
Population Genetic Structure and Hybridization of Schistosoma haematobium in Nigeria

 ,
,  , ,
, ,  ,
,
Abstract
:1. Introduction
2. Results
2.1. Schistosome Genotyping Using Cox1 and ITS2
2.2. Cox1 Phylogenetic Trees
2.3. Microsatellite Analysis
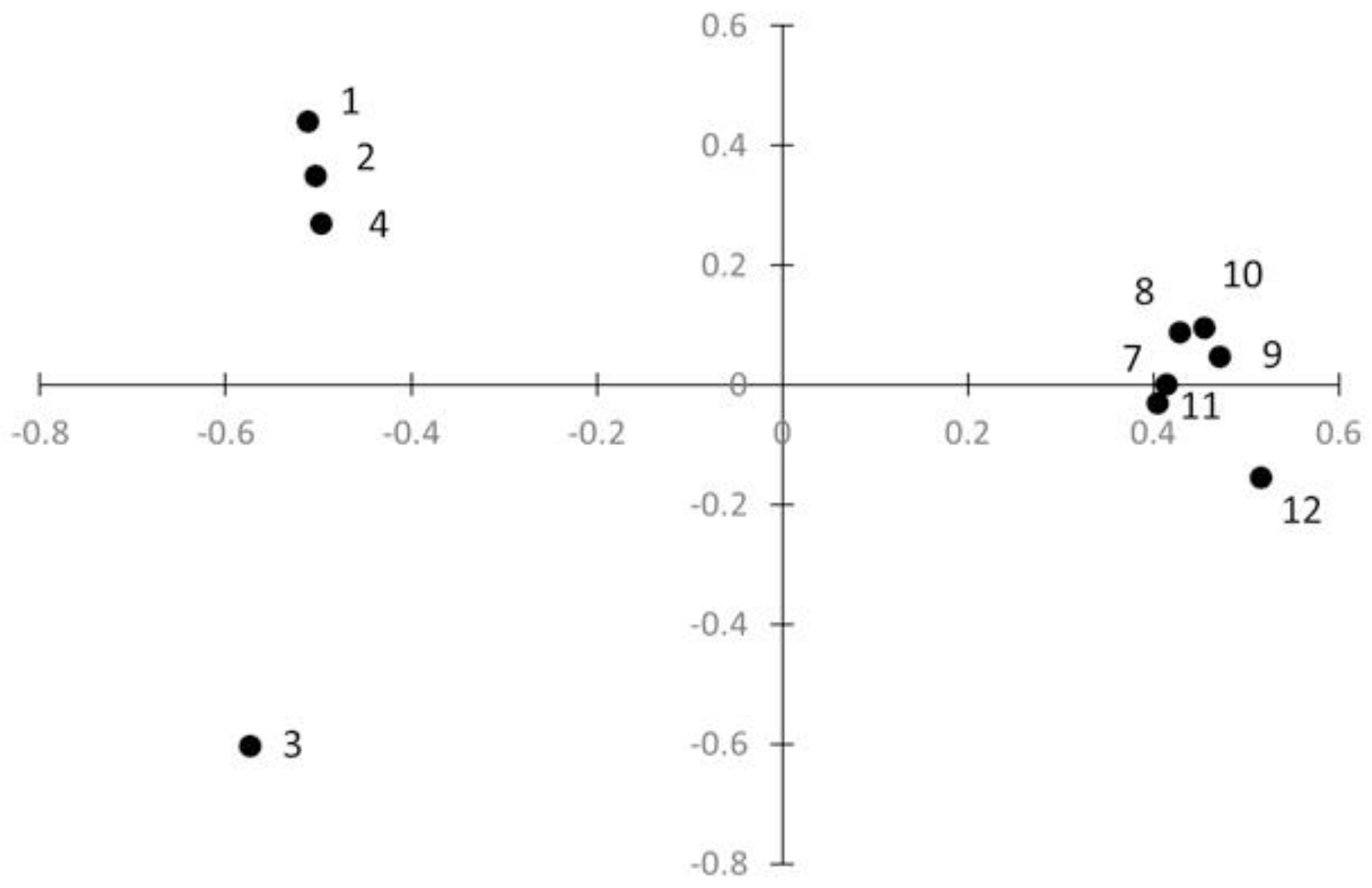
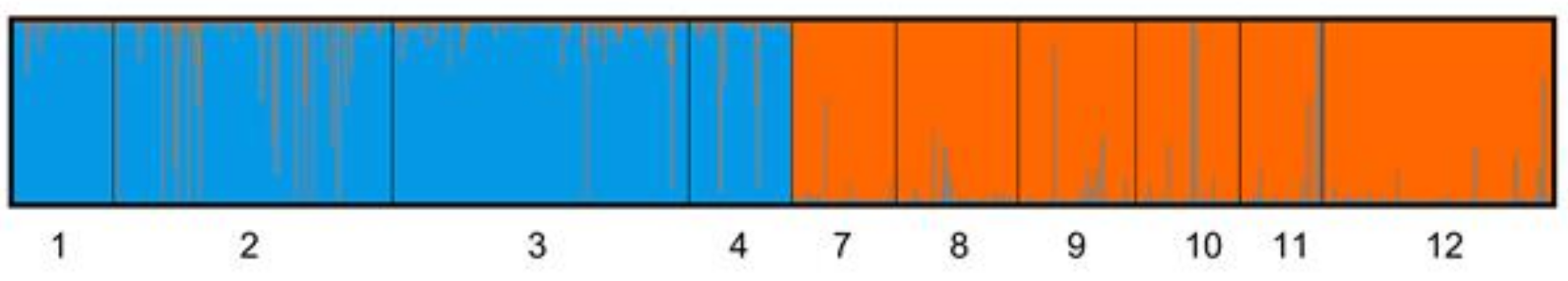
2.4. Population Genetic Structure
3. Discussion
4. Materials and Methods
4.1. Parasitological Survey and Sampling Collection
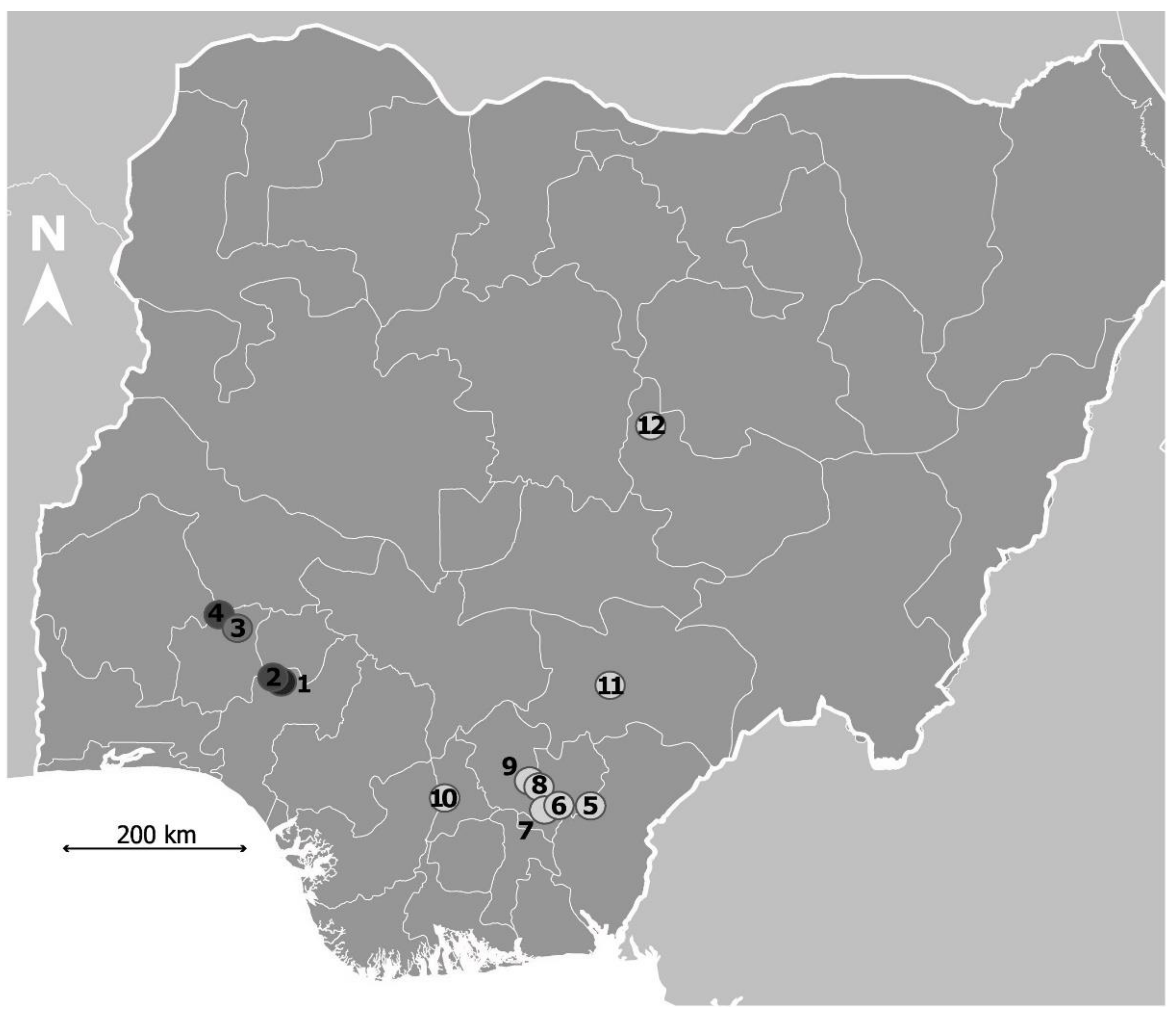
4.1.1. Study Area and Study Population
4.1.2. Urine Sample Collection and Miracidia Sampling
4.2. Genomic Analysis
4.2.1. DNA Extraction
4.2.2. Estimation of Hybrid Prevalence by Mitochondrial DNA Identification
4.2.3. Mitochondrial DNA (Cox1) and Nuclear Internal Transcribed Spacer II (ITS2) Sequencing
4.2.4. Sequence Analysis
4.2.5. Microsatellite Genotyping
4.2.6. Population Genetic Structure
