
Cell-Type Apoptosis in Lung during SARS-CoV-2 Infection

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
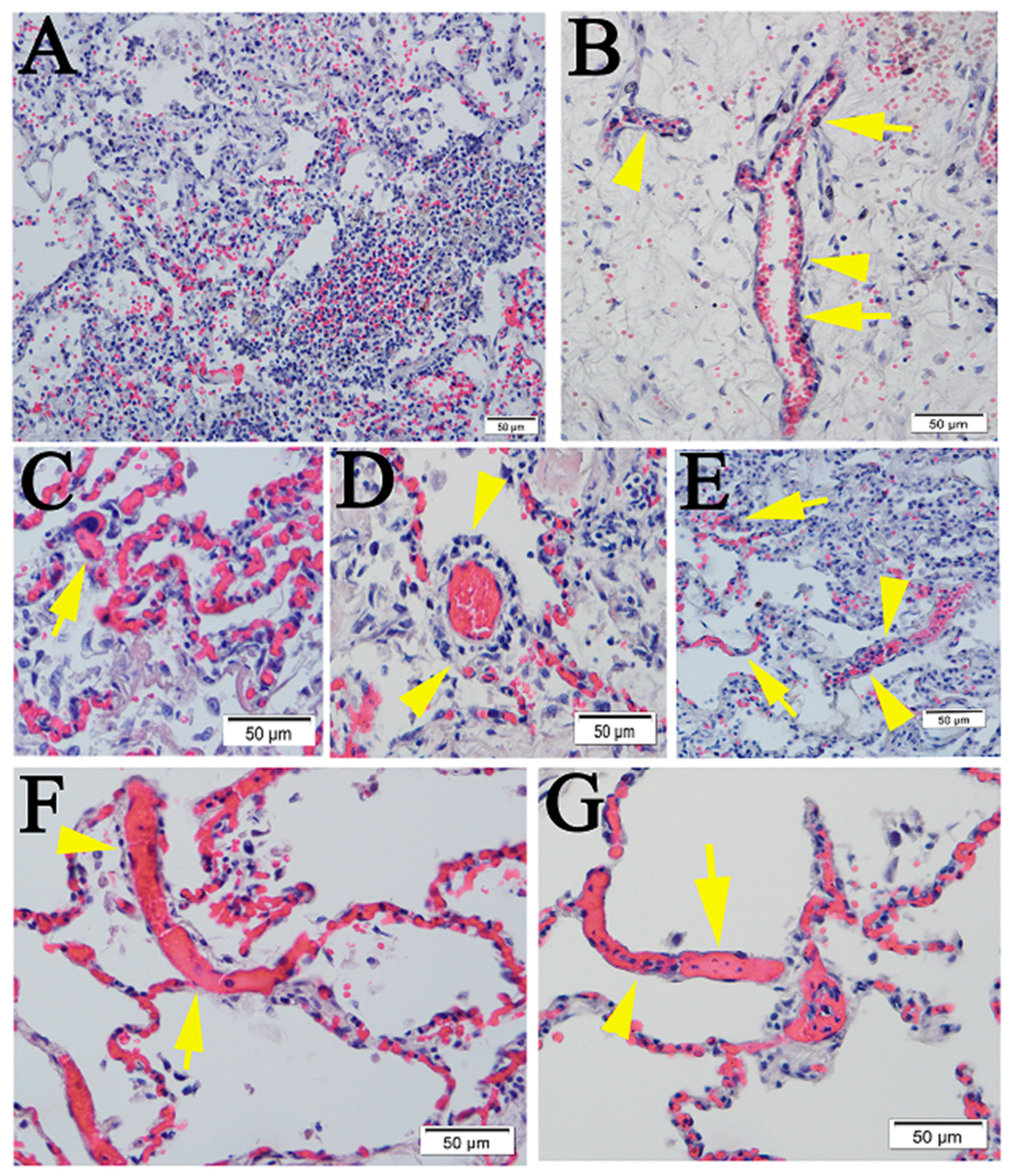
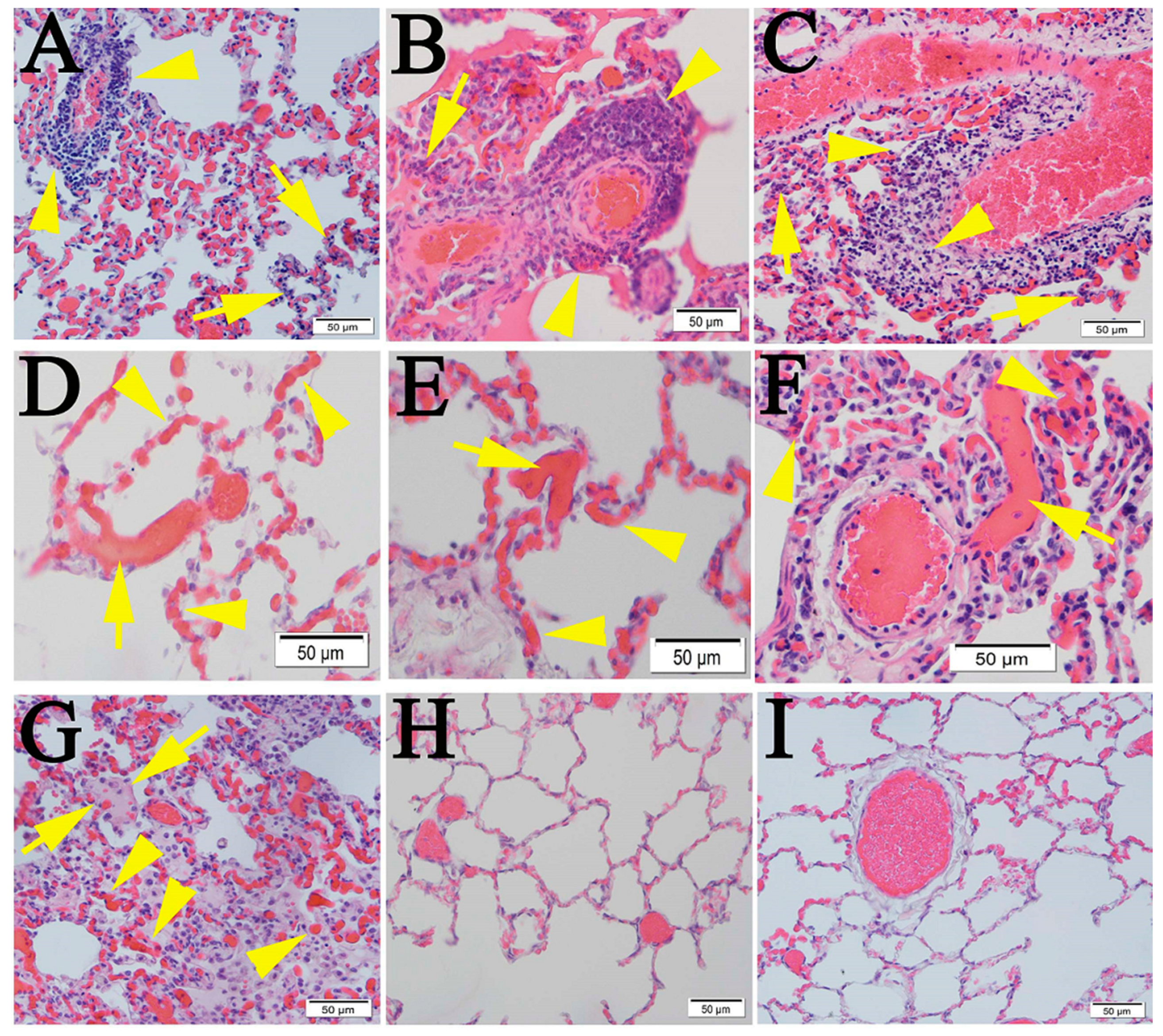
2.1. Similar Pulmonary Pathology Was Observed in Humans and Non-Human Primates Following SARS-CoV-2 Infection
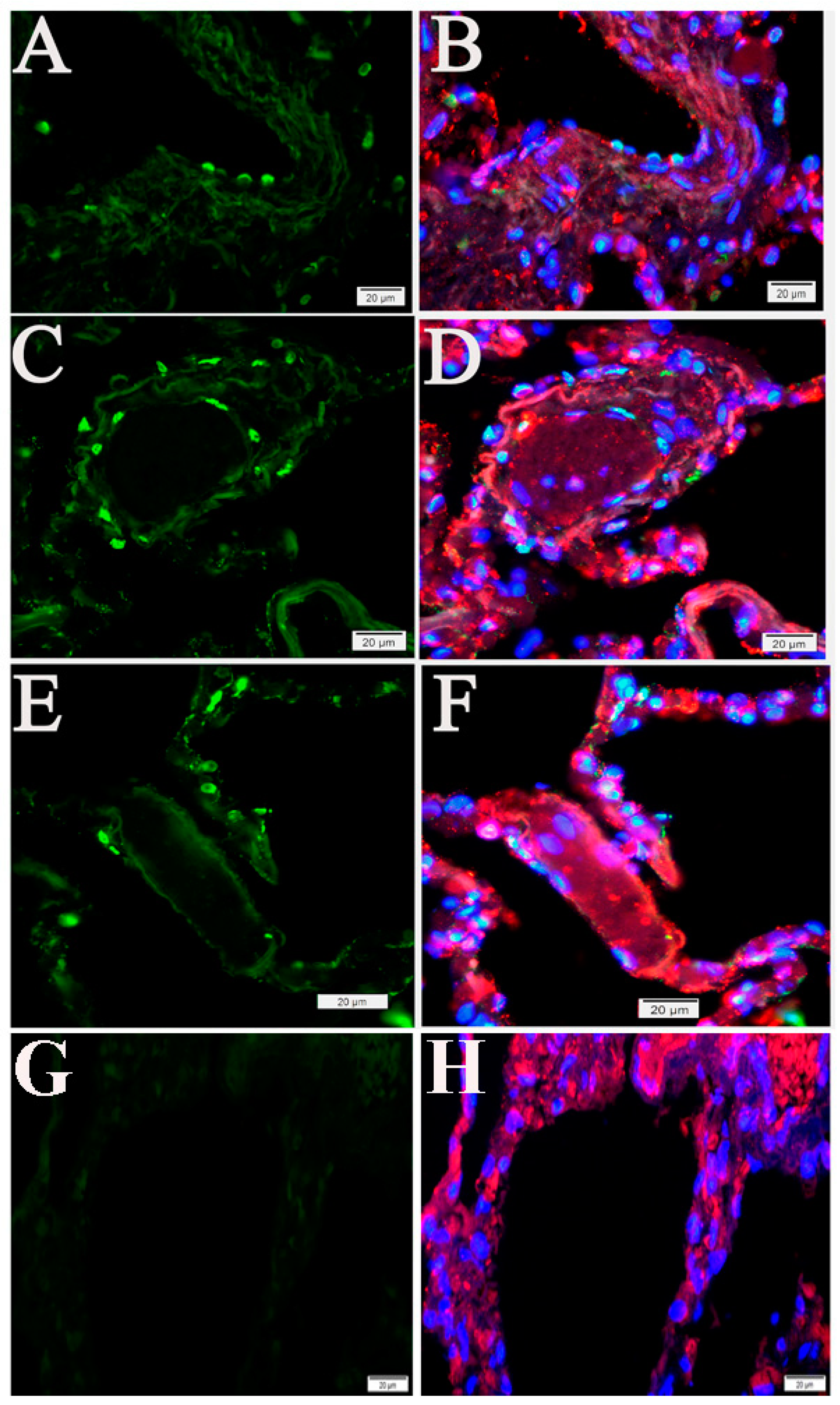
2.2. Extensive Apoptotic Signals Were Detected in the Lung Tissues of Humans and NHPs Following SARS-CoV-2 Infections
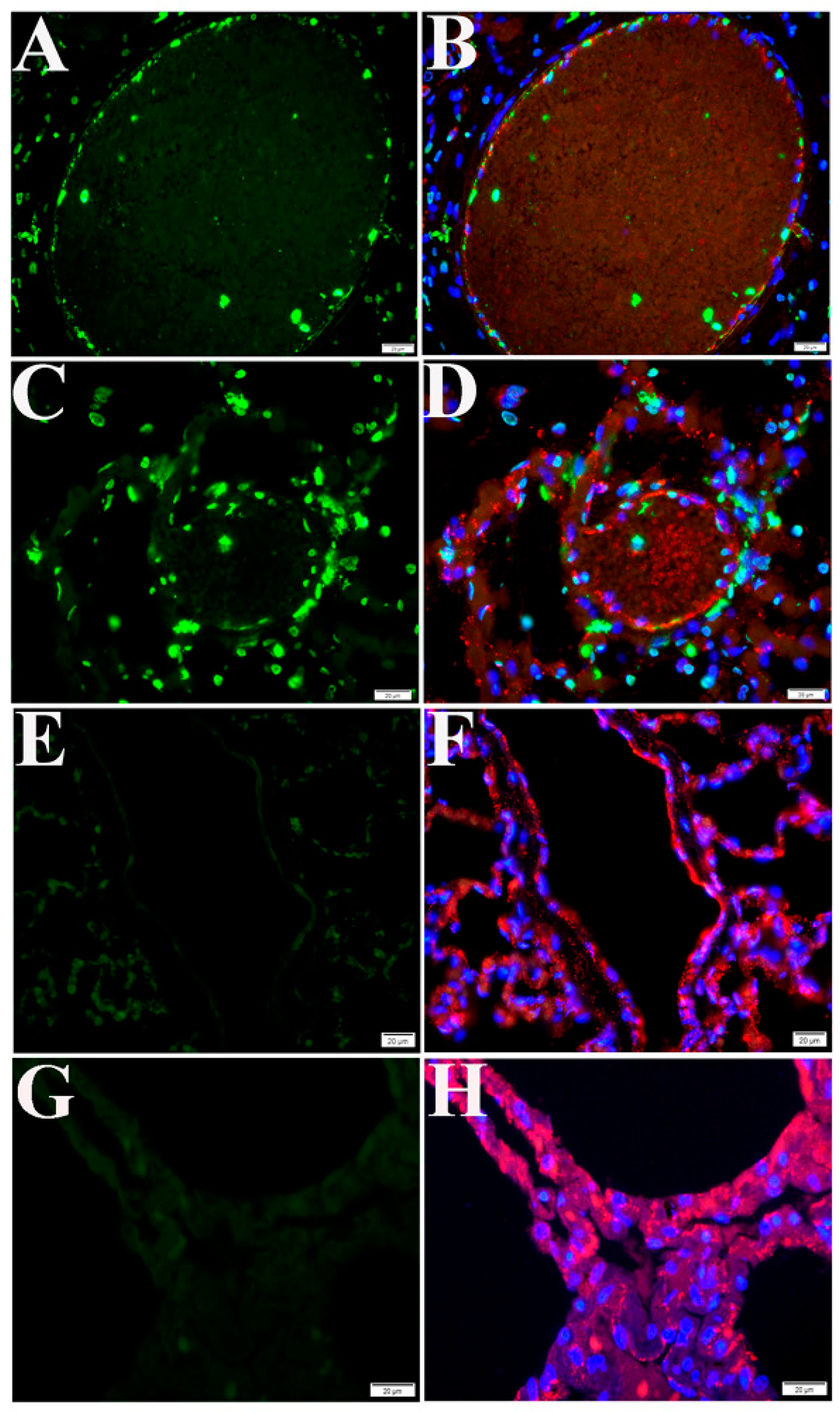
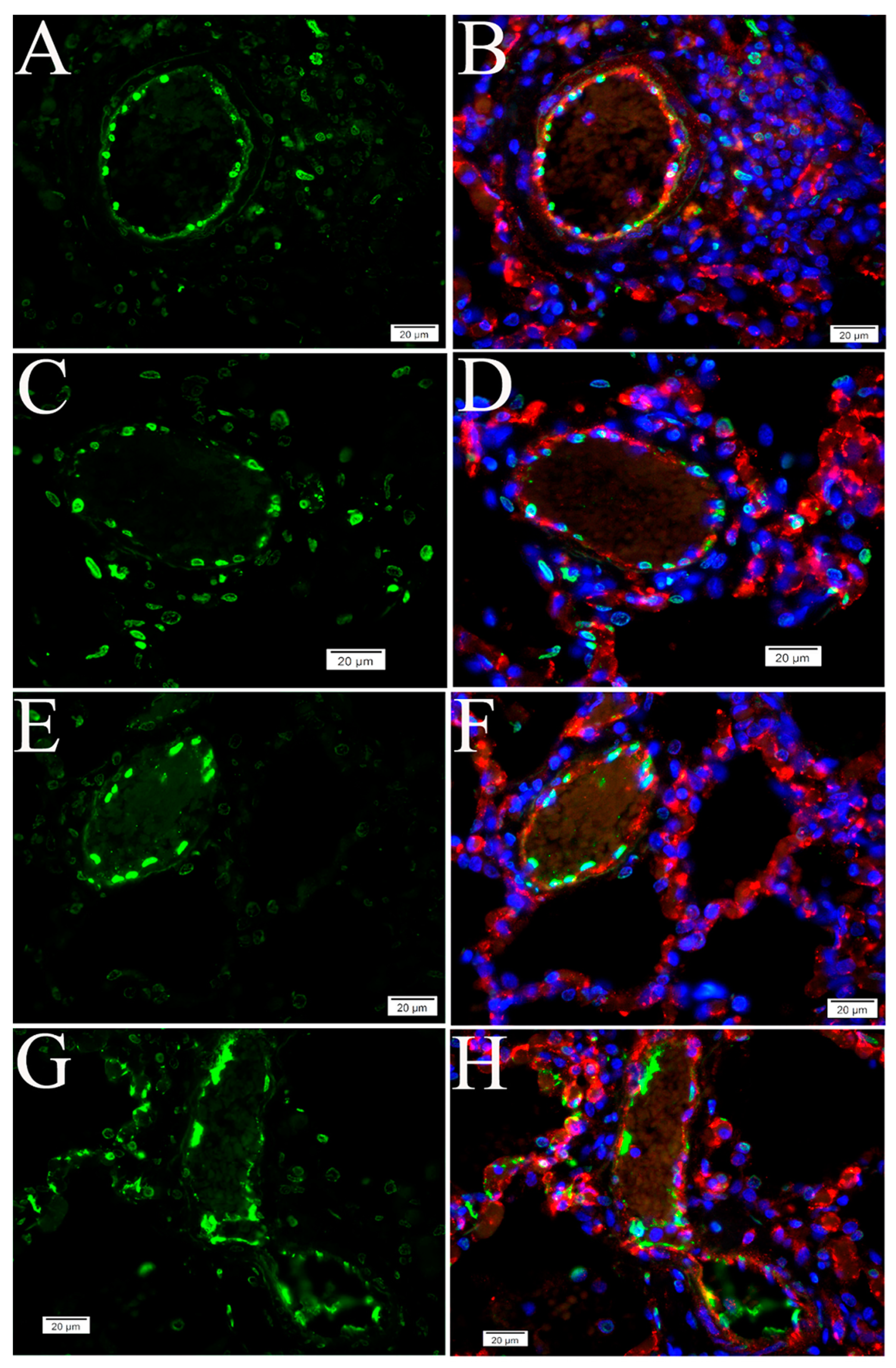
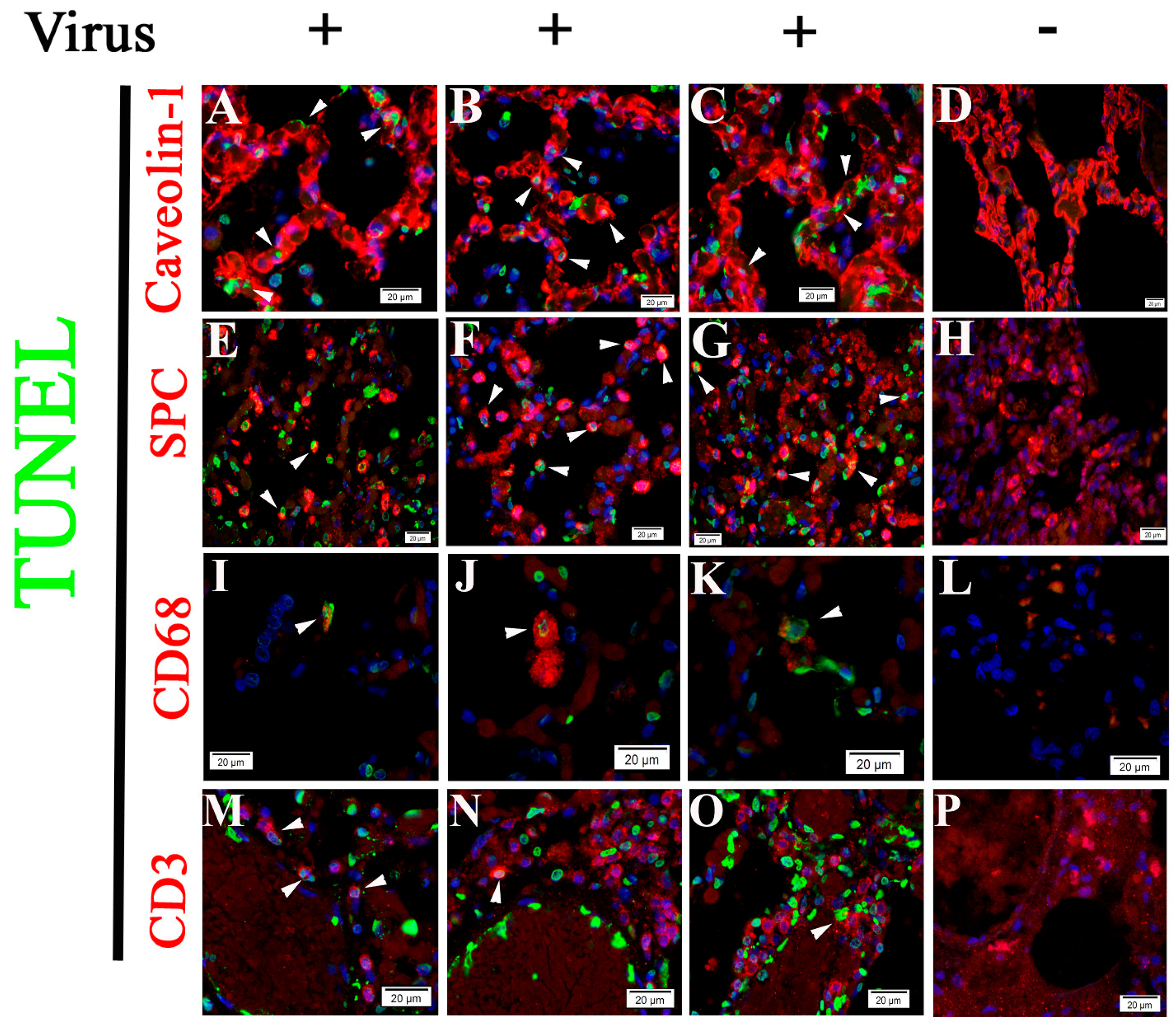
2.3. Cell Type Identification of Apoptosis in NHP Lung Following SARS-CoV-2 Infection
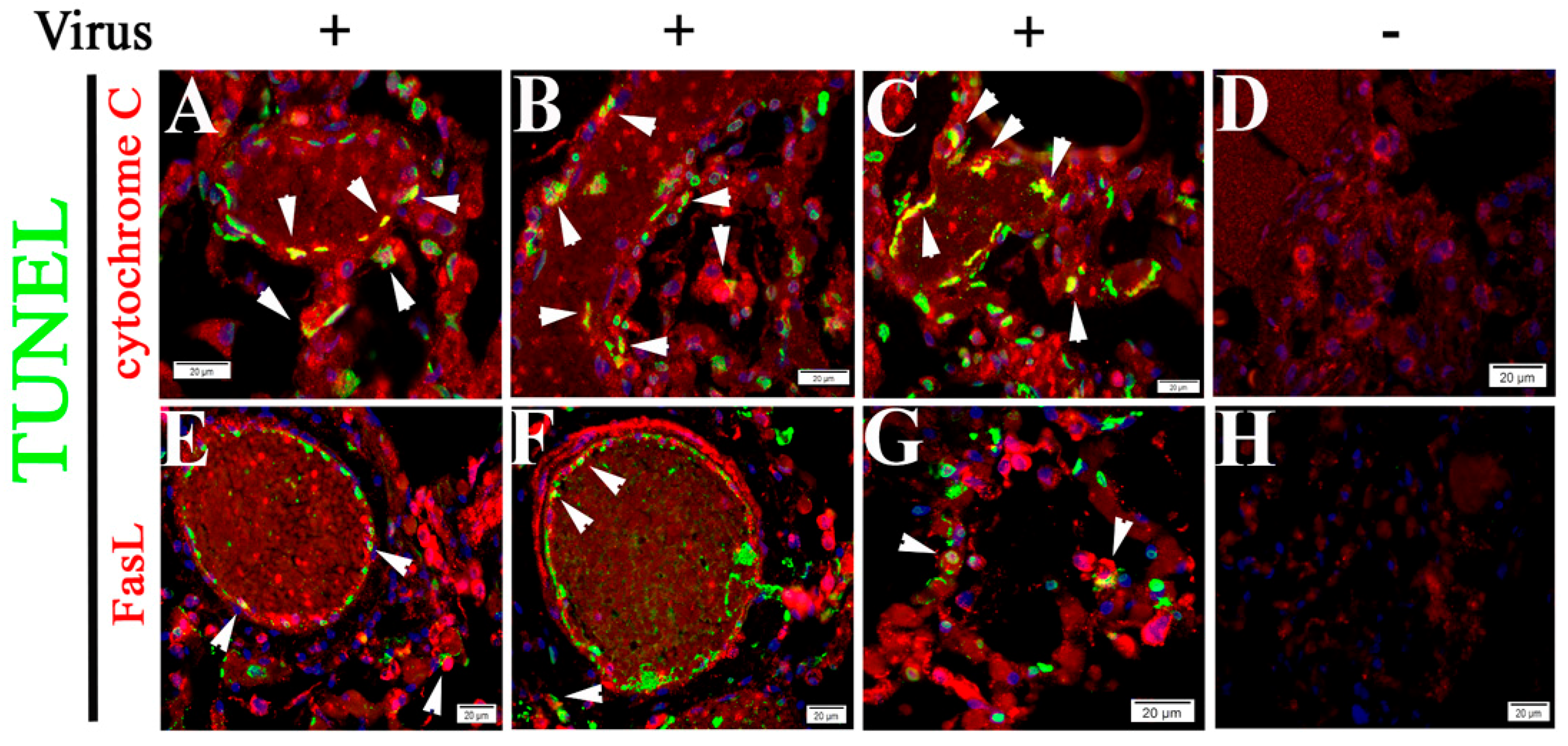
2.4. Both Intrinsic and Extrinsic Apoptotic Pathways Are Activated Following SARS-CoV-2 Infection
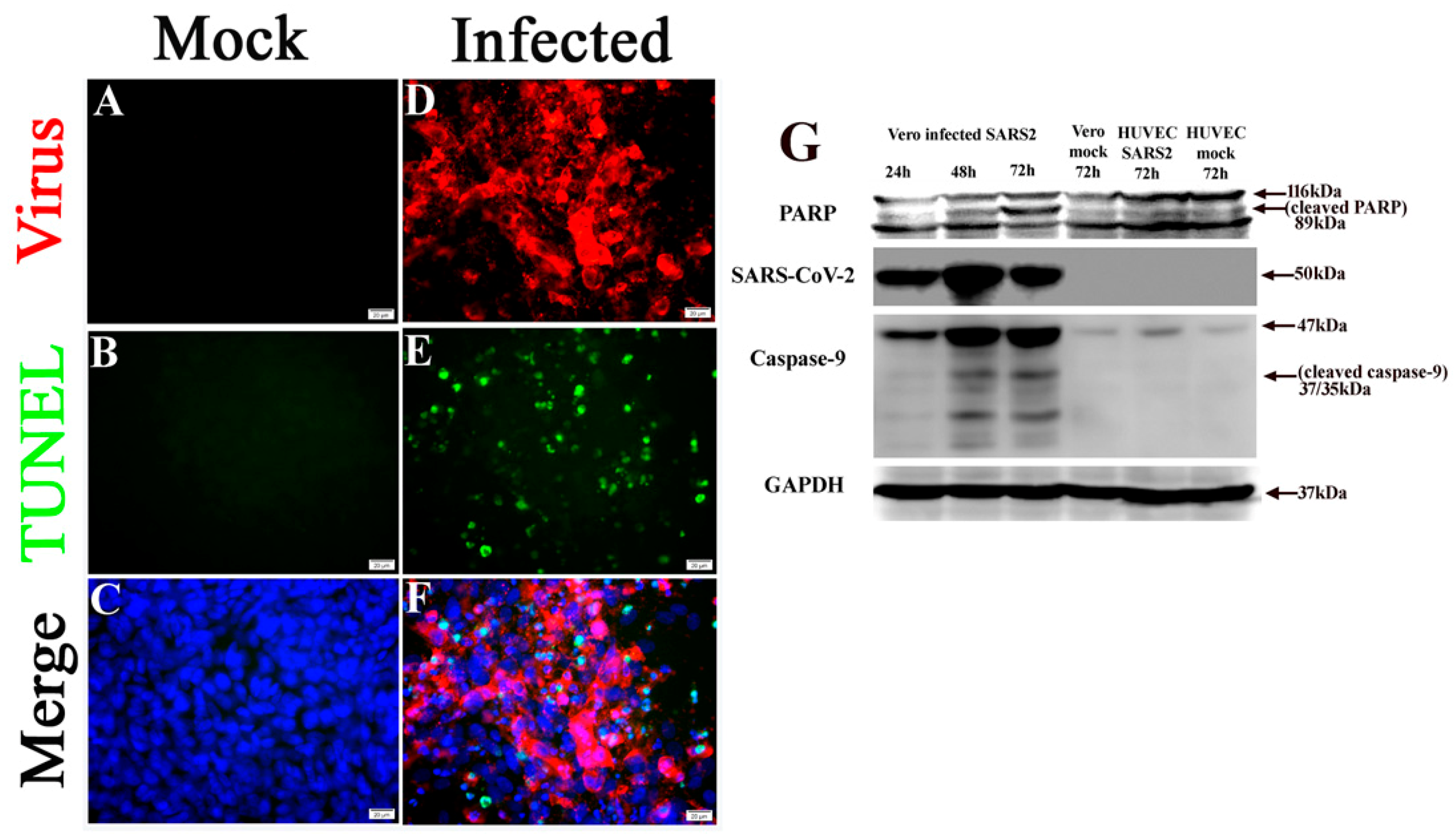
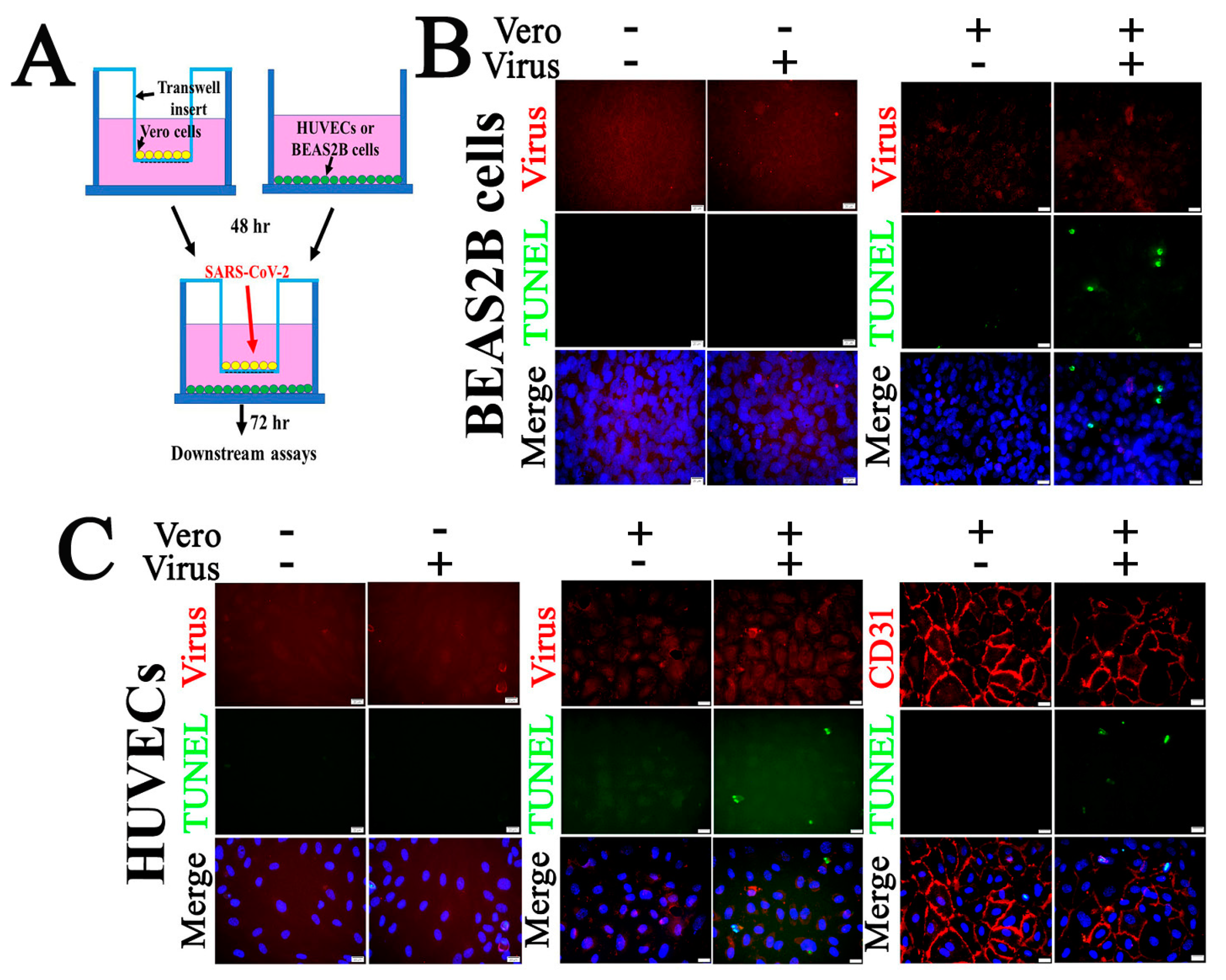
2.5. SARS-CoV-2 Infection Triggers Apoptosis in Non-Permissive Cells
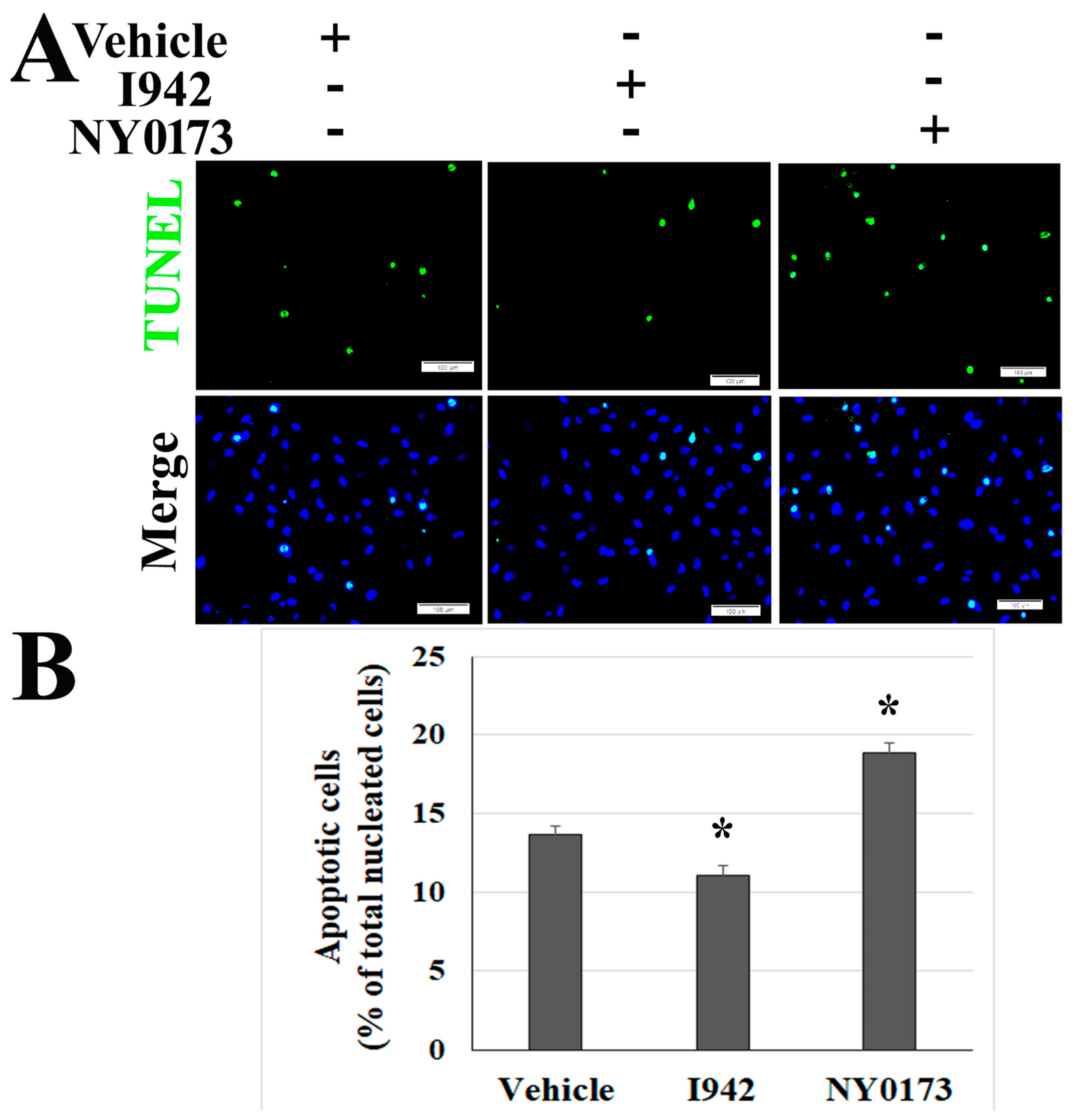
2.6. Pharmacological Activators of EPAC1 Reduce Endothelial Apoptosis in Vero Cells and HUVECs co-Culture Following Infection with SARS-CoV-2
3. Discussion
4. Materials and Methods
4.1. Clinical Specimens
4.2. Biosafety Level and Study Subjects
4.3. Co-Culture of Vero Cells and HUVECs or Vero Cells and BEAS2B Cells
4.4. In Situ TUNEL Staining and Quantitative Analysis of TUNEL Signals
4.5. IF-TUNEL Dual Staining
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Munster, V.J.; Feldmann, F.; Williamson, B.N.; van Doremalen, N.; Pérez-Pérez, L.; Schulz, J.; Meade-White, K.; Okumura, A.; Callison, J.; Brumbaugh, B.; et al. Respiratory disease in rhesus macaques inoculated with SARS-CoV-2. Nature 2020. [Google Scholar] [CrossRef] [PubMed]
- Attaway, A.H.; Scheraga, R.G.; Bhimraj, A.; Biehl, M.; Hatipoğlu, U. Severe covid-19 pneumonia: Pathogenesis and clinical management. BMJ 2021, 372, n436. [Google Scholar] [CrossRef] [PubMed]
- Bompard, F.; Monnier, H.; Saab, I.; Tordjman, M.; Abdoul, H.; Fournier, L.; Sanchez, O.; Lorut, C.; Chassagnon, G.; Revel, M.P. Pulmonary embolism in patients with Covid-19 pneumonia. Eur. Respir. J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ma, X. Acute respiratory failure in COVID-19: Is it “typical” ARDS? Crit. Care 2020, 24, 198. [Google Scholar] [CrossRef]
- Hariri, L.; Hardin, C.C. Covid-19, Angiogenesis, and ARDS Endotypes. N. Engl. J. Med. 2020, 383, 182–183. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020. [Google Scholar] [CrossRef]
- Chandrashekar, A.; Liu, J.; Martinot, A.J.; McMahan, K.; Mercado, N.B.; Peter, L.; Tostanoski, L.H.; Yu, J.; Maliga, Z.; Nekorchuk, M.; et al. SARS-CoV-2 infection protects against rechallenge in rhesus macaques. Science 2020, 369, 812–817. [Google Scholar] [CrossRef]
- Jose, R.J.; Manuel, A. COVID-19 cytokine storm: The interplay between inflammation and coagulation. Lancet Respir. Med. 2020, 8, e46–e47. [Google Scholar] [CrossRef]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J.; HLH Across Speciality Collaboration, U.K. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Cortese, M.; Lee, J.Y.; Cerikan, B.; Neufeldt, C.J.; Oorschot, V.M.J.; Köhrer, S.; Hennies, J.; Schieber, N.L.; Ronchi, P.; Mizzon, G.; et al. Integrative Imaging Reveals SARS-CoV-2-Induced Reshaping of Subcellular Morphologies. Cell Host Microbe 2020. [Google Scholar] [CrossRef]
- Karki, R.; Sharma, B.R.; Tuladhar, S.; Williams, E.P.; Zalduondo, L.; Samir, P.; Zheng, M.; Sundaram, B.; Banoth, B.; Malireddi, R.K.S.; et al. Synergism of TNF-α and IFN-γ Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell 2020. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, T.S.; de Sá, K.S.G.; Ishimoto, A.Y.; Becerra, A.; Oliveira, S.; Almeida, L.; Gonçalves, A.V.; Perucello, D.B.; Andrade, W.A.; Castro, R.; et al. Inflammasomes are activated in response to SARS-CoV-2 infection and are associated with COVID-19 severity in patients. J. Exp. Med. 2021, 218. [Google Scholar] [CrossRef] [PubMed]
- Sardu, C.; Gambardella, J.; Morelli, M.B.; Wang, X.; Marfella, R.; Santulli, G. Hypertension, Thrombosis, Kidney Failure, and Diabetes: Is COVID-19 an Endothelial Disease? A Comprehensive Evaluation of Clinical and Basic Evidence. J. Clin. Med. 2020, 9, 1417. [Google Scholar] [CrossRef] [PubMed]
- Rowley, A.H.; Shulman, S.T.; Arditi, M. Immune pathogenesis of COVID-19-related multisystem inflammatory syndrome in children. J. Clin. Investig. 2020, 130, 5619–5621. [Google Scholar] [CrossRef]
- Bao, L.; Deng, W.; Huang, B.; Gao, H.; Liu, J.; Ren, L.; Wei, Q.; Yu, P.; Xu, Y.; Qi, F.; et al. The pathogenicity of SARS-CoV-2 in hACE2 transgenic mice. Nature 2020, 583, 830–833. [Google Scholar] [CrossRef]
- Sauler, M.; Bazan, I.S.; Lee, P.J. Cell Death in the Lung: The Apoptosis-Necroptosis Axis. Annu. Rev. Physiol. 2019, 81, 375–402. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Aid, M.; Busman-Sahay, K.; Vidal, S.J.; Maliga, Z.; Bondoc, S.; Starke, C.; Terry, M.; Jacobson, C.A.; Wrijil, L.; Ducat, S.; et al. Vascular Disease and Thrombosis in SARS-CoV-2-Infected Rhesus Macaques. Cell 2020. [Google Scholar] [CrossRef]
- Veras, F.P.; Pontelli, M.C.; Silva, C.M.; Toller-Kawahisa, J.E.; de Lima, M.; Nascimento, D.C.; Schneider, A.H.; Caetité, D.; Tavares, L.A.; Paiva, I.M.; et al. SARS-CoV-2-triggered neutrophil extracellular traps mediate COVID-19 pathology. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Sheahan, T.P.; Sims, A.C.; Zhou, S.; Graham, R.L.; Pruijssers, A.J.; Agostini, M.L.; Leist, S.R.; Schäfer, A.; Dinnon, K.H.; Stevens, L.J.; et al. An orally bioavailable broad-spectrum antiviral inhibits SARS-CoV-2 in human airway epithelial cell cultures and multiple coronaviruses in mice. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.F.; Zhang, A.J.; Yuan, S.; Poon, V.K.; Chan, C.C.; Lee, A.C.; Chan, W.M.; Fan, Z.; Tsoi, H.W.; Wen, L.; et al. Simulation of the clinical and pathological manifestations of Coronavirus Disease 2019 (COVID-19) in golden Syrian hamster model: Implications for disease pathogenesis and transmissibility. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhang, Y.; Guan, Z.; Li, H.; Ye, M.; Chen, X.; Shen, J.; Zhou, Y.; Shi, Z.L.; Zhou, P.; et al. SARS-CoV-2 triggers inflammatory responses and cell death through caspase-8 activation. Signal Transduct. Target Ther. 2020, 5, 235. [Google Scholar] [CrossRef] [PubMed]
- Rockx, B.; Kuiken, T.; Herfst, S.; Bestebroer, T.; Lamers, M.M.; Oude Munnink, B.B.; de Meulder, D.; van Amerongen, G.; van den Brand, J.; Okba, N.M.A.; et al. Comparative pathogenesis of COVID-19, MERS, and SARS in a nonhuman primate model. Science 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, D.K.; Singh, B.; Ganatra, S.R.; Gazi, M.; Cole, J.; Thippeshappa, R.; Alfson, K.J.; Clemmons, E.; Gonzalez, O.; Escobedo, R.; et al. Responses to acute infection with SARS-CoV-2 in the lungs of rhesus macaques, baboons and marmosets. Nat. Microbiol. 2020. [Google Scholar] [CrossRef]
