
Altered Nasal Microbiota Composition Associated with Development of Polyserositis by Mycoplasma hyorhinis

 ,
,  ,
,  , and
, and 
Abstract
:1. Introduction
2. Results
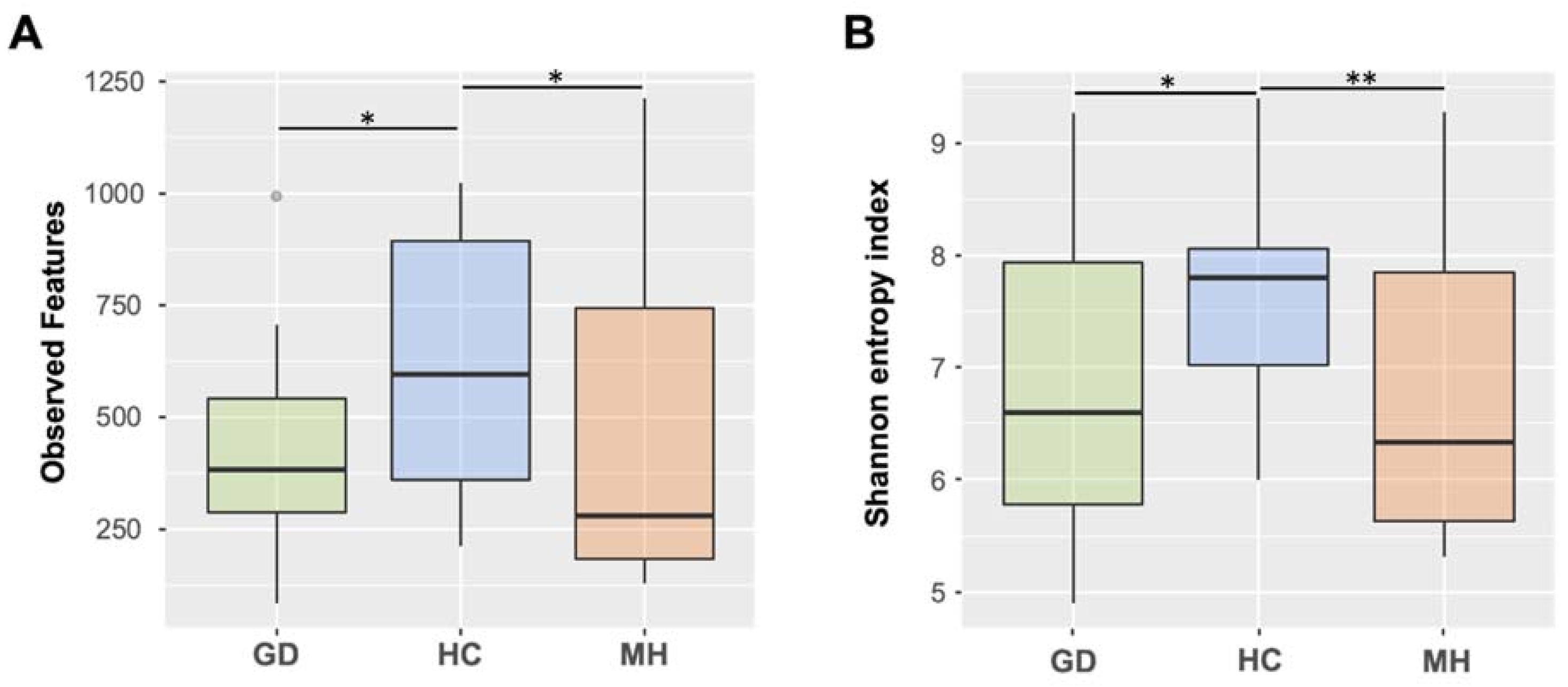
2.1. Alpha Diversity Is Significantly Reduced in the Nasal Microbiota of Piglets from Farms with Polyserositis
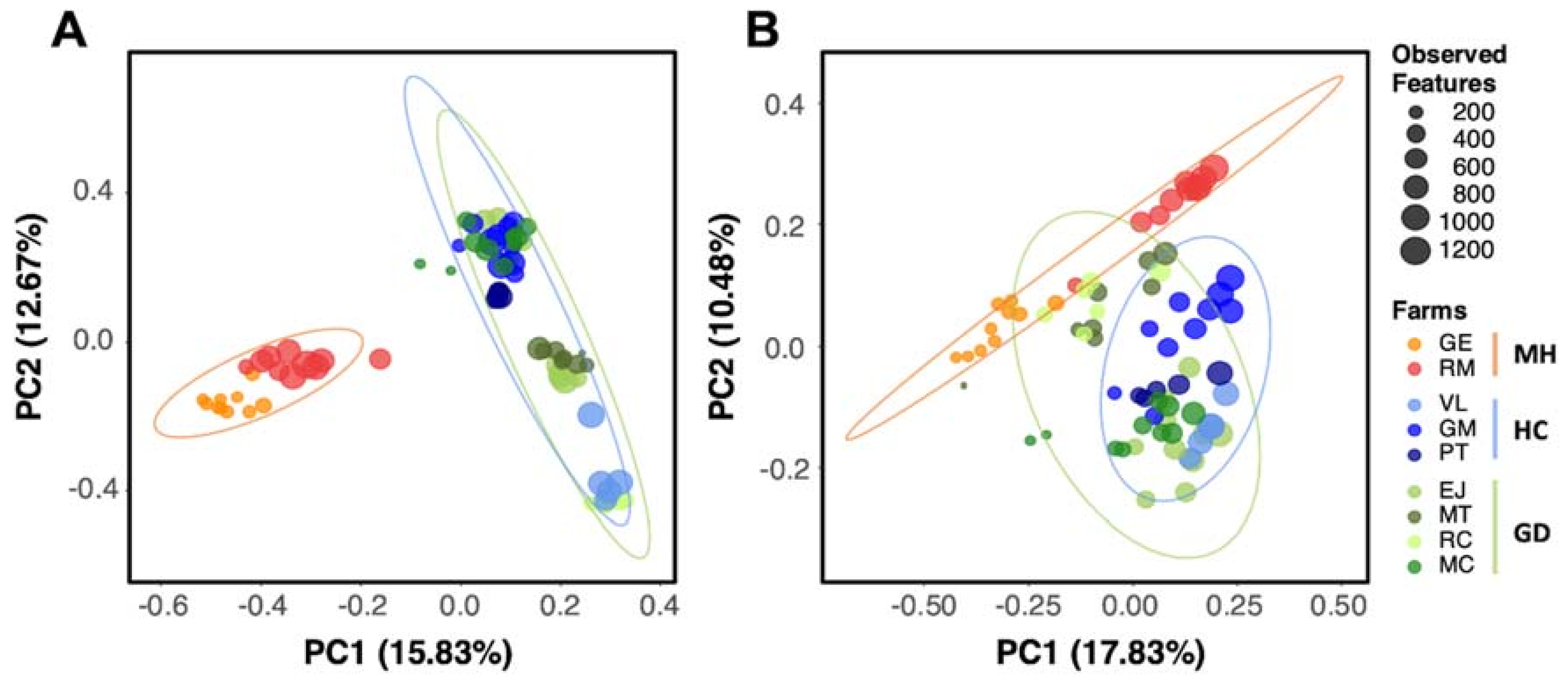
2.2. Piglets from Farms with Polyserositis by M. hyorhinis Showed Significatively Different Nasal Microbiota Composition Compared to Piglets from HC and GD Farms
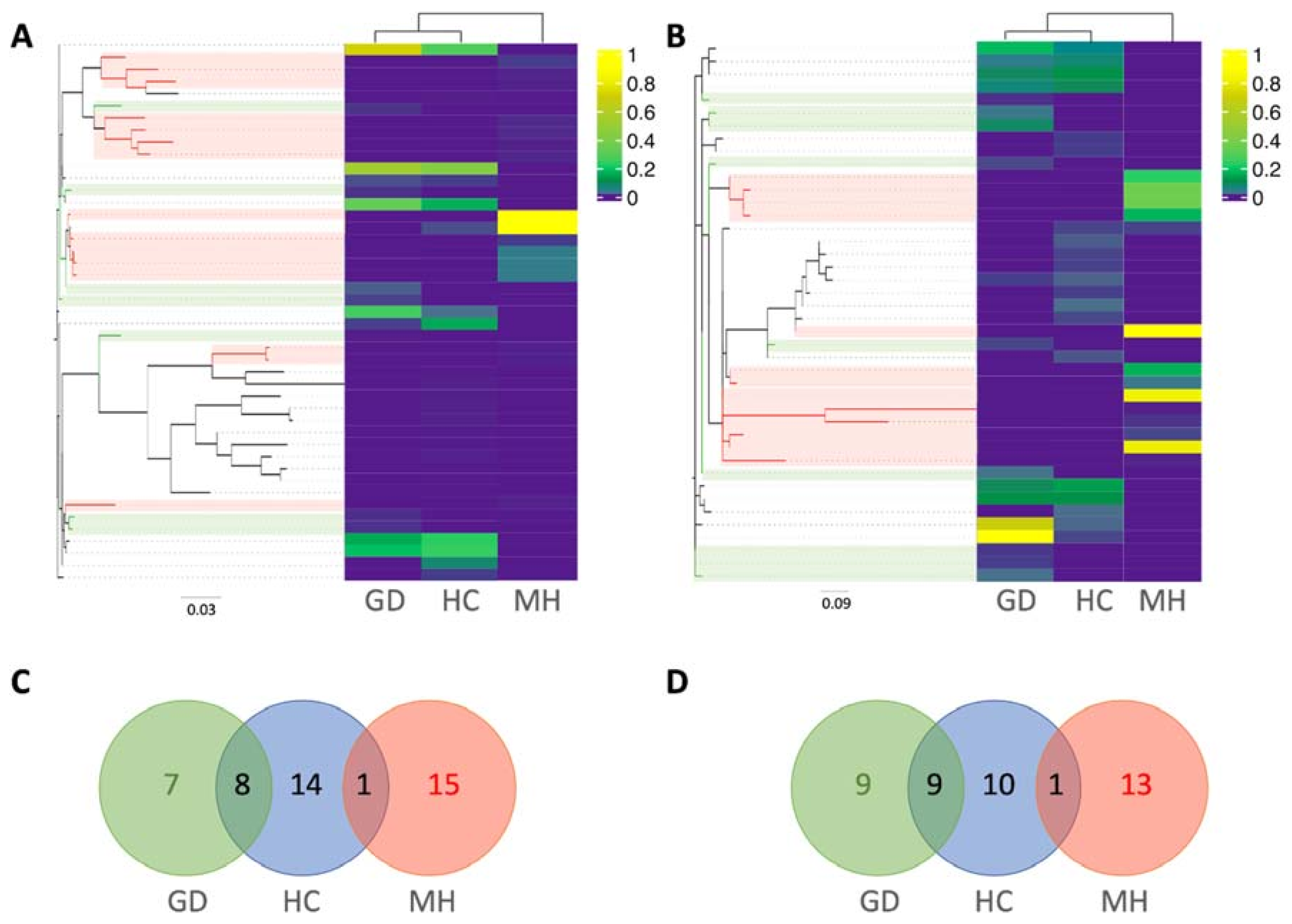
2.3. Taxonomic Assignment of the ASVs
Differential Abundance Analysis
3. Discussion
4. Materials and Methods
4.1. Farm Selection and Sampling
4.2. DNA Extraction and 16S rRNA Gene Amplicon Sequencing
4.3. Microbiota In-Silico Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- NIH HMP Working Group; Peterson, J.; Garges, S.; Giovanni, M.; McInnes, P.; Wang, L.; Schloss, J.A.; Bonazzi, V.; McEwen, J.E.; Wetterstrand, K.A.; et al. The NIH Human Microbiome Project. Genome Res. 2009, 19, 2317–2323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, D.; Liwinski, T.; Elinav, E. Interaction between microbiota and immunity in health and disease. Cell Res. 2020, 30, 492–506. [Google Scholar] [CrossRef] [PubMed]
- Han, G.G.; Lee, J.-Y.; Jin, G.-D.; Park, J.; Choi, Y.H.; Chae, B.J.; Kim, E.B.; Choi, Y.-J. Evaluating the association between body weight and the intestinal microbiota of weaned piglets via 16S rRNA Sequencing. Appl. Microbiol. Biotechnol. 2017, 101, 5903–5911. [Google Scholar] [CrossRef] [PubMed]
- McCormack, U.M.; Curião, T.; Buzoianu, S.G.; Prieto, M.L.; Ryan, T.; Varley, P.; Crispie, F.; Magowan, E.; Metzler-Zebeli, B.U.; Berry, D.; et al. Exploring a Possible Link between the Intestinal Microbiota and Feed Efficiency in Pigs. Appl. Environ. Microbiol. 2017, 83. [Google Scholar] [CrossRef] [Green Version]