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hotez, P.J.; Alvarado, M.; Basanez, M.G.; Bolliger, I.; Bourne, R.; Boussinesq, M.; Brooker, S.J.; Brown, A.S.; Buckle, G.; Budke, C.M.; et al. The global burden of disease study 2010: Interpretation and implications for the neglected tropical diseases. PLoS Negl. Trop Dis. 2014, 8, e2865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boissier, J.; Mouahid, G.; Moné, H. Schistosoma spp. (Robertson, L (eds) Part 4 Helminths); Rose, J.B., Jiménez-Cisneros, B., Eds.; Michigan State University: E. Lansing, MI, USA, 2019. [Google Scholar]
- Panzner, U.; Boissier, J. Natural Intra- and Interclade Human Hybrid Schistosomes in Africa with Considerations on Prevention through Vaccination. Microorganisms 2021, 9, 1465. [Google Scholar] [CrossRef]
- Leger, E.; Webster, J.P. Hybridizations within the Genus Schistosoma: Implications for evolution, epidemiology and control. Parasitology 2017, 144, 65–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mone, H.; Mouahid, G.; Morand, S. The distribution of Schistosoma bovis Sonsino, 1876 in relation to intermediate host mollusc-parasite relationships. Adv. Parasitol. 1999, 44, 99–138. [Google Scholar] [PubMed]
- Djuikwo-Teukeng, F.; Kouam Simo, A.; Alliene, J.F.; Rey, O.; Njayou Ngapagna, A.; Tchuem Tchuente, L.A.; Boissier, J. Population genetic structure of Schistosoma bovis in Cameroon. Parasit Vectors 2019, 12, 56–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huyse, T.; Webster, B.L.; Geldof, S.; Stothard, J.R.; Diaw, O.T.; Polman, K.; Rollinson, D. Bidirectional introgressive hybridization between a cattle and human schistosome species. PLoS Pathog. 2009, 5, e1000571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sene-Wade, M.; Marchand, B.; Rollinson, D.; Webster, B.L. Urogenital schistosomiasis and hybridization between Schistosoma haematobium and Schistosoma bovis in adults living in Richard-Toll, Senegal. