
Clinico-Genomic Analysis Reveals Mutations Associated with COVID-19 Disease Severity: Possible Modulation by RNA Structure

 ,
,  , , and
, , and 
Abstract
:1. Introduction
2. Results
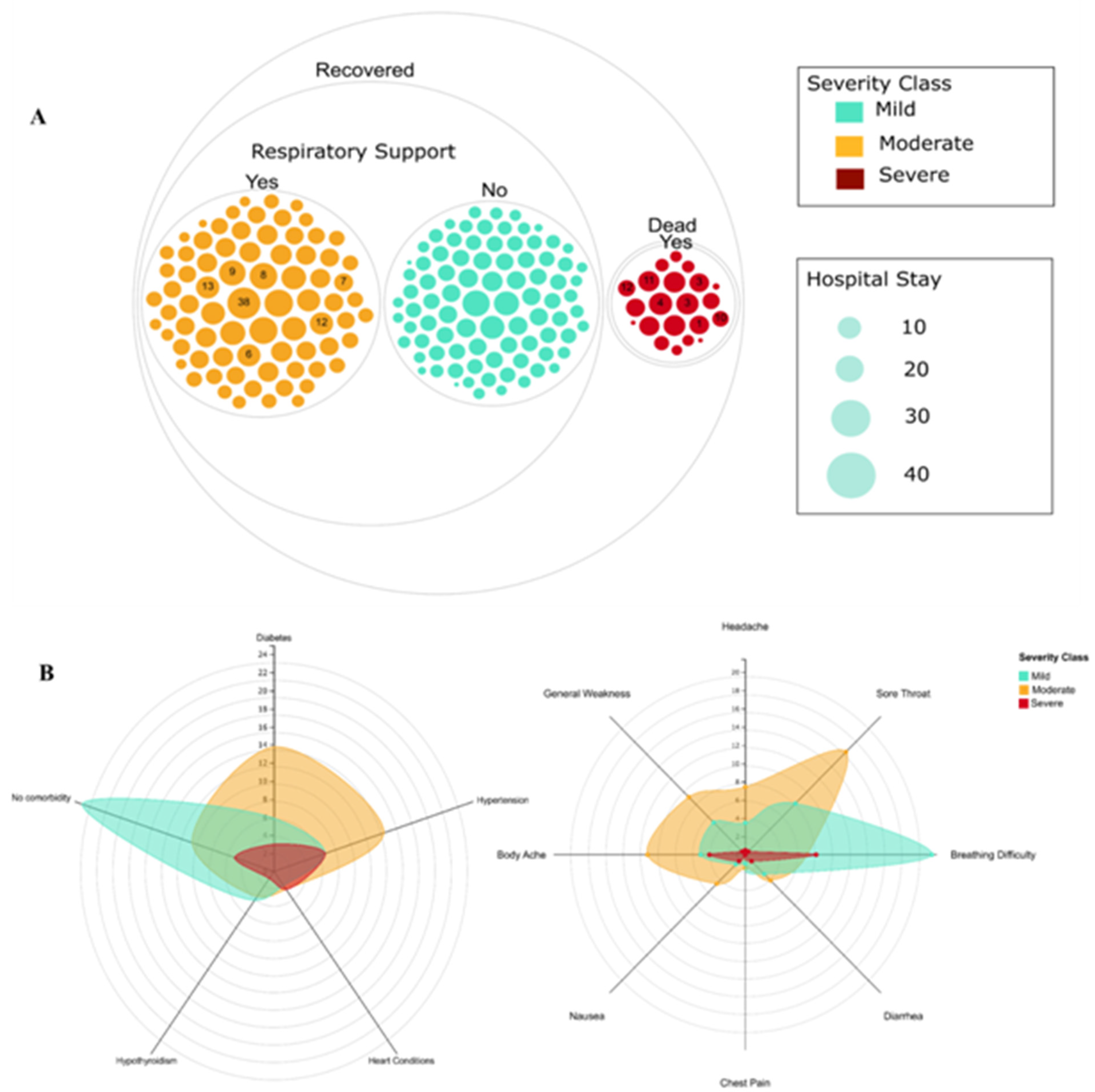
2.1. Demographics and Diversity of Clinical Features and Severity in COVID-19 Patients
