
Specific Secondary Bile Acids Control Chicken Necrotic Enteritis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
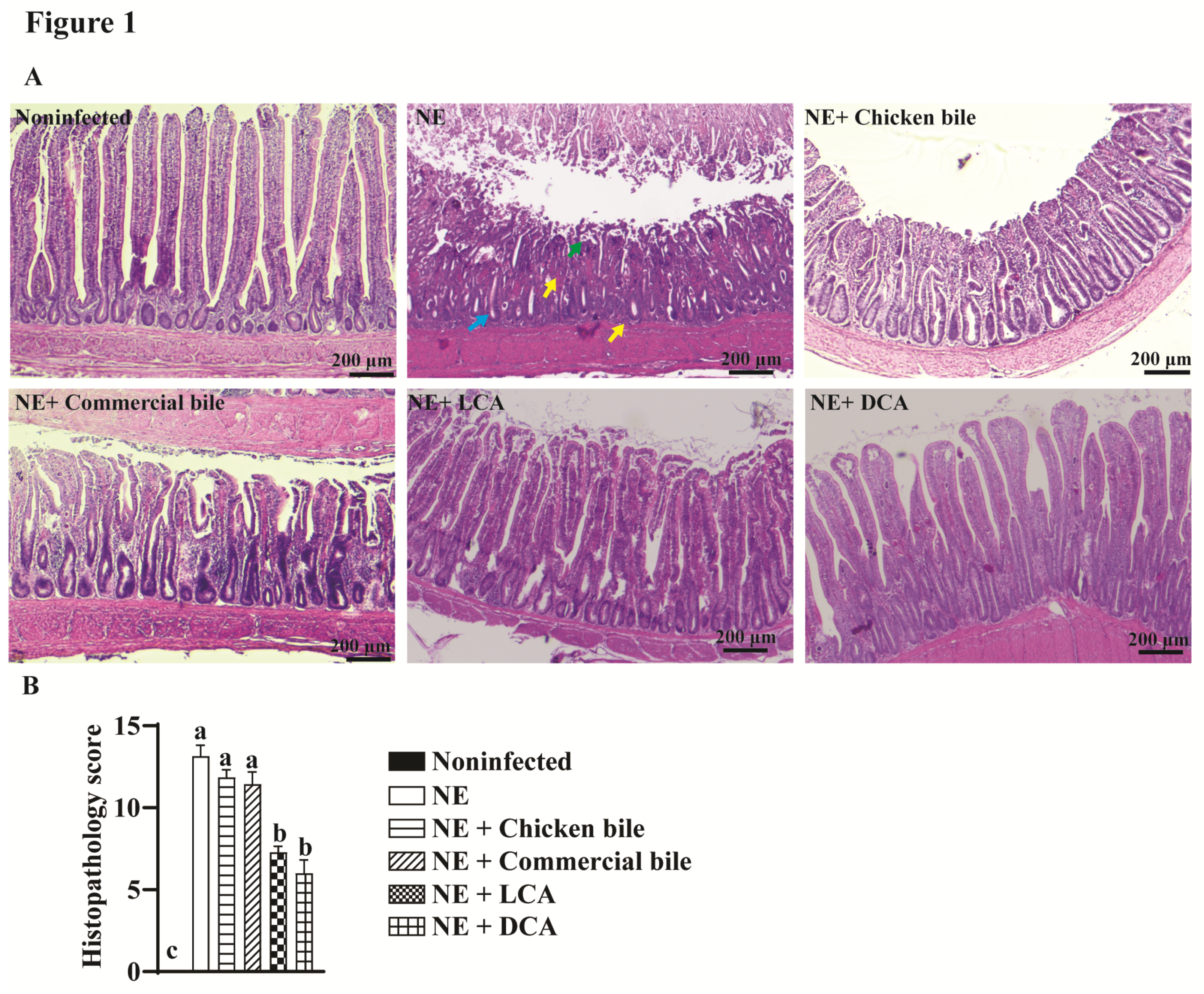
2.1. DCA and LCA Attenuated Clinical NE-Induced Ileitis
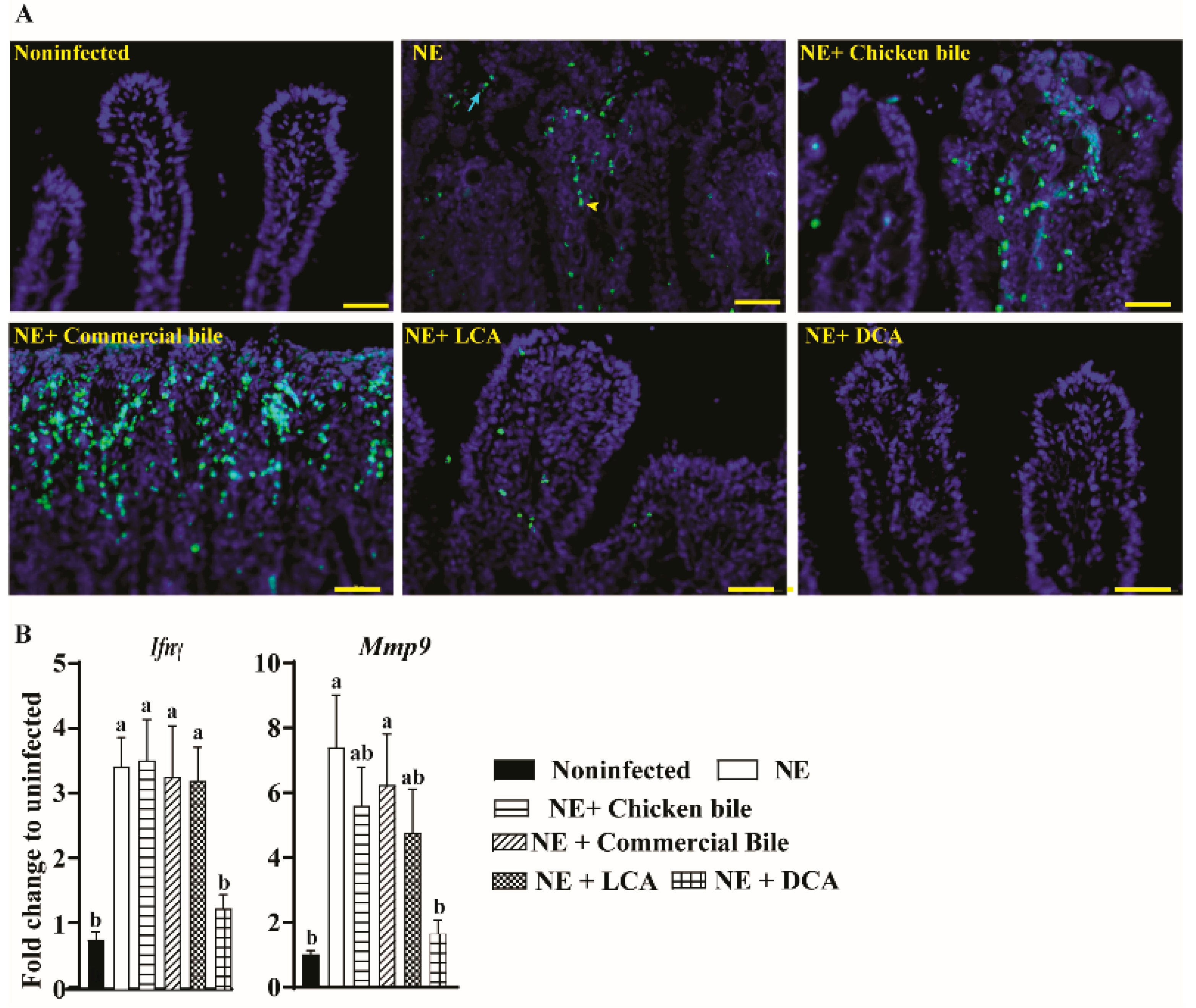
2.2. DCA Attenuated Clinical NE-Induced Ileal Inflammation
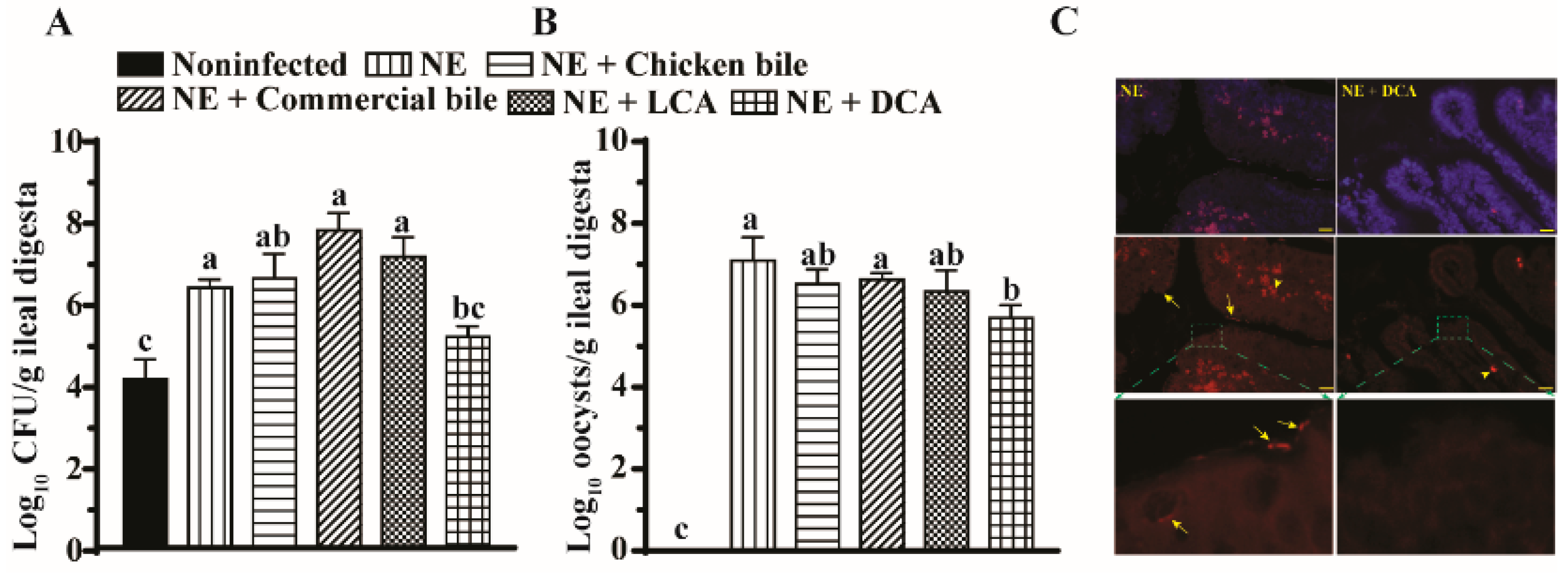
2.3. DCA Reduced C. difficils and E. maxima Colonization in the Ileum
2.4. DCA Reduced Clinical NE-Induced BWG Loss
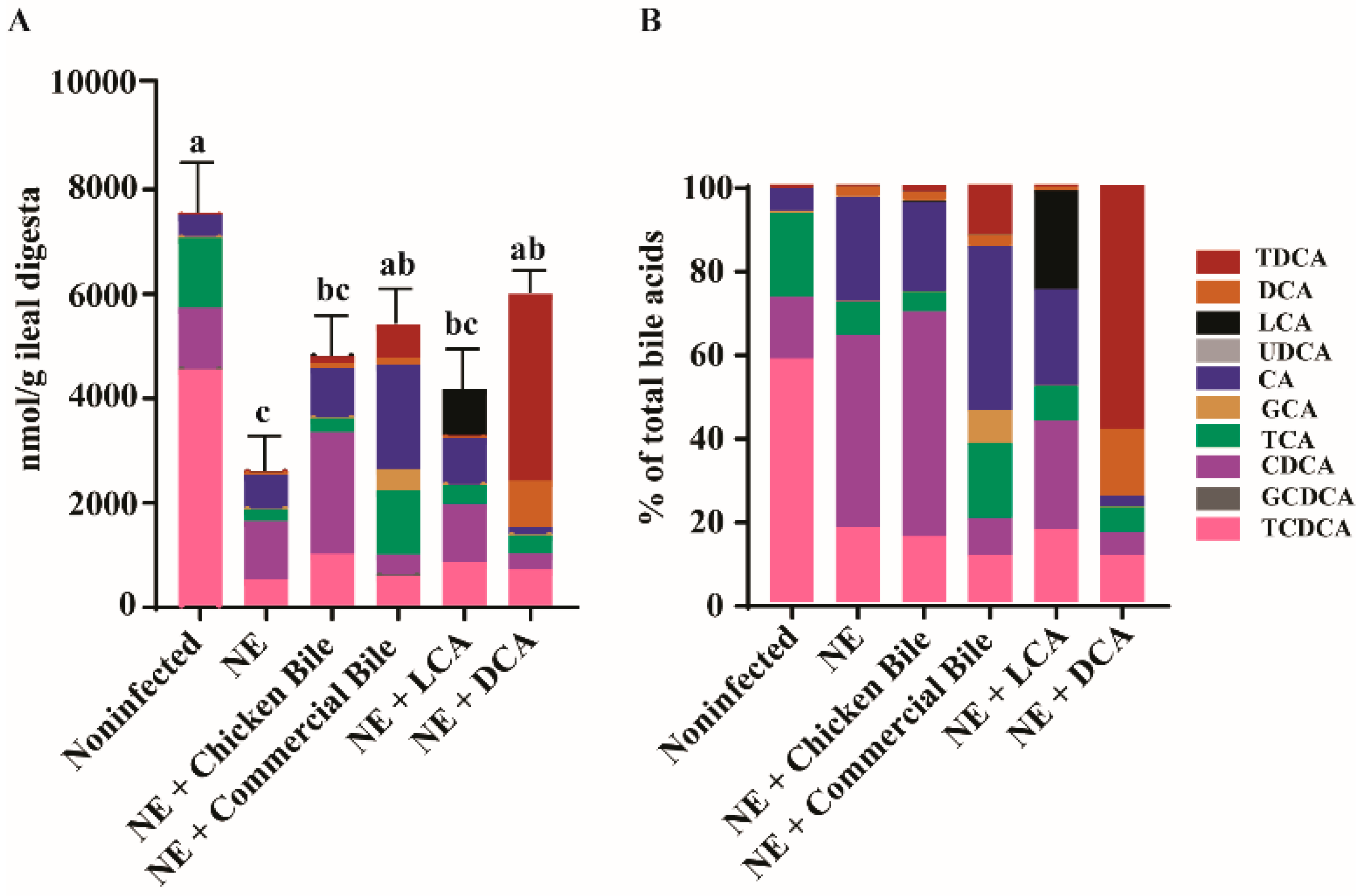
2.5. Dietary Bile Increased NE-Induced Total Bile Acid Level Reduction
3. Discussion
4. Materials and Methods
4.1. Chicken Experiment
4.2. Histopathology Analysis of Intestinal Inflammation
4.3. C. perfringens and E. maxima Colonization in Ileal Lumen Using Real Time PCR and FISH
4.4. Host Inflammatory Response Using a Real Time PCR and a Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling (TUNEL) Assay
4.5. Quantification of Ileal Bile Acids Using Targeted Metabolomics
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, H.; Latorre, J.D.; Bansal, M.; Abraha, M.; Al-Rubaye, B.; Tellez-Isaias, G.; Hargis, B.; Sun, X. Microbial metabolite deoxycholic acid controls Clostridium perfringens-induced chicken necrotic enteritis through attenuating inflammatory cyclooxygenase signaling. Sci. Rep. 2019, 9, 14541. [Google Scholar] [CrossRef] [Green Version]
- Van Immerseel, F.; De Buck, J.; Pasmans, F.; Huyghebaert, G.; Haesebrouck, F.; Ducatelle, R. Clostridium perfringens in poultry: An emerging threat for animal and public health. Avian. Pathol. 2004, 33, 537–549. [Google Scholar] [CrossRef]
- Bansal, M.; Fu, Y.; Alrubaye, B.; Abraha, M.; Almansour, A.; Gupta, A.; Liyanage, R.; Wang, H.; Hargis, B.; Sun, X. A secondary bile acid from microbiota metabolism attenuates ileitis and bile acid reduction in subclinical necrotic enteritis in chickens. J. Anim. Sci. Biotechnol. 2020, 11, 37. [Google Scholar] [CrossRef] [Green Version]