- Imai, M.; Iwatsuki-Horimoto, K.; Hatta, M.; Loeber, S.; Halfmann, P.J.; Nakajima, N.; Watanabe, T.; Ujie, M.; Takahashi, K.; Ito, M.; et al. Syrian hamsters as a small animal model for SARS-CoV-2 infection and countermeasure development. Proc. Natl. Acad. Sci. USA 2020, 117, 16587–16595. [Google Scholar] [CrossRef]
- Sia, S.F.; Yan, L.M.; Chin, A.W.H.; Fung, K.; Choy, K.T.; Wong, A.Y.L.; Kaewpreedee, P.; Perera, R.A.P.M.; Poon, L.L.M.; Nicholls, J.M.; et al. Pathogenesis and transmission of SARS-CoV-2 in golden hamsters. Nature 2020, 583, 834–838. [Google Scholar] [CrossRef]
- Osterrieder, N.; Bertzbach, L.D.; Dietert, K.; Abdelgawad, A.; Vladimirova, D.; Kunec, D.; Hoffmann, D.; Beer, M.; Gruber, A.D.; Trimpert, J. Age-Dependent Progression of SARS-CoV-2 Infection in Syrian Hamsters. Viruses 2020, 12, 779. [Google Scholar] [CrossRef]
- Tostanoski, L.H.; Wegmann, F.; Martinot, A.J.; Loos, C.; McMahan, K.; Mercado, N.B.; Yu, J.; Chan, C.N.; Bondoc, S.; Starke, C.E.; et al. Ad26 vaccine protects against SARS-CoV-2 severe clinical disease in hamsters. Nat. Med. 2020. [Google Scholar] [CrossRef]
- Kreye, J.; Reincke, S.M.; Kornau, H.C.; Sánchez-Sendin, E.; Corman, V.M.; Liu, H.; Yuan, M.; Wu, N.C.; Zhu, X.; Lee, C.D.; et al. A Therapeutic Non-self-reactive SARS-CoV-2 Antibody Protects from Lung Pathology in a COVID-19 Hamster Model. Cell 2020. [Google Scholar] [CrossRef]
- Leist, S.R.; Dinnon, K.H.; Schäfer, A.; Tse, L.V.; Okuda, K.; Hou, Y.J.; West, A.; Edwards, C.E.; Sanders, W.; Fritch, E.J.; et al. A Mouse-Adapted SARS-CoV-2 Induces Acute Lung Injury and Mortality in Standard Laboratory Mice. Cell 2020. [Google Scholar] [CrossRef]
- Winkler, E.S.; Bailey, A.L.; Kafai, N.M.; Nair, S.; McCune, B.T.; Yu, J.; Fox, J.M.; Chen, R.E.; Earnest, J.T.; Keeler, S.P.; et al. SARS-CoV-2 infection of human ACE2-transgenic mice causes severe lung inflammation and impaired function. Nat. Immunol. 2020, 21, 1327–1335. [Google Scholar] [CrossRef] [PubMed]