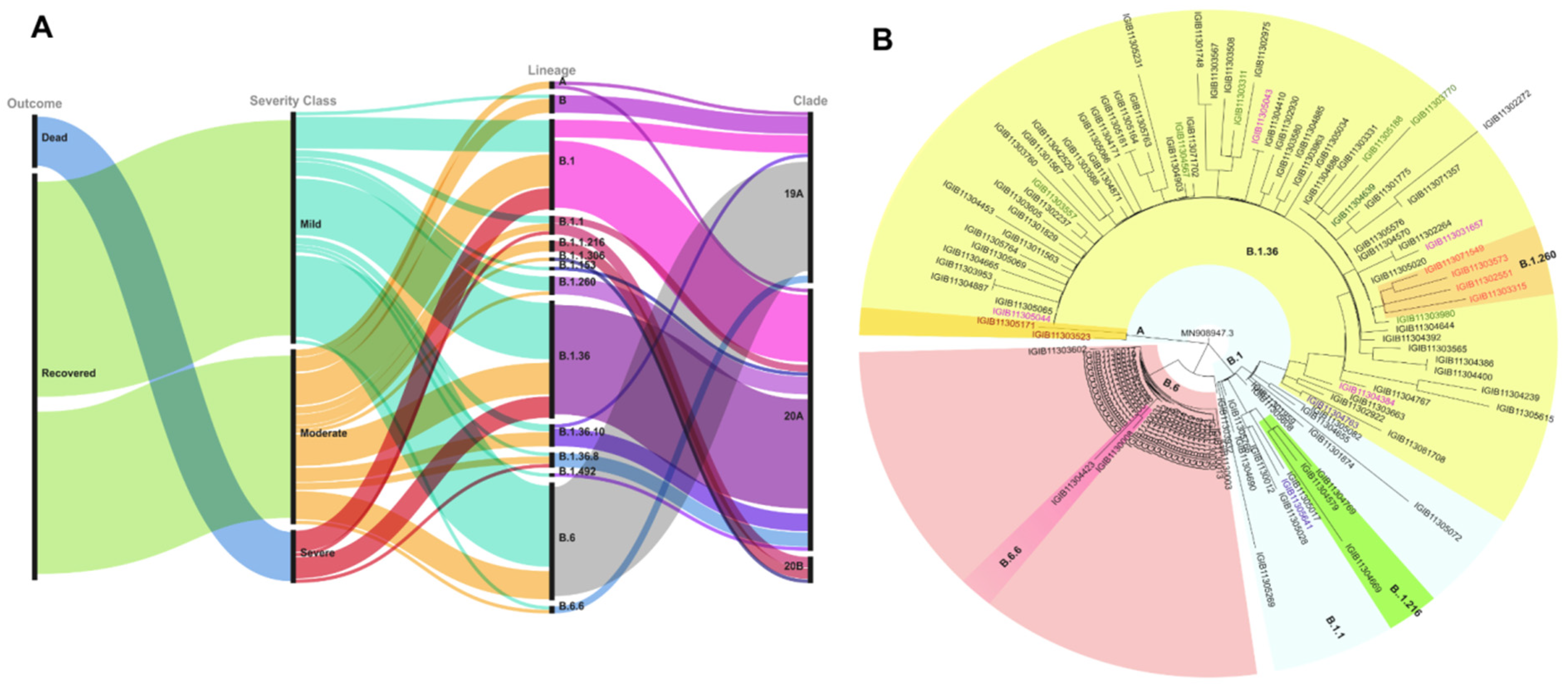
2.2. Phylogenetic and Mutation Variation of SARS-CoV-2
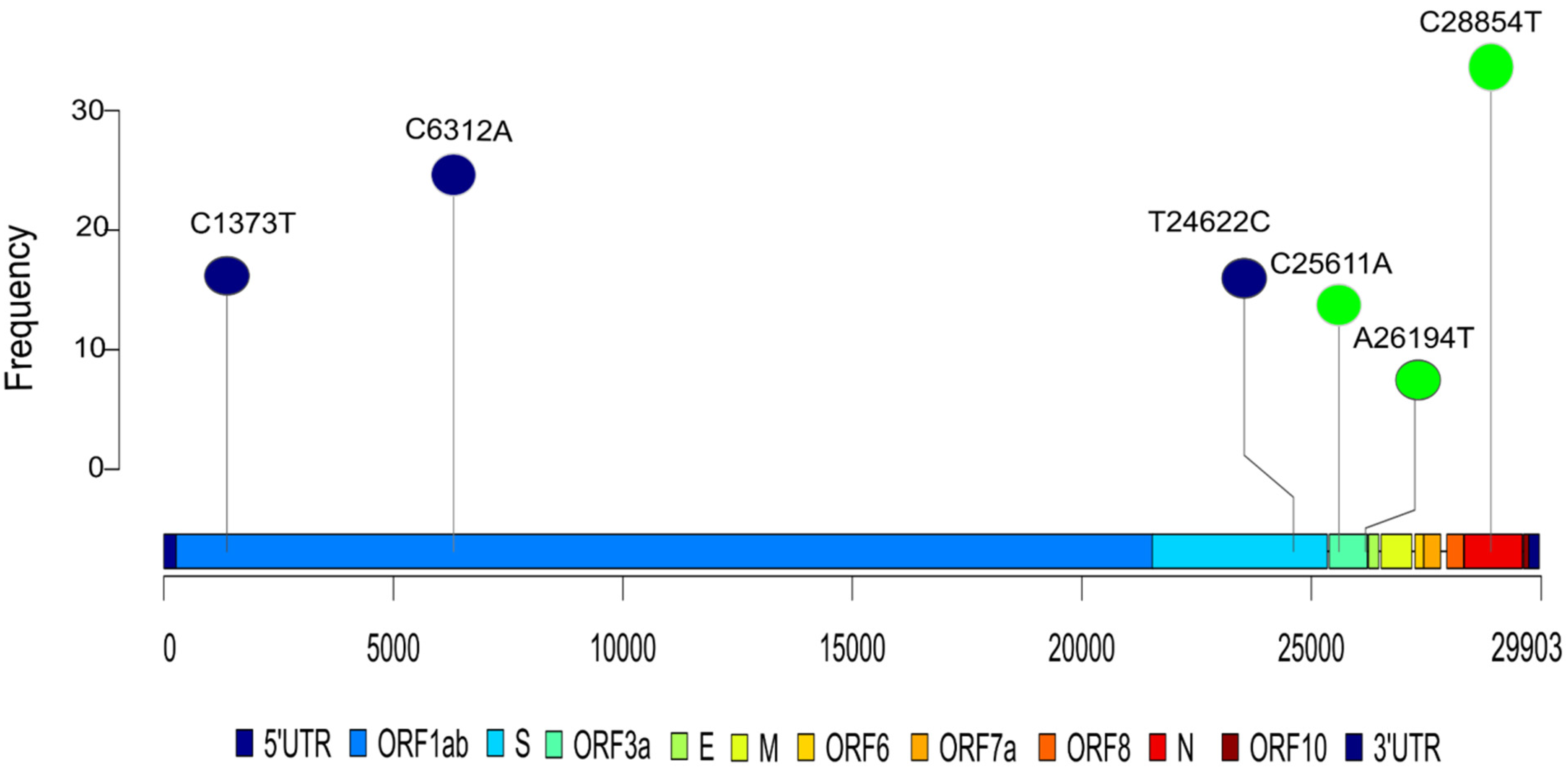
2.3. Association of Mutation with Disease Severity
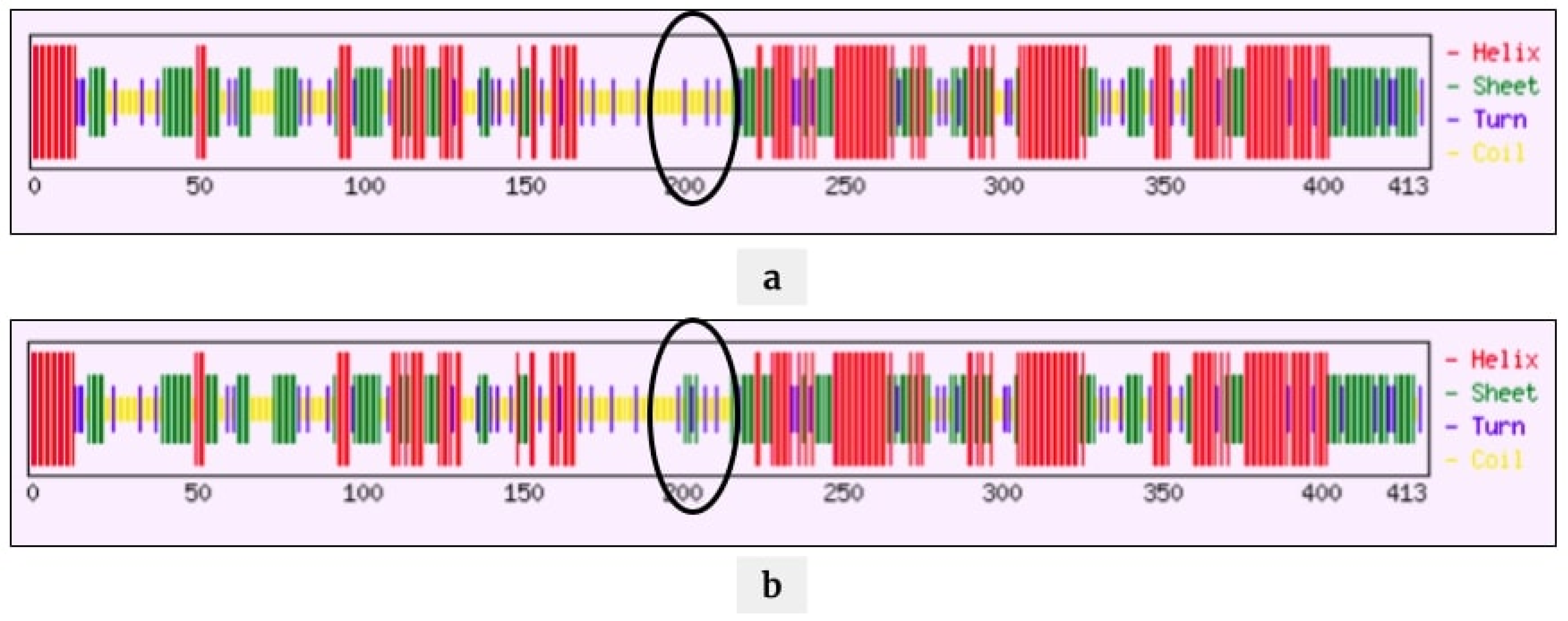
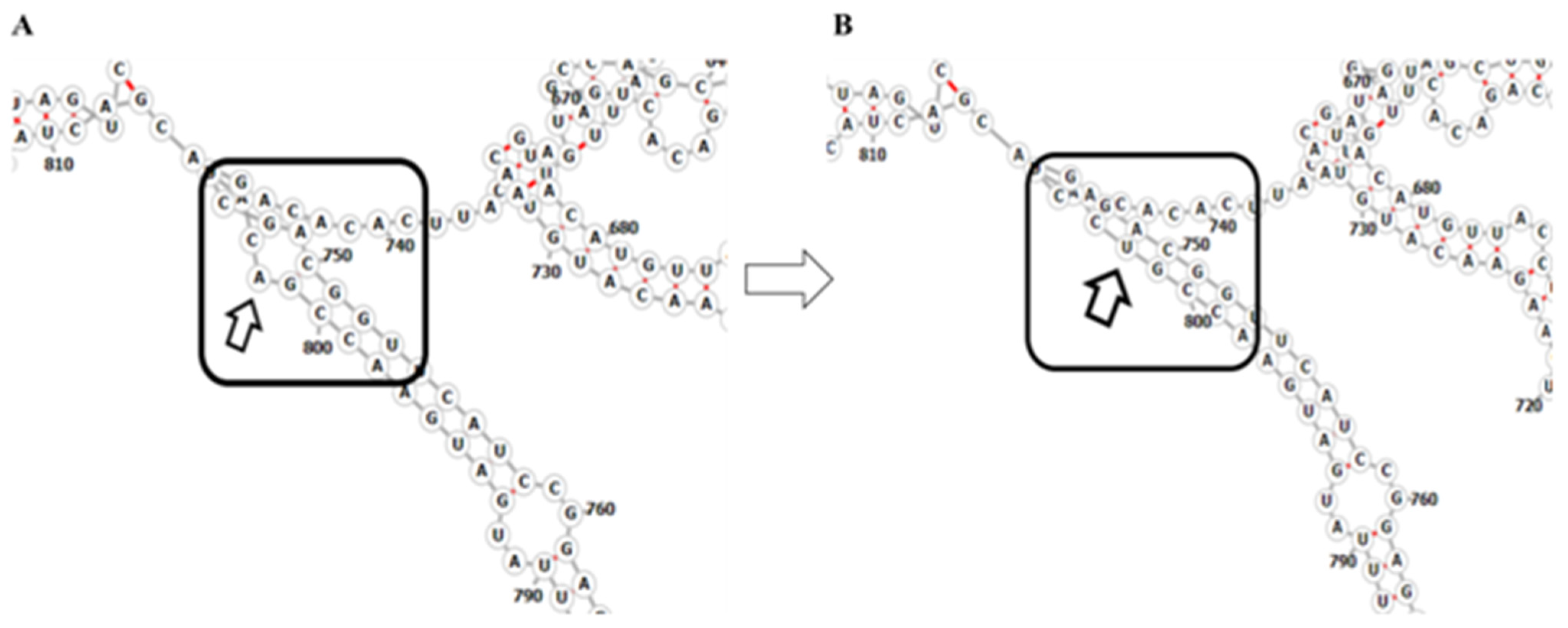