- Rood, J.I.; Keyburn, A.L.; Moore, R.J. NetB and necrotic enteritis: The hole movable story. Avian. Pathol. 2016, 45, 295–301. [Google Scholar] [CrossRef] [Green Version]
- Kaldhusdal, M.; Benestad, S.L.; Lovland, A. Epidemiologic aspects of necrotic enteritis in broiler chickens - disease occurrence and production performance. Avian Pathol. 2016, 45, 271–274. [Google Scholar] [CrossRef]
- Williams, R.B. Intercurrent coccidiosis and necrotic enteritis of chickens: Rational, integrated disease management by maintenance of gut integrity. Avian Pathol. 2005, 34, 159–180. [Google Scholar] [CrossRef]
- Gholamiandehkordi, A.R.; Timbermont, L.; Lanckriet, A.; Van Den Broeck, W.; Pedersen, K.; Dewulf, J.; Pasmans, F.; Haesebrouck, F.; Ducatelle, R.; Van Immerseel, F. Quantification of gut lesions in a subclinical necrotic enteritis model. Avian Pathol. 2007, 36, 375–382. [Google Scholar] [CrossRef] [Green Version]
- Paiva, D.M.; Walk, C.L.; McElroy, A.P. Influence of dietary calcium level, calcium source, and phytase on bird performance and mineral digestibility during a natural necrotic enteritis episode. Poult. Sci. 2013, 92, 3125–3133. [Google Scholar] [CrossRef]
- Calik, A.; Omara, I.I.; White, M.B.; Evans, N.P.; Karnezos, T.P.; Dalloul, R.A. Dietary Non-Drug Feed Additive as an Alternative for Antibiotic Growth Promoters for Broilers During a Necrotic Enteritis Challenge. Microorganisms 2019, 7. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.B.; Rodgers, N.; Choct, M. Optimized necrotic enteritis model producing clinical and subclinical infection of Clostridium perfringens in broiler chickens. Avian Dis. 2010, 54, 1058–1065. [Google Scholar] [CrossRef]