- Man, W.H.; de Steenhuijsen Piters, W.A.A.; Bogaert, D. The microbiota of the respiratory tract: Gatekeeper to respiratory health. Nat. Rev. Microbiol. 2017, 15, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Correa-Fiz, F.; Fraile, L.; Aragon, V. Piglet nasal microbiota at weaning may influence the development of Glässer’s disease during the rearing period. BMC Genom. 2016, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mycoplasma Hyorhinis—An Overview | ScienceDirect Topics. Available online: https://www.sciencedirect.com/topics/agricultural-and-biological-sciences/mycoplasma-hyorhinis (accessed on 15 March 2021).
- Clavijo, M.J.; Davies, P.; Morrison, R.; Bruner, L.; Olson, S.; Rosey, E.; Rovira, A. Temporal patterns of colonization and infection with Mycoplasma hyorhinis in two swine production systems in the USA. Vet. Microbiol. 2019, 234, 110–118. [Google Scholar] [CrossRef]
- Roos, L.R.; Surendran Nair, M.; Rendahl, A.K.; Pieters, M. Mycoplasma hyorhinis and Mycoplasma hyosynoviae Dual Detection Patterns in Dams and Piglets. PLoS ONE 2019, 14, e0209975. [Google Scholar] [CrossRef] [Green Version]
- Cerdà-Cuéllar, M.; Naranjo, J.F.; Verge, A.; Nofrarías, M.; Cortey, M.; Olvera, A.; Segalés, J.; Aragon, V. Sow Vaccination Modulates the Colonization of Piglets by Haemophilus parasuis. Vet. Microbiol. 2010, 145, 315–320. [Google Scholar] [CrossRef]
- Costa-Hurtado, M.; Barba-Vidal, E.; Maldonado, J.; Aragon, V. Update on Glässer’s Disease: How to Control the Disease under Restrictive Use of Antimicrobials. Vet. Microbiol. 2020, 242, 108595. [Google Scholar] [CrossRef]