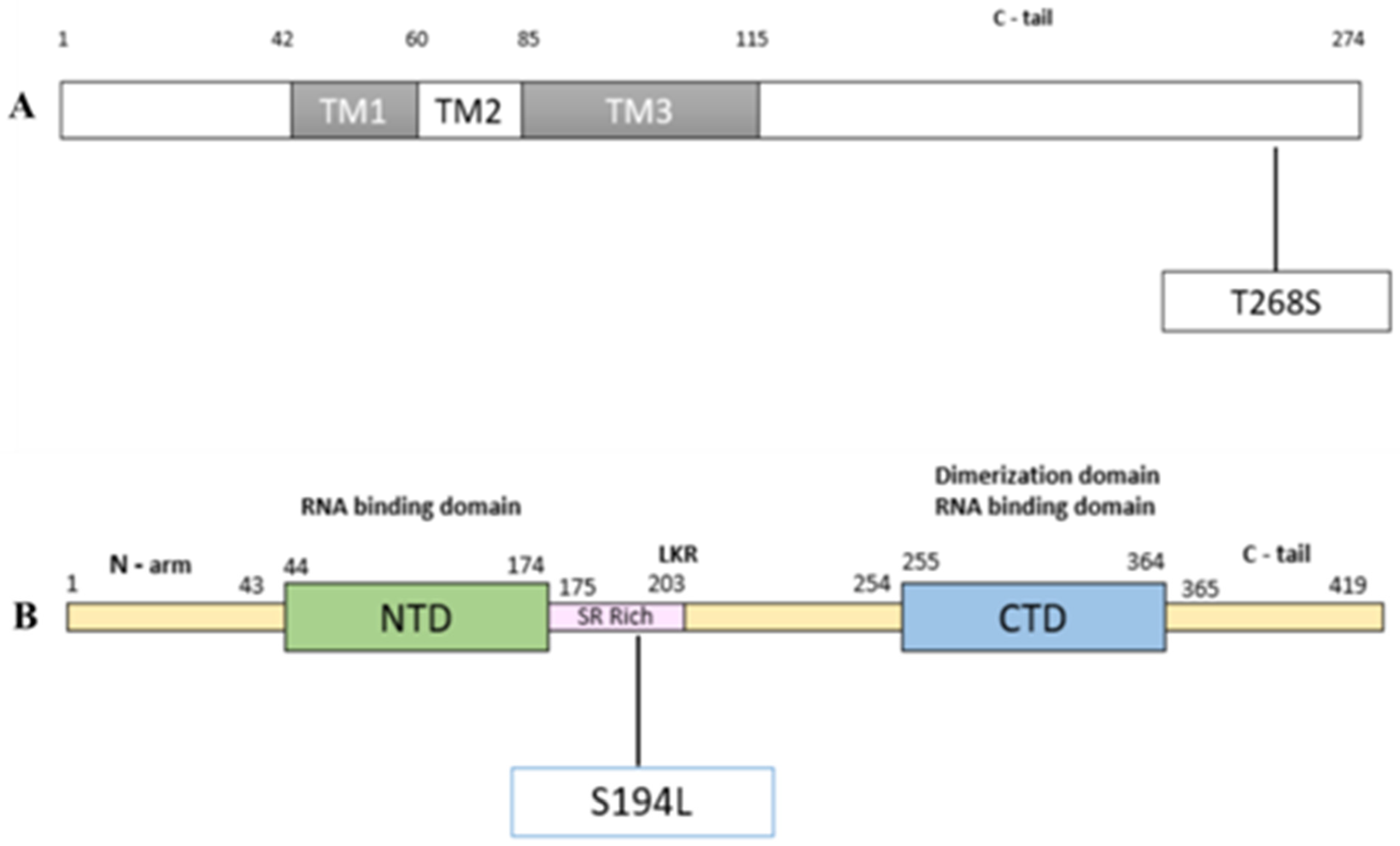
2.4. Mutations Modulating the Structure
3. Discussion
4. Materials and Methods
4.1. Sample Collection and Sequencing
4.2. Phylogenetic and Mutation Analysis
4.3. Statistical Analysis
4.4. Mutation Association Study
4.5. Data Preparation
4.6. Mutation Selection
- The mutation is in the top 15 most correlated mutations with respect to both disease severity and disease mortality. We only consider mutations that are highly correlated with both severity and mortality as it highlights the significance of the mutation and helps avoid outliers.
