animal model for HEV study. Rev. Med. Virol. 2018, 28, e1961. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Schielke, A.; Sachs, K.; Lierz, M.; Appel, B.; Jansen, A.; Johne, R. Detection of hepatitis E virus in wild boars of rural and urban regions in Germany and whole genome characterization of an endemic strain. Virol. J. 2009, 6, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, D.B.; Simmonds, P.; Izopet, J.; Oliveira-Filho, E.F.; Ulrich, R.G.; Johne, R.; Koenig, M.; Jameel, S.; Harrison, T.J.; Meng, X.-J.; et al. Proposed reference sequences for hepatitis E virus subtypes. J. Gen. Virol. 2016, 97, 537–542. [Google Scholar] [CrossRef]
- Nicot, F.; Dimeglio, C.; Migueres, M.; Jeanne, N.; Latour, J.; Abravanel, F.; Ranger, N.; Harter, A.; Dubois, M.; Lameiras, S.; et al. Classification of the Zoonotic Hepatitis E Virus Genotype 3 Into Distinct Subgenotypes. Front. Microbiol. 2020, 11, 634430. [Google Scholar] [CrossRef]
- Brayne, A.B.; Dearlove, B.L.; Lester, J.S.; Kosakovsky Pond, S.L.; Frost, S.D.W. Genotype-Specific Evolution of Hepatitis E Virus. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Mahar, J.E.; Jenckel, M.; Huang, N.; Smertina, E.; Holmes, E.C.; Strive, T.; Hall, R.N. Frequent Intergenotypic Recombination Between the Non-Structural and Structural Genes is a Major Driver of Epidemiological Fitness in Caliciviruses. Virus Evol. 1093. [Google Scholar] [CrossRef] [PubMed]
- Mahar, J.E.; Hall, R.N.; Peacock, D.; Kovaliski, J.; Piper, M.; Mourant, R.; Huang, N.; Campbell, S.; Gu, X.; Read, A.; et al. Rabbit hemorrhagic disease virus 2 (RHDV2; GI.2) is replacing endemic strains of RHDV in the Australian landscape within 18 months of its arrival. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahar, J.E.; Read, A.J.; Gu, X.; Urakova, N.; Mourant, R.; Piper, M.; Haboury, S.; Holmes, E.C.; Strive, T.; Hall, R.N. Detection and Circulation of a Novel Rabbit Hemorrhagic Disease Virus in Australia. Emerg. Infect. Dis. 2018, 24, 22–31. [Google Scholar] [CrossRef]
- Kozakiewicz, C.P.; Burridge, C.P.; Funk, W.C.; Craft, M.E.; Crooks, K.R.; Fisher, R.N.; Fountain-Jones, N.M.; Jennings, M.K.; Kraberger, S.J.; Lee, J.S.; et al. Does the virus cross the road? Viral phylogeographic patterns among bobcat populations reflect a history of urban development. Evol Appl 2020, 13, 1806–1817. [Google Scholar] [CrossRef] [Green Version]
- Luk, K.C.; Coller, K.E.; Dawson, G.J.; Cloherty, G.A. Identification of a putative novel genotype 3/rabbit hepatitis E virus (HEV) recombinant. PLoS ONE 2018, 13, e0203618. [Google Scholar] [CrossRef] [PubMed]
- O’Keefe, J.; Tracy, L.; Yuen, L.; Bonanzinga, S.; Li, X.; Chong, B.; Nicholson, S.; Jackson, K. Autochthonous and Travel Acquired Hepatitis E Virus in Australia. Front. Microbiol. 2021, 12, 640325. [Google Scholar] [CrossRef] [PubMed]
- Yapa, C.M.; Furlong, C.; Rosewell, A.; Ward, K.A.; Adamson, S.; Shadbolt, C.; Kok, J.; Tracy, S.L.; Bowden, S.; Smedley, E.J.; et al. First reported outbreak of locally acquired hepatitis E virus infection in Australia. Med. J. Aust. 2016, 204, 274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, G.; Kruger, I. Farming Meat Rabbits in NSW. In Primefacts; NSW Department of Primary Industries: Wagga Wagga, Australia, 2006. [Google Scholar]
- Cierniak, F.; von Arnim, F.; Heckel, G.; Ulrich, R.G.; Groschup, M.H.; Eiden, M. A Putative Novel Hepatitis E Virus Genotype 3 Subtype Identified in Rabbit, Germany 2016. Viruses 2021, 13, 1065. [Google Scholar] [CrossRef] [PubMed]
- Eden, J.S.; Kovaliski, J.; Duckworth, J.A.; Swain, G.; Mahar, J.E.; Strive, T.; Holmes, E.C. Comparative Phylodynamics of Rabbit Hemorrhagic Disease Virus in Australia and New Zealand. J. Virol. 2015, 89, 9548–9558. [Google Scholar] [CrossRef] [Green Version]
- Hall, R.N.; Mahar, J.E.; Read, A.J.; Mourant, R.; Piper, M.; Huang, N.; Strive, T. A strain-specific multiplex RT-PCR for Australian rabbit haemorrhagic disease viruses uncovers a new recombinant virus variant in rabbits and hares. Transbound. Emerg. Dis. 2018, 65, e444–e456. [Google Scholar] [CrossRef]
- Jothikumar, N.; Cromeans, T.L.; Robertson, B.H.; Meng, X.J.; Hill, V.R. A broadly reactive one-step real-time RT-PCR assay for rapid and sensitive detection of hepatitis E virus. J. Virol. Methods 2006, 131, 65–71. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [Green Version]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [Green Version]
- Ramsey, D.S.L.; Cox, T.; Strive, T.; Forsyth, D.M.; Stuart, I.; Hall, R.; Elsworth, P.; Campbell, S.; Knutie, S. Emerging RHDV2 suppresses the impact of endemic and novel strains of RHDV on wild rabbit populations. J. Appl. Ecol. 2020, 57, 630–641. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Campitelli, E. ggnewscale: Multiple Fill and Colour Scales in ’ggplot2’, R package version 0.4.3.