- Mossel, E.C.; Huang, C.; Narayanan, K.; Makino, S.; Tesh, R.B.; Peters, C.J. Exogenous ACE2 expression allows refractory cell lines to support severe acute respiratory syndrome coronavirus replication. J. Virol. 2005, 79, 3846–3850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, S.; Yokoyama, U.; Abe, T.; Kiyonari, H.; Yamashita, N.; Kato, Y.; Kurotani, R.; Sato, M.; Okumura, S.; Ishikawa, Y. Differential roles of Epac in regulating cell death in neuronal and myocardial cells. J. Biol. Chem. 2010, 285, 24248–24259. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, K.A.; Gao, Z.G. Adenosine receptors as therapeutic targets. Nat. Rev. Drug Discov. 2006, 5, 247–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, C.; Oliveira, R.C.; Serra, P.; Baptista, J.P.; Sousa, E.; Casanova, P.; Pimentel, J.; Carvalho, L. Pathophysiology of acute fibrinous and organizing pneumonia—Clinical and morphological spectra. Pathophysiology 2019, 26, 213–217. [Google Scholar] [CrossRef]
- Yan, H.; Xiao, G.; Zhang, J.; Hu, Y.; Yuan, F.; Cole, D.K.; Zheng, C.; Gao, G.F. SARS coronavirus induces apoptosis in Vero E6 cells. J. Med. Virol. 2004, 73, 323–331. [Google Scholar] [CrossRef]
- Gong, B.; Asimakis, G.K.; Chen, Z.; Albrecht, T.B.; Boor, P.J.; Pappas, T.C.; Bell, B.; Motamedi, M. Whole-body hyperthermia induces up-regulation of vascular endothelial growth factor accompanied by neovascularization in cardiac tissue. Life Sci. 2006, 79, 1781–1788. [Google Scholar] [CrossRef]
- Fuchs, S.; Hollins, A.J.; Laue, M.; Schaefer, U.F.; Roemer, K.; Gumbleton, M.; Lehr, C.M. Differentiation of human alveolar epithelial cells in primary culture: Morphological characterization and synthesis of caveolin-1 and surfactant protein-C. Cell Tissue Res. 2003, 311, 31–45. [Google Scholar] [CrossRef]
- Soong, L.; Wang, H.; Shelite, T.R.; Liang, Y.; Mendell, N.L.; Sun, J.; Gong, B.; Valbuena, G.A.; Bouyer, D.H.; Walker, D.H. Strong type 1, but impaired type 2, immune responses contribute to Orientia tsutsugamushi-induced pathology in mice. PLoS Negl. Trop. Dis. 2014, 8, e3191. [Google Scholar] [CrossRef]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef]
- Ren, Y.; Shu, T.; Wu, D.; Mu, J.; Wang, C.; Huang, M.; Han, Y.; Zhang, X.Y.; Zhou, W.; Qiu, Y.; et al. The ORF3a protein of SARS-CoV-2 induces apoptosis in cells. Cell. Mol. Immunol. 2020, 17, 881–883. [Google Scholar] [CrossRef]