- The mutation has a statistically significant correlation (p < 0.05) with either disease severity or mortality.
- The mutation is positively correlated with severity, i.e., the presence of mutation increases the severity of the disease. We consider a mutation to be positively correlated if the proportion of severe patients with the mutation is higher than the average.
4.7. Structural Analysis
Orf3a and N Protein Sequence Collection
4.8. Secondary Structure Analysis of RNA
4.9. Protein Disorder Prediction
4.10. Protein Structural Analysis of Orf3a and N Protein of SARS-CoV-2
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Duffy, S. Why Are RNA Virus Mutation Rates so Damn High? PLoS Biol. 2018, 16, e3000003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pachetti, M.; Marini, B.; Benedetti, F.; Giudici, F.; Mauro, E.; Storici, P.; Masciovecchio, C.; Angeletti, S.; Ciccozzi, M.; Gallo, R.C.; et al. Emerging SARS-CoV-2 Mutation Hot Spots Include a Novel RNA-Dependent-RNA Polymerase Variant. J. Transl. Med. 2020, 18, 179. [Google Scholar] [CrossRef] [Green Version]
- Symptoms of COVID-19|CDC. Available online: http://www.cdc.gov/coronavirus/2019-ncov/symptoms-testing/symptoms.html (accessed on 3 August 2021).
- Zhou, S.; Wang, Y.; Zhu, T.; Xia, L. CT Features of Coronavirus Disease 2019 (COVID-19) Pneumonia in 62 Patients in Wuhan, China. Am. J.Roentgenol. 2020, 214, 1287–1294. [Google Scholar] [CrossRef]
- Azer, S.A. COVID-19: Pathophysiology, Diagnosis, Complications and Investigational Therapeutics. New Microbes New Infect. 2020, 37, 100738. [Google Scholar] [CrossRef]
- Xiong, T.-Y.; Redwood, S.; Prendergast, B.; Chen, M. Coronaviruses and the Cardiovascular System: Acute and Long-Term Implications. Eur. Heart J. 2020, 41, 1798–1800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, X.-W.; Xu, D.; Zhang, H.; Zhou, W.; Wang, L.-H.; Cui, X.-G. Identification of a Potential Mechanism of Acute Kidney Injury during the COVID-19 Outbreak: A Study Based on Single-Cell Transcriptome Analysis. Intensive Care Med. 2020, 46, 1114–1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kordzadeh-Kermani, E.; Khalili, H.; Karimzadeh, I. Pathogenesis, Clinical Manifestations and Complications of Coronavirus Disease 2019 (COVID-19). Future Microbiol. 2020, 15, 1287–1305. [Google Scholar] [CrossRef]
- Pennisi, M.; Lanza, G.; Falzone, L.; Fisicaro, F.; Ferri, R.; Bella, R. SARS-CoV-2 and the Nervous System: From Clinical Features to Molecular Mechanisms. Int. J. Mol. Sci. 2020, 21, 21. [Google Scholar] [CrossRef] [PubMed]
- Rello, J.; Storti, E.; Belliato, M.; Serrano, R. Clinical Phenotypes of SARS-CoV-2: Implications for Clinicians and Researchers. Eur. Respir. J. 2020, 55, 55. [Google Scholar] [CrossRef]
- Richardson, S.; Hirsch, J.S.; Narasimhan, M.; Crawford, J.M.; McGinn, T.; Davidson, K.W.; The Northwell COVID-19 Research Consortium; Barnaby, D.P.; Becker, L.B.; Chelico, J.D.; et al. Presenting Characteristics, Comorbidities, and Outcomes Among 5700 Patients Hospitalized With COVID-19 in the New York City Area. JAMA 2020, 323, 2052–2059. [Google Scholar] [CrossRef]
- Wu, Z.; McGoogan, J.M. Characteristics of and Important Lessons from the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72 314 Cases From the Chinese Center for Disease Control and Prevention. JAMA 2020, 323, 1239–1242. [Google Scholar] [CrossRef] [PubMed]
- Falzone, L.; Gattuso, G.; Tsatsakis, A.; Spandidos, D.A.; Libra, M. Current and Innovative Methods for the Diagnosis of COVID-19 Infection (Review). Int. J. Mol. Med. 2021, 47, 1–23. [Google Scholar] [CrossRef] [PubMed]
- FnCas9-Based CRISPR Diagnostic for Rapid and Accurate Detection of Major SARS-CoV-2 Variants on a Paper Strip | ELife. Available online: https://elifesciences.org/articles/67130 (accessed on 7 August 2021).