- Mahmmod, Y.S.; Correa-Fiz, F.; Aragon, V. Variations in association of nasal microbiota with virulent and non-virulent strains of Glaesserella (Haemophilus) parasuis in weaning piglets. Vet. Res. 2020, 51, 7. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Cai, R.; Huang, A.; Wang, X.; Qu, W.; Shi, L.; Li, C.; Yan, H. Comparison of oropharyngeal microbiota in healthy piglets and piglets with respiratory disease. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Valeris-Chacin, R.; Sponheim, A.; Fano, E.; Isaacson, R.; Singer, R.S.; Nerem, J.; Leite, F.L.; Pieters, M. Relationships among fecal, air, oral, and tracheal microbial communities in pigs in a respiratory infection disease model. Microorganisms 2021, 9, 252. [Google Scholar] [CrossRef]
- Ojetti, V.; Gigante, G.; Ainora, M.E.; Fiore, F.; Barbaro, F.; Gasbarrini, A. Microflora imbalance and gastrointestinal diseases. Dig. Liver Dis. Suppl. 2009, 3, 35–39. [Google Scholar] [CrossRef]
- Prehn-Kristensen, A.; Zimmermann, A.; Tittmann, L.; Lieb, W.; Schreiber, S.; Baving, L.; Fischer, A. Reduced microbiome alpha diversity in young patients with ADHD. PLoS ONE 2018, 13. [Google Scholar] [CrossRef]
- Pirolo, M.; Espinosa-Gongora, C.; Bogaert, D.; Guardabassi, L. The porcine respiratory microbiome: Recent insights and future challenges. Anim. Microbiome 2021, 3, 9. [Google Scholar] [CrossRef]
- Amat, S.; Lantz, H.; Munyaka, P.M.; Willing, B.P. Prevotella in Pigs: The positive and negative associations with production and health. Microorganisms 2020, 8, 1584. [Google Scholar] [CrossRef]
- Quan, J.; Cai, G.; Ye, J.; Yang, M.; Ding, R.; Wang, X.; Zheng, E.; Fu, D.; Li, S.; Zhou, S.; et al. A global comparison of the microbiome compositions of three gut Locations in commercial pigs with extreme feed conversion ratios. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- Palzer, A.; Haedke, K.; Heinritzi, K.; Zoels, S.; Ladinig, A.; Ritzmann, M. Associations among Haemophilus parasuis, Mycoplasma hyorhinis, and porcine reproductive and respiratory syndrome virus infections in pigs with polyserositis. Can. Vet. J. 2015, 56, 285–287. [Google Scholar]