- Shojadoost, B.; Vince, A.R.; Prescott, J.F. The successful experimental induction of necrotic enteritis in chickens by Clostridium perfringens: A critical review. Vet. Res. 2012, 43, 74. [Google Scholar] [CrossRef] [Green Version]
- Emami, N.K.; Dalloul, R.A. Centennial Review: Recent developments in host-pathogen interactions during necrotic enteritis in poultry. Poult. Sci. 2021, 100, 101330. [Google Scholar] [CrossRef]
- Latorre, J.D.; Adhikari, B.; Park, S.H.; Teague, K.D.; Graham, L.E.; Mahaffey, B.D.; Baxter, M.F.A.; Hernandez-Velasco, X.; Kwon, Y.M.; Ricke, S.C.; et al. Evaluation of the Epithelial Barrier Function and Ileal Microbiome in an Established Necrotic Enteritis Challenge Model in Broiler Chickens. Front. Vet. Sci. 2018, 5, 199. [Google Scholar] [CrossRef]
- Navarro, M.A.; McClane, B.A.; Uzal, F.A. Mechanisms of Action and Cell Death Associated with Clostridium perfringens Toxins. Toxins 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Freedman, J.C.; Theoret, J.R.; Wisniewski, J.A.; Uzal, F.A.; Rood, J.I.; McClane, B.A. Clostridium perfringens type A-E toxin plasmids. Res. Microbiol. 2015, 166, 264–279. [Google Scholar] [CrossRef] [Green Version]
- Uzal, F.A.; Freedman, J.C.; Shrestha, A.; Theoret, J.R.; Garcia, J.; Awad, M.M.; Adams, V.; Moore, R.J.; Rood, J.I.; McClane, B.A. Towards an understanding of the role of Clostridium perfringens toxins in human and animal disease. Future Microbiol. 2014, 9, 361–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uzal, F.A.; Vidal, J.E.; McClane, B.A.; Gurjar, A.A. Clostridium Perfringens Toxins Involved in Mammalian Veterinary Diseases. Open Toxinology J. 2010, 2, 24–42. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.C.; Lin, C.H.; Sung, C.T.; Fang, J.Y. Antibacterial activities of bacteriocins: Application in foods and pharmaceuticals. Front. Microbiol. 2014, 5, 241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, T.G.; Smyth, J.A. Prevalence of netB among some clinical isolates of Clostridium perfringens from animals in the United States. Vet. Microbiol. 2009, 136, 202–205. [Google Scholar] [CrossRef] [PubMed]
- Geremia, A.; Biancheri, P.; Allan, P.; Corazza, G.R.; Di Sabatino, A. Innate and adaptive immunity in inflammatory bowel disease. Autoimmun Rev. 2014, 13, 3–10. [Google Scholar] [CrossRef]
- Tokuhira, N.; Kitagishi, Y.; Suzuki, M.; Minami, A.; Nakanishi, A.; Ono, Y.; Kobayashi, K.; Matsuda, S.; Ogura, Y. PI3K/AKT/PTEN pathway as a target for Crohn’s disease therapy (Review). Int. J. Mol. Med. 2015, 35, 10–16. [Google Scholar] [CrossRef] [Green Version]
- Muzes, G.; Molnar, B.; Tulassay, Z.; Sipos, F. Changes of the cytokine profile in inflammatory bowel diseases. World J. Gastroenterol. 2012, 18, 5848–5861. [Google Scholar] [CrossRef] [Green Version]
- Neish, A.S.; Gewirtz, A.T.; Zeng, H.; Young, A.N.; Hobert, M.E.; Karmali, V.; Rao, A.S.; Madara, J.L. Prokaryotic regulation of epithelial responses by inhibition of IkappaB-alpha ubiquitination. Science 2000, 289, 1560–1563. [Google Scholar] [CrossRef]
- Rock, K.L.; Kono, H. The inflammatory response to cell death. Annu. Rev. Pathol. 2008, 3, 99–126. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Watkins, J.B. Lipid digestion and absorption. Pediatrics 1985, 75, 151–156. [Google Scholar]
- Ridlon, J.M.; Harris, S.C.; Bhowmik, S.; Kang, D.J.; Hylemon, P.B. Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes 2016, 7, 22–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuda, N. Deconjugation of bile salts by Bacteroids and Clostridium. Microbiol. Immunol. 1981, 25, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Buffie, C.G.; Bucci, V.; Stein, R.R.; McKenney, P.T.; Ling, L.; Gobourne, A.; No, D.; Liu, H.; Kinnebrew, M.; Viale, A.; et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 2015, 517, 205–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Winglee, K.; Gharaibeh, R.Z.; Gauthier, J.; He, Z.; Tripathi, P.; Avram, D.; Bruner, S.; Fodor, A.; Jobin, C. Microbiota-Derived Metabolic Factors Reduce Campylobacteriosis in Mice. Gastroenterology 2018, 154, 1751–1763.e2. [Google Scholar] [CrossRef] [PubMed]