- Yu, G. scatterpie: Scatter Pie Plot, R package version 0.1.6.
- Hijmans, R.J. raster: Geographic Data Analysis and Modeling, R package version 3.4-13.
- Wang, L.G.; Lam, T.T.; Xu, S.; Dai, Z.; Zhou, L.; Feng, T.; Guo, P.; Dunn, C.W.; Jones, B.R.; Bradley, T.; et al. Treeio: An R Package for Phylogenetic Tree Input and Output with Richly Annotated and Associated Data. Mol. Biol. Evol. 2020, 37, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.-Y. ggtree: An r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Revell, L.J. phytools: An R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 2012, 3, 217–223. [Google Scholar] [CrossRef]
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4. [Google Scholar] [CrossRef]
- Wilke, C.O. cowplot: Streamlined Plot Theme and Plot Annotations for ’ggplot2’; R package version 1.1.1.
- Neuwirth, E. RColorBrewer: ColorBrewer Palettes, R package version 1.1-2.
- Wickham, H.; Seidel, D. scales: Scale Functions for Visualization, R package version 1.1.1.
- Dowle, M.; Srinivasan, A. data.table: Extension of ‘data.frame‘, R package version 1.14.0.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site | State * | Detected % (Number of Samples) | Not Detected % (Number of Samples) | Indeterminate % (Number of Samples) |
|---|---|---|---|---|
| Oaky Creek | NSW | 10 (2/20) | 75 (15/20) | 15 (3/20) |
| Coorong | SA | 5.43 (5/92) | 91.30 (84/92) | 3.26 (3/92) |
| Scobie | SA | 22.09 (25/86) | 72.09 (68/86) | 5.81 (11/86) |
| Wallangarra East | QLD | 0 (0/24) | 100 (24/24) | 0 (0/24) |
| Avalon | VIC | 5 (1/20) | 95 (19/20) | 0 (0/20) |
| Pyramid Hill | VIC | 5 (1/20) | 75 (15/20) | 20 (4/20) |
| Drummonds | WA | 5 (1/20) | 85 (17/20) | 10 (2/20) |
| Nelsons | WA | 10 (2/20) | 90 (18/20) | 0 (0/20) |
| Eurolie | NSW | 0 (0/20) | 100 (20/20) | 0 (0/20) |
| Gudgenby | ACT | 0 (0/20) | 100 (20/20) | 0 (0/20) |
| Wallangarra West | QLD | 10 (2/20) | 85 (17/20) | 5 (1/20) |
| Total (wild rabbits) | 9.11 (33/362) | 85.91 (311/362) | 4.97 (18/362) | |
| domestic rabbits | 11.25 (9/80) | 81.25 (65/80) | 7.5 (6/80) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jenckel, M.; Smith, I.; King, T.; West, P.; Taggart, P.L.; Strive, T.; Hall, R.N. Distribution and Genetic Diversity of Hepatitis E Virus in Wild and Domestic Rabbits in Australia. Pathogens 2021, 10, 1637. https://doi.org/10.3390/pathogens10121637
Jenckel M, Smith I, King T, West P, Taggart PL, Strive T, Hall RN. Distribution and Genetic Diversity of Hepatitis E Virus in Wild and Domestic Rabbits in Australia. Pathogens. 2021; 10(12):1637. https://doi.org/10.3390/pathogens10121637
Chicago/Turabian StyleJenckel, Maria, Ina Smith, Tegan King, Peter West, Patrick L. Taggart, Tanja Strive, and Robyn N. Hall. 2021. "Distribution and Genetic Diversity of Hepatitis E Virus in Wild and Domestic Rabbits in Australia" Pathogens 10, no. 12: 1637. https://doi.org/10.3390/pathogens10121637
APA StyleJenckel, M., Smith, I., King, T., West, P., Taggart, P. L., Strive, T., & Hall, R. N. (2021). Distribution and Genetic Diversity of Hepatitis E Virus in Wild and Domestic Rabbits in Australia. Pathogens, 10(12), 1637. https://doi.org/10.3390/pathogens10121637