- Drelich, A.; Judy, B.; He, X.; Chang, Q.; Yu, S.; Li, X.; Lu, F.; Wakamiya, M.; Popov, V.; Zhou, J.; et al. Exchange Protein Directly Activated by cAMP Modulates Ebola Virus Uptake into Vascular Endothelial Cells. Viruses 2018, 10, 563. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Mason, R.J. Pathogenesis of COVID-19 from a cell biology perspective. Eur. Respir. J. 2020, 55. [Google Scholar] [CrossRef] [Green Version]
- Ivanisenko, N.V.; Seyrek, K.; Kolchanov, N.A.; Ivanisenko, V.A.; Lavrik, I.N. The role of death domain proteins in host response upon SARS-CoV-2 infection: Modulation of programmed cell death and translational applications. Cell Death Discov. 2020, 6, 101. [Google Scholar] [CrossRef]
- Huertas, A.; Montani, D.; Savale, L.; Pichon, J.; Tu, L.; Parent, F.; Guignabert, C.; Humbert, M. Endothelial cell dysfunction: A major player in SARS-CoV-2 infection (COVID-19)? Eur. Respir. J. 2020, 56. [Google Scholar] [CrossRef]
- Imre, G. Cell death signalling in virus infection. Cell. Signal. 2020, 76, 109772. [Google Scholar] [CrossRef]
- Zheng, M.; Williams, E.P.; Malireddi, R.K.S.; Karki, R.; Banoth, B.; Burton, A.; Webby, R.; Channappanavar, R.; Jonsson, C.B.; Kanneganti, T.D. Impaired NLRP3 inflammasome activation/pyroptosis leads to robust inflammatory cell death via caspase-8/RIPK3 during coronavirus infection. J. Biol. Chem. 2020, 295, 14040–14052. [Google Scholar] [CrossRef] [PubMed]
- Hoagland, D.A.; Møller, R.; Uhl, S.A.; Oishi, K.; Frere, J.; Golynker, I.; Horiuchi, S.; Panis, M.; Blanco-Melo, D.; Sachs, D.; et al. Leveraging the antiviral type I interferon system as a first line of defense against SARS-CoV-2 pathogenicity. Immunity 2021, 54, 557–570.e555. [Google Scholar] [CrossRef]
- Chaudhry, M.Z.; Casalegno-Garduno, R.; Sitnik, K.M.; Kasmapour, B.; Pulm, A.K.; Brizic, I.; Eiz-Vesper, B.; Moosmann, A.; Jonjic, S.; Mocarski, E.S.; et al. Cytomegalovirus inhibition of extrinsic apoptosis determines fitness and resistance to cytotoxic CD8 T cells. Proc. Natl. Acad. Sci. USA 2020, 117, 12961–12968. [Google Scholar] [CrossRef] [PubMed]
- Halder, S.K.; Milner, R. Chronic mild hypoxia accelerates recovery from preexisting EAE by enhancing vascular integrity and apoptosis of infiltrated monocytes. Proc. Natl. Acad. Sci. USA 2020, 117, 11126–11135. [Google Scholar] [CrossRef]