- Galofré-Milà, N.; Correa-Fiz, F.; Lacouture, S.; Gottschalk, M.; Strutzberg-Minder, K.; Bensaid, A.; Pina-Pedrero, S.; Aragon, V. A robust PCR for the differentiation of potential virulent strains of Haemophilus parasuis. BMC Vet. Res. 2017, 13, 124. [Google Scholar] [CrossRef] [Green Version]
- Brockmeier, S.L.; Loving, C.L.; Mullins, M.A.; Register, K.B.; Nicholson, T.L.; Wiseman, B.S.; Baker, R.B.; Kehrli, M.E. Virulence, transmission, and heterologous protection of four isolates of Haemophilus parasuis. Clin. Vaccine Immunol. 2013, 20, 1466–1472. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.H.; Chen, S.P.; Yeh, K.S.; Weng, C.N. Mycoplasma hyorhinis in Taiwan: Diagnosis and isolation of swine pneumonia pathogen. Vet. Microbiol. 2006, 115, 111–116. [Google Scholar] [CrossRef]
- Clavijo, M.J.; Oliveira, S.; Zimmerman, J.; Rendahl, A.; Rovira, A. Field evaluation of a quantitative polymerase chain reaction assay for Mycoplasma hyorhinis. J. Vet. Diagn Investig. 2014, 26, 755–760. [Google Scholar] [CrossRef] [Green Version]
- Olvera, A.; Pina, S.; Macedo, N.; Oliveira, S.; Aragon, V.; Bensaid, A. Identification of potentially virulent strains of Haemophilus parasuis using a multiplex PCR for virulence-associated autotransporters (vtaA). Vet. J. 2012, 191, 213–218. [Google Scholar] [CrossRef]
- Ishida, S.; Tien, L.H.T.; Osawa, R.; Tohya, M.; Nomoto, R.; Kawamura, Y.; Takahashi, T.; Kikuchi, N.; Kikuchi, K.; Sekizaki, T. Development of an appropriate PCR system for the reclassification of Streptococcus suis. J. Microbiol. Methods 2014, 107, 66–70. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution rample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012, 6, 610–618. [Google Scholar] [CrossRef]
- Diaz, S.; Escobar, J.S.; William-Avila, F. Identification and removal of potential contaminants in 16S rRNA gene sequence datasets from low microbial biomass samples: An example from the mosquito gut. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Shannon, C.; Weaver, W. The Mathematical Theory of Communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef] [Green Version]
- Eren, M.I.; Chao, A.; Hwang, W.-H.; Colwell, R.K. Estimating the Richness of a Population When the Maximum Number of Classes Is Fixed: A Nonparametric Solution to an Archaeological Problem. PLoS ONE 2012, 7, e34179. [Google Scholar] [CrossRef] [Green Version]
- Jaccard, P. Nouvelles Recherches sur la Distribution Florale; Rouge: Lausanne, Switzerland, 1908. [Google Scholar]
- Sørensen, T.J. A Method of Establishing Groups of Equal Amplitude in Plant Sociology Based on Similarity of Species Content and Its Application to Analyses of the Vegetation on Danish Commons; I kommission hos E. Munksgaard: København, Demark, 1948. [Google Scholar]
- Lozupone, C.; Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Z.-Z.; Chen, G.; Alekseyenko, A.V. PERMANOVA-S: Association test for microbial community composition that accommodates confounders and multiple distances. Bioinformatics 2016, 32, 2618–2625. [Google Scholar] [CrossRef]
- Anderson, M.J. A new Method for non-parametric multivariate analysis of variance. Austral. Ecol. 2001, 26, 32–46. [Google Scholar] [CrossRef]
- Jari Oksanen, F.; Blanchet, G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Peter, R.; Minchin, R.B.; O’Hara, G.; Simpson, L.; et al. Stevens, Eduard Szoecs and Helene Wagner. Vegan: Community Ecology Package. R Package Version 2.5-7. 2020. Available online: https://CRAN.R-project.org/package=vegan (accessed on 15 February 2021).