- Kumar, P.; Pandey, R.; Sharma, P.; Dhar, M.S.; Vivekanand, A.; Uppili, B.; Vashisht, H.; Wadhwa, S.; Tyagi, N.; Fatihi, S.; et al. Integrated Genomic View of SARS-CoV-2 in India. Wellcome Open Res. 2020, 5, 184. [Google Scholar] [CrossRef]
- Dhar, M.S.; Asokachandran, V.; Uppili, B.; Tyagi, N.; Sharma, P.; Tiwari, S.; Srinivasan, R.V.; Marwal, R.; Kanakan, A.; Khan, A.M.; et al. Reinfection or Reactivation: Genome-Based Two Distinct SNP Profile of SARS-CoV2 Repositivity in an Indian Case. J. Med. Virol. 2021, 93, 4152–4155. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, I.; Datir, R.; Papa, G.; Kemp, S.; Meng, B.; Rakshit, P.; Singh, S.; Pandey, R.; Ponnusamy, K.; Radhakrishnan, V.S.; et al. SARS-CoV-2 B.1.617 Emergence and Sensitivity to Vaccine-Elicited Antibodies. BioRxiv 2021. [Google Scholar] [CrossRef]
- Kale, P.; Gupta, E.; Bihari, C.; Patel, N.; Rooge, S.; Pandey, A.; Bajpai, M.; Khillan, V.; Chattopadhyay, P.C.; Devi, P.; et al. Clinicogenomic Analysis of Breakthrough Infections by SARS CoV2 Variants after ChAdOx1 NCoV- 19 Vaccination in Healthcare Workers. medRxiv 2021. [Google Scholar] [CrossRef]
- Meredith, L.W.; Hamilton, W.L.; Warne, B.; Houldcroft, C.J.; Hosmillo, M.; Jahun, A.S.; Curran, M.D.; Parmar, S.; Caller, L.G.; Caddy, S.L.; et al. Rapid Implementation of SARS-CoV-2 Sequencing to Investigate Cases of Health-Care Associated COVID-19: A Prospective Genomic Surveillance Study. Lancet Infect. Dis. 2020, 20, 1263–1271. [Google Scholar] [CrossRef]
- Khailany, R.A.; Safdar, M.; Ozaslan, M. Genomic Characterization of a Novel SARS-CoV-2. Gene Rep. 2020, 19, 100682. [Google Scholar] [CrossRef]
- Nagy, Á.; Pongor, S.; Győrffy, B. Different Mutations in SARS-CoV-2 Associate with Severe and Mild Outcome. Int. J. Antimicrob. Agents 2021, 57, 106272. [Google Scholar] [CrossRef]
- Young, B.E.; Fong, S.-W.; Chan, Y.-H.; Mak, T.-M.; Ang, L.W.; Anderson, D.E.; Lee, C.Y.-P.; Amrun, S.N.; Lee, B.; Goh, Y.S.; et al. Effects of a Major Deletion in the SARS-CoV-2 Genome on the Severity of Infection and the Inflammatory Response: An Observational Cohort Study. Lancet 2020, 396, 603–611. [Google Scholar] [CrossRef]
- Lin, J.-W.; Tang, C.; Wei, H.-C.; Du, B.; Chen, C.; Wang, M.; Zhou, Y.; Yu, M.-X.; Cheng, L.; Kuivanen, S.; et al. Genomic Monitoring of SARS-CoV-2 Uncovers an Nsp1 Deletion Variant That Modulates Type I Interferon Response. Cell Host Microbe 2021, 29, 489–502.e8. [Google Scholar] [CrossRef]
- Esper, F.P.; Cheng, Y.-W.; Adhikari, T.M.; Tu, Z.J.; Li, D.; Li, E.A.; Farkas, D.H.; Procop, G.W.; Ko, J.S.; Chan, T.A.; et al. Genomic Epidemiology of SARS-CoV-2 Infection During the Initial Pandemic Wave and Association With Disease Severity. JAMA Netw. Open 2021, 4, e217746. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Li, P.; Ju, X.; Rao, J.; Huang, W.; Ren, L.; Zhang, S.; Xiong, T.; Xu, K.; Zhou, X.; et al. In Vivo Structural Characterization of the SARS-CoV-2 RNA Genome Identifies Host Proteins Vulnerable to Repurposed Drugs. Cell 2021, 184, 1865–1883. [Google Scholar] [CrossRef] [PubMed]
- Reuter, J.S.; Mathews, D.H. RNAstructure: Software for RNA Secondary Structure Prediction and Analysis. BMC Bioinform. 2010, 11, 129. [Google Scholar] [CrossRef] [Green Version]
- Sanyaolu, A.; Okorie, C.; Marinkovic, A.; Patidar, R.; Younis, K.; Desai, P.; Hosein, Z.; Padda, I.; Mangat, J.; Altaf, M. Comorbidity and Its Impact on Patients with COVID-19. SN Compr. Clin. Med. 2020, 2, 1069–1076. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; She, Z.-G.; Cheng, X.; Qin, J.-J.; Zhang, X.-J.; Cai, J.; Lei, F.; Wang, H.; Xie, J.; Wang, W.; et al. Association of Blood Glucose Control and Outcomes in Patients with COVID-19 and Pre-Existing Type 2 Diabetes. Cell Metab. 2020, 31, 1068–1077.e3. [Google Scholar] [CrossRef] [PubMed]