- Lanzini, A.; Lanzarotto, F. Review article: The ‘mechanical pumps’ and the enterohepatic circulation of bile acids--defects in coeliac disease. Aliment. Pharm. 2000, 14 (Suppl. 2), 58–61. [Google Scholar] [CrossRef] [PubMed]
- Hagenbuch, B.; Dawson, P. The sodium bile salt cotransport family SLC10. Pflug. Arch. 2004, 447, 566–570. [Google Scholar] [CrossRef] [PubMed]
- Gothe, F.; Beigel, F.; Rust, C.; Hajji, M.; Koletzko, S.; Freudenberg, F. Bile acid malabsorption assessed by 7 alpha-hydroxy-4-cholesten-3-one in pediatric inflammatory bowel disease: Correlation to clinical and laboratory findings. J. Crohns Colitis 2014, 8, 1072–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitek, L. Bile acid malabsorption in inflammatory bowel disease. Inflamm Bowel. Dis. 2015, 21, 476–483. [Google Scholar] [CrossRef]
- Jung, D.; Fantin, A.C.; Scheurer, U.; Fried, M.; Kullak-Ublick, G.A. Human ileal bile acid transporter gene ASBT (SLC10A2) is transactivated by the glucocorticoid receptor. Gut 2004, 53, 78–84. [Google Scholar] [CrossRef] [Green Version]
- Mot, D.; Timbermont, L.; Haesebrouck, F.; Ducatelle, R.; Van Immerseel, F. Progress and problems in vaccination against necrotic enteritis in broiler chickens. Avian Pathol. 2014, 43, 290–300. [Google Scholar] [CrossRef] [Green Version]
- Macdonald, I.A.; Hutchison, D.M.; Forrest, T.P.; Bokkenheuser, V.D.; Winter, J.; Holdeman, L.V. Metabolism of primary bile acids by Clostridium perfringens. J. Steroid Biochem. 1983, 18, 97–104. [Google Scholar] [CrossRef]
- Knarreborg, A.; Engberg, R.M.; Jensen, S.K.; Jensen, B.B. Quantitative determination of bile salt hydrolase activity in bacteria isolated from the small intestine of chickens. Appl. Environ. Microbiol. 2002, 68, 6425–6428. [Google Scholar] [CrossRef] [Green Version]
- Sorg, J.A.; Sonenshein, A.L. Bile salts and glycine as cogerminants for Clostridium difficile spores. J. Bacteriol. 2008, 190, 2505–2512. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Paredes-Sabja, D.; Sarker, M.R.; McClane, B.A. Clostridium perfringens Sporulation and Sporulation-Associated Toxin Production. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [Green Version]
- Hickey, C.S.; Johnson, M.G. Effects of pH shifts, bile salts, and glucose on sporulation of Clostridium perfringens NCTC 8798. Appl. Environ. Microbiol. 1981, 41, 124–129. [Google Scholar] [CrossRef] [Green Version]
- Fickert, P.; Fuchsbichler, A.; Marschall, H.U.; Wagner, M.; Zollner, G.; Krause, R.; Zatloukal, K.; Jaeschke, H.; Denk, H.; Trauner, M. Lithocholic acid feeding induces segmental bile duct obstruction and destructive cholangitis in mice. Am. J. Pathol 2006, 168, 410–422. [Google Scholar] [CrossRef] [Green Version]
- Wallace, J.L. Prostaglandin biology in inflammatory bowel disease. Gastroenterol. Clin. N. Am. 2001, 30, 971–980. [Google Scholar] [CrossRef]
- Tessner, T.G.; Muhale, F.; Riehl, T.E.; Anant, S.; Stenson, W.F. Prostaglandin E2 reduces radiation-induced epithelial apoptosis through a mechanism involving AKT activation and bax translocation. J. Clin. Investig. 2004, 114, 1676–1685. [Google Scholar] [CrossRef] [Green Version]
- Martin-Venegas, R.; Roig-Perez, S.; Ferrer, R.; Moreno, J.J. Arachidonic acid cascade and epithelial barrier function during Caco-2 cell differentiation. J. Lipid Res. 2006, 47, 1416–1423. [Google Scholar] [CrossRef] [Green Version]
- Zamuner, S.R.; Warrier, N.; Buret, A.G.; MacNaughton, W.K.; Wallace, J.L. Cyclooxygenase 2 mediates post-inflammatory colonic secretory and barrier dysfunction. Gut 2003, 52, 1714–1720. [Google Scholar] [CrossRef]