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference Sequence (RefSeq) Database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [Green Version]
- Mandal, S.; Van Treuren, W.; White, R.A.; Eggesbø, M.; Knight, R.; Peddada, S.D. Analysis of composition of microbiomes: A novel method for studying microbial composition. Microb. Ecol. Health Dis. 2015, 26, 27663. [Google Scholar] [CrossRef] [Green Version]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.D.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- RStudio|Open Source & Professional Software for Data Science Teams. Available online: https://rstudio.com/ (accessed on 15 February 2021).
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009; ISBN 978-0-387-98141-3. [Google Scholar]
- Gu, Z.; Eils, R.; Schlesner, M. Complex Heatmaps Reveal Patterns and Correlations in Multidimensional Genomic Data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [Green Version]
- Gao, C.-H. Gaospecial/GgVennDiagram. Available online: https://github.com/gaospecial/ggVennDiagram (accessed on 15 February 2021).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Farm (n *) | Group | Health Status | Production System | Size ** | Treatments *** | Reference |
|---|---|---|---|---|---|---|
| RM (10) | MH | Polyserositis by M. hyorhinis | Multi-site | 650 | Amx | This study |
| GE (10) | MH | Polyserositis by M. hyorhinis | Multi-site | 800 | Amx | This study |
| VL (5) | HC | Healthy | Farrow to finish | 700 | Tlt-Ceft | [6] |
| PT (5) | HC | Healthy | Multi-site | 1000 | NA | [6] |
| GM (10) | HC | Healthy | Multi-site | 1200 | Amx | [6] |
| MT (8) | GD | Glässer’s disease | Multi-site | 3300 | Pen-Strep | [6] |
| MC (10) | GD | Glässer’s disease | Farrow to finish | 480 | Ceft | [6] |
| RC (6) | GD | Glässer’s disease | Multi-site | 1400 | Ceft | [6] |
| EJ (10) | GD | Glässer’s disease | Multi-site | 2000 | Enro | [6] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blanco-Fuertes, M.; Correa-Fiz, F.; Fraile, L.; Sibila, M.; Aragon, V. Altered Nasal Microbiota Composition Associated with Development of Polyserositis by Mycoplasma hyorhinis. Pathogens 2021, 10, 603. https://doi.org/10.3390/pathogens10050603
Blanco-Fuertes M, Correa-Fiz F, Fraile L, Sibila M, Aragon V. Altered Nasal Microbiota Composition Associated with Development of Polyserositis by Mycoplasma hyorhinis. Pathogens. 2021; 10(5):603. https://doi.org/10.3390/pathogens10050603
Chicago/Turabian StyleBlanco-Fuertes, Miguel, Florencia Correa-Fiz, Lorenzo Fraile, Marina Sibila, and Virginia Aragon. 2021. "Altered Nasal Microbiota Composition Associated with Development of Polyserositis by Mycoplasma hyorhinis" Pathogens 10, no. 5: 603. https://doi.org/10.3390/pathogens10050603
APA StyleBlanco-Fuertes, M., Correa-Fiz, F., Fraile, L., Sibila, M., & Aragon, V. (2021). Altered Nasal Microbiota Composition Associated with Development of Polyserositis by Mycoplasma hyorhinis. Pathogens, 10(5), 603. https://doi.org/10.3390/pathogens10050603