- Saghir, S.A.M.; AlGabri, N.A.; Alagawany, M.M.; Attia, Y.A.; Alyileili, S.R.; Elnesr, S.S.; Shafi, M.E.; Al-Shargi, O.Y.A.; Al-Balagi, N.; Alwajeeh, A.S.; et al. Chloroquine and Hydroxychloroquine for the Prevention and Treatment of COVID-19: A Fiction, Hope or Hype? An Updated Review. Ther. Clin. Risk Manag. 2021, 17, 371–387. [Google Scholar] [CrossRef]
- Chong, Y.M.; Sam, I.-C.; Chong, J.; Kahar Bador, M.; Ponnampalavanar, S.; Syed Omar, S.F.; Kamarulzaman, A.; Munusamy, V.; Wong, C.K.; Jamaluddin, F.H.; et al. SARS-CoV-2 Lineage, B.6 Was the Major Contributor to Early Pandemic Transmission in Malaysia. PLoS Negl. Trop. Dis. 2020, 14, e0008744. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence That D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef] [PubMed]
- Issa, E.; Merhi, G.; Panossian, B.; Salloum, T.; Tokajian, S. SARS-CoV-2 and ORF3a: Nonsynonymous Mutations, Functional Domains, and Viral Pathogenesis. mSystems 2020, 5, e00266-20. [Google Scholar] [CrossRef]
- Tylor, S.; Andonov, A.; Cutts, T.; Cao, J.; Grudesky, E.; Van Domselaar, G.; Li, X.; He, R. The SR-Rich Motif in SARS-CoV Nucleocapsid Protein Is Important for Virus Replication. Can. J. Microbiol. 2009, 55, 254–260. [Google Scholar] [CrossRef]
- Azad, G.K.; Khan, P.K. Variations in Orf3a Protein of SARS-CoV-2 Alter Its Structure and Function. Biochem. Biophys. Rep. 2021, 26, 100933. [Google Scholar] [CrossRef]
- Letko, M.; Marzi, A.; Munster, V. Functional Assessment of Cell Entry and Receptor Usage for SARS-CoV-2 and Other Lineage B Betacoronaviruses. Nat. Microbiol. 2020, 5, 562–569. [Google Scholar] [CrossRef] [Green Version]
- Majumdar, P.; Niyogi, S. ORF3a Mutation Associated with Higher Mortality Rate in SARS-CoV-2 Infection. Epidemiol. Infect. 2020, 148, e262. [Google Scholar] [CrossRef] [PubMed]
- Pandey, R.; Agrawal, A. Next Generation Sequencing for Pandemic Preparedness. Indian Chem. Eng. 2020, 62, 351–358. [Google Scholar] [CrossRef]
- Shastri, J.; Parikh, S.; Agrawal, S.; Chatterjee, N.; Pathak, M.; Chaudhary, S.; Sharma, C.; Kanakan, A.; A, V.; Srinivasa Vasudevan, J.; et al. Clinical, Serological, Whole Genome Sequence Analyses to Confirm SARS-CoV-2 Reinfection in Patients From Mumbai, India. Front. Med. 2021, 8, 631769. [Google Scholar] [CrossRef]
- Mehta, P.; Sahni, S.; Siddiqui, S.; Mishra, N.; Sharma, P.; Sharma, S.; Tyagi, A.; Chattopadhyay, P.; Vivekanand, A.; Devi, P.; et al. Respiratory Co-Infections: Modulators of SARS-CoV-2 Patients’ Clinical Sub-Phenotype. Front. Microbiol. 2021, 12, 653399. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Chernomor, O.; von Haeseler, A.; Minh, B.Q. Terrace Aware Data Structure for Phylogenomic Inference from Supermatrices. Syst. Biol. 2016, 65, 997–1008. [Google Scholar] [CrossRef] [Green Version]
- Hahne, F.; Ivanek, R. Visualizing Genomic Data Using Gviz and Bioconductor. Methods Mol. Biol. 2016, 1418, 335–351. [Google Scholar] [CrossRef] [PubMed]
- Ou, J.; Zhu, L.J. TrackViewer: A Bioconductor Package for Interactive and Integrative Visualization of Multi-Omics Data. Nat. Methods 2019, 16, 453–454. [Google Scholar] [CrossRef] [PubMed]
- Schöler, U. Inkscape: Der WegZurProfessionellenVektorgrafik; Carl Hanser Verlag GmbH & Co. KG: München, Germany, 2014; ISBN 978-3-446-43865-1. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Total (N = 196) | Recovered (N = 174) | Dead (N = 22) | p Value |
|---|---|---|---|---|
| Age | 54(36–65) | 52(32–64) | 65(55–70) | <0.001a |
| Gender F/M | 58/138 | 52/122 | 6/16 | 0.8 b |
| Signs & Symptoms | ||||
| Fever | 150(76%) | 137(79%) | 13(59%) | 0.04 b |
| Cough | 93(47%) | 86(49%) | 7(32%) | 0.119 b |
| Sore Throat | 49(25%) | 48(28%) | 1(5%) | 0.018 b |
| Headache | 23(12%) | 22(13%) | 1(5%) | 0.266 b |
| Loss of Taste and Smell | 2(1%) | 2(11%) | 0 | - |
| Breathing Difficulty | 72(37%) | 56(32%) | 16(72%) | <0.001 b |
| Chest Pain | 5(2%) | 5(3%) | 0 | - |