- Gomez-Cambronero, J.; Horn, J.; Paul, C.C.; Baumann, M.A. Granulocyte-macrophage colony-stimulating factor is a chemoattractant cytokine for human neutrophils: Involvement of the ribosomal p70 S6 kinase signaling pathway. J. Immunol. 2003, 171, 6846–6855. [Google Scholar] [CrossRef] [Green Version]
- Janes, M.R.; Fruman, D.A. Immune regulation by rapamycin: Moving beyond T cells. Sci. Signal. 2009, 2, pe25. [Google Scholar] [CrossRef]
- Weichhart, T.; Saemann, M.D. The PI3K/Akt/mTOR pathway in innate immune cells: Emerging therapeutic applications. Ann. Rheum Dis. 2008, 67 (Suppl. 3), iii70–iii74. [Google Scholar] [CrossRef]
. |





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bansal, M.; Alenezi, T.; Fu, Y.; Almansour, A.; Wang, H.; Gupta, A.; Liyanage, R.; Graham, D.B.; Hargis, B.M.; Sun, X. Specific Secondary Bile Acids Control Chicken Necrotic Enteritis. Pathogens 2021, 10, 1041. https://doi.org/10.3390/pathogens10081041
Bansal M, Alenezi T, Fu Y, Almansour A, Wang H, Gupta A, Liyanage R, Graham DB, Hargis BM, Sun X. Specific Secondary Bile Acids Control Chicken Necrotic Enteritis. Pathogens. 2021; 10(8):1041. https://doi.org/10.3390/pathogens10081041
Chicago/Turabian StyleBansal, Mohit, Tahrir Alenezi, Ying Fu, Ayidh Almansour, Hong Wang, Anamika Gupta, Rohana Liyanage, Danielle B. Graham, Billy M. Hargis, and Xiaolun Sun. 2021. "Specific Secondary Bile Acids Control Chicken Necrotic Enteritis" Pathogens 10, no. 8: 1041. https://doi.org/10.3390/pathogens10081041
APA StyleBansal, M., Alenezi, T., Fu, Y., Almansour, A., Wang, H., Gupta, A., Liyanage, R., Graham, D. B., Hargis, B. M., & Sun, X. (2021). Specific Secondary Bile Acids Control Chicken Necrotic Enteritis. Pathogens, 10(8), 1041. https://doi.org/10.3390/pathogens10081041