- O’Neill, K.; Olson, B.J.; Huang, N.; Unis, D.; Clem, R.J. Rapid selection against arbovirus-induced apoptosis during infection of a mosquito vector. Proc. Natl. Acad. Sci. USA 2015, 112, E1152–E1161. [Google Scholar] [CrossRef] [Green Version]
- Clarke, P.; Tyler, K.L. Apoptosis in animal models of virus-induced disease. Nat. Rev. Microbiol. 2009, 7, 144–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bombeli, T.; Karsan, A.; Tait, J.F.; Harlan, J.M. Apoptotic vascular endothelial cells become procoagulant. Blood 1997, 89, 2429–2442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paone, S.; Baxter, A.A.; Hulett, M.D.; Poon, I.K.H. Endothelial cell apoptosis and the role of endothelial cell-derived extracellular vesicles in the progression of atherosclerosis. Cell. Mol. Life Sci. 2019, 76, 1093–1106. [Google Scholar] [CrossRef] [PubMed]
- Belhadjer, Z.; Méot, M.; Bajolle, F.; Khraiche, D.; Legendre, A.; Abakka, S.; Auriau, J.; Grimaud, M.; Oualha, M.; Beghetti, M.; et al. Acute Heart Failure in Multisystem Inflammatory Syndrome in Children in the Context of Global SARS-CoV-2 Pandemic. Circulation 2020, 142, 429–436. [Google Scholar] [CrossRef]
- Fox, S.E.; Lameira, F.S.; Rinker, E.B.; Vander Heide, R.S. Cardiac Endotheliitis and Multisystem Inflammatory Syndrome after COVID-19. Ann. Intern. Med. 2020, 173, 1025–1027. [Google Scholar] [CrossRef]
- Beavo, J.A.; Brunton, L.L. Cyclic nucleotide research—Still expanding after half a century. Nat. Rev. Mol. Cell Biol. 2002, 3, 710–718. [Google Scholar] [CrossRef]
- Tao, X.; Mei, F.; Agrawal, A.; Peters, C.J.; Ksiazek, T.G.; Cheng, X.; Tseng, C.T. Blocking of exchange proteins directly activated by cAMP leads to reduced replication of Middle East respiratory syndrome coronavirus. J. Virol. 2014, 88, 3902–3910. [Google Scholar] [CrossRef] [Green Version]
- Kooistra, M.R.; Corada, M.; Dejana, E.; Bos, J.L. Epac1 regulates integrity of endothelial cell junctions through VE-cadherin. FEBS Lett. 2005, 579, 4966–4972. [Google Scholar] [CrossRef] [Green Version]