| General Weakness | 29(15%) | 28(16%) | 1(5%) | 0.15 b |
| Body Ache | 40(20%) | 33(19%) | 8(36%) | 0.058 b |
| Diarrhoea | 16(8%) | 14(8%) | 2(9%) | 0.866 b |
| Nausea | 14(7%) | 12(7%) | 2(9%) | 0.7 b |
| Hospital Stays | 11(7–16) | 11(7–15) | 12(6–17) | 0.71 a |
| Respiratory Support | 98(50%) | 76(44%) | 22(100%) | <0.001 b |
| Ventilator Support | 17(4–12) | 9(7–13) | 8(2.5–8.75) | 0.034 a |
| Ct Values | ||||
| E | 25.05(21.5–27.5) | 25.17(21.6–27.5) | 23.47(19.6–27.0) | 0.33 a |
| RdRp | 26.40(22.6–29.2) | 26.53(22.7–29.5) | 25.41(22.1–28.06) | 0.211 a |
| Comorbidities | ||||
| Diabetes | 46(23%) | 40(23%) | 6(27%) | 0.655 b |
| Hypertension | 54(27%) | 41(24%) | 13(59%) | <0.001 b |
| Heart Conditions | 14(7%) | 9(5%) | 5(23%) | 0.0025 b |
| Hypothyroidism | 17(9%) | 15(9%) | 2(9%) | 0.941 b |
| No Co-morbidities | 80(40.81%) | 73(41.9%) | 7(31.81%) | 0.065 b |
| Treatment | ||||
| Antiviral | 61(31%) | 57(33%) | 4(18%) | 0.164 b |
| Steroid | 66(34%) | 61(35%) | 5(23%) | 0.248 b |
| Hydroxychloroquine (HCQ) | 93(50%) | 93(47%) | 5(23%) | 0.006 b |
| Position/SNP | Gene | Amino Acid Change | Frequency (n = 196) |
|---|---|---|---|
| C14408T | ORF1b | P314L | 75.0 |
| A23403G | S | D614G | 63.8 |
| C18877T | ORF1b | - | 54.1 |
| C26735T | M | - | 51.5 |
| G25563T | ORF3a | Q57H | 51.0 |
| C3037T | ORF1a | - | 50.5 |
| G11083T | ORF1a | L3606F | 43.9 |
| C22444T | S | - | 39.3 |
| C28854T | N | S194L | 36.2 |
| C6312A | Nsp3 | T2016K | 29.1 |
| C28311T | N | P13L | 28.1 |
| Mutation | p-Value (Mortality) | p-Value (Severity) | Frequency (n = 196) | Locus | Amino Acid Change | Severity Rate | Mortality Rate | Frequency (%) |
|---|---|---|---|---|---|---|---|---|
| C631A | 0.1203 | 0.0236 | 33 | Orf1a:122 | - | 0.3030 | 0.0303 | 16.83 |
| C1373T | 0.0949 | 0.0077 | 36 | Orf1a | C370R | 0.2778 | 0.0278 | 18.36 |
| C3037T | 0.0928 | 0.1317 | 99 | Orf1a:924 | - | 0.5758 | 0.1717 | 50.51 |
| C6312A | 0.0192 | 0.0243 | 57 | Orf1ab | T2016K | 0.3509 | 0.0175 | 29.08 |
| T24622C | 0.0335 | 0.0395 | 34 | S:1020 | - | 0.3235 | 0.0000 | 17.34 |
| C25611A | 0.1340 | 0.0158 | 29 | Orf3a:74 | - | 0.7241 | 0.2069 | 14.79 |
| A26194T | 0.0028 | 0.0023 | 13 | Orf3a | T268S | 0.9231 | 0.3846 | 6.63 |
| C28854T | 0.0366 | 0.1228 | 71 | N | S194L | 0.5915 | 0.1972 | 36.22 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mehta, P.; Alle, S.; Chaturvedi, A.; Swaminathan, A.; Saifi, S.; Maurya, R.; Chattopadhyay, P.; Devi, P.; Chauhan, R.; Kanakan, A.; et al. Clinico-Genomic Analysis Reveals Mutations Associated with COVID-19 Disease Severity: Possible Modulation by RNA Structure. Pathogens 2021, 10, 1109. https://doi.org/10.3390/pathogens10091109
Mehta P, Alle S, Chaturvedi A, Swaminathan A, Saifi S, Maurya R, Chattopadhyay P, Devi P, Chauhan R, Kanakan A, et al. Clinico-Genomic Analysis Reveals Mutations Associated with COVID-19 Disease Severity: Possible Modulation by RNA Structure. Pathogens. 2021; 10(9):1109. https://doi.org/10.3390/pathogens10091109
Chicago/Turabian StyleMehta, Priyanka, Shanmukh Alle, Anusha Chaturvedi, Aparna Swaminathan, Sheeba Saifi, Ranjeet Maurya, Partha Chattopadhyay, Priti Devi, Ruchi Chauhan, Akshay Kanakan, and et al. 2021. "Clinico-Genomic Analysis Reveals Mutations Associated with COVID-19 Disease Severity: Possible Modulation by RNA Structure" Pathogens 10, no. 9: 1109. https://doi.org/10.3390/pathogens10091109
APA StyleMehta, P., Alle, S., Chaturvedi, A., Swaminathan, A., Saifi, S., Maurya, R., Chattopadhyay, P., Devi, P., Chauhan, R., Kanakan, A., Vasudevan, J. S., Sethuraman, R., Chidambaram, S., Srivastava, M., Chakravarthi, A., Jacob, J., Namagiri, M., Konala, V., Jha, S., ... Pandey, R. (2021). Clinico-Genomic Analysis Reveals Mutations Associated with COVID-19 Disease Severity: Possible Modulation by RNA Structure. Pathogens, 10(9), 1109. https://doi.org/10.3390/pathogens10091109