- Choi, E.J.; Ren, Y.; Chen, Y.; Liu, S.; Wu, W.; Ren, J.; Wang, P.; Garofalo, R.P.; Zhou, J.; Bao, X. Exchange Proteins Directly Activated by cAMP and Their Roles in Respiratory Syncytial Virus Infection. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, B.; Shelite, T.; Mei, F.C.; Ha, T.; Hu, Y.; Xu, G.; Chang, Q.; Wakamiya, M.; Ksiazek, T.G.; Boor, P.J.; et al. Exchange protein directly activated by cAMP plays a critical role in bacterial invasion during fatal rickettsioses. Proc. Natl. Acad. Sci. USA 2013, 110, 19615–19620. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Tang, K.; Levin, M.; Irfan, O.; Morris, S.K.; Wilson, K.; Klein, J.D.; Bhutta, Z.A. COVID-19 and multisystem inflammatory syndrome in children and adolescents. Lancet Infect. Dis. 2020, 20, e276–e288. [Google Scholar] [CrossRef]
- Gruber, C.N.; Patel, R.S.; Trachtman, R.; Lepow, L.; Amanat, F.; Krammer, F.; Wilson, K.M.; Onel, K.; Geanon, D.; Tuballes, K.; et al. Mapping Systemic Inflammation and Antibody Responses in Multisystem Inflammatory Syndrome in Children (MIS-C). Cell 2020, 183, 982–995.e914. [Google Scholar] [CrossRef] [PubMed]
- Hanley, B.; Lucas, S.B.; Youd, E.; Swift, B.; Osborn, M. Autopsy in suspected COVID-19 cases. J. Clin. Pathol. 2020, 73, 239–242. [Google Scholar] [CrossRef] [Green Version]
- Daniel, B.; DeCoster, M.A. Quantification of sPLA2-induced early and late apoptosis changes in neuronal cell cultures using combined TUNEL and DAPI staining. Brain Res. Brain Res. Protoc. 2004, 13, 144–150. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Garron, T.M.; Chang, Q.; Su, Z.; Zhou, C.; Qiu, Y.; Gong, E.C.; Zheng, J.; Yin, Y.W.; Ksiazek, T.; et al. Cell-Type Apoptosis in Lung during SARS-CoV-2 Infection. Pathogens 2021, 10, 509. https://doi.org/10.3390/pathogens10050509
Liu Y, Garron TM, Chang Q, Su Z, Zhou C, Qiu Y, Gong EC, Zheng J, Yin YW, Ksiazek T, et al. Cell-Type Apoptosis in Lung during SARS-CoV-2 Infection. Pathogens. 2021; 10(5):509. https://doi.org/10.3390/pathogens10050509
Chicago/Turabian StyleLiu, Yakun, Tania M. Garron, Qing Chang, Zhengchen Su, Changcheng Zhou, Yuan Qiu, Eric C. Gong, Junying Zheng, Y. Whitney Yin, Thomas Ksiazek, and et al. 2021. "Cell-Type Apoptosis in Lung during SARS-CoV-2 Infection" Pathogens 10, no. 5: 509. https://doi.org/10.3390/pathogens10050509
APA StyleLiu, Y., Garron, T. M., Chang, Q., Su, Z., Zhou, C., Qiu, Y., Gong, E. C., Zheng, J., Yin, Y. W., Ksiazek, T., Brasel, T., Jin, Y., Boor, P., Comer, J. E., & Gong, B. (2021). Cell-Type Apoptosis in Lung during SARS-CoV-2 Infection. Pathogens, 10(5), 509. https://doi.org/10.3390/pathogens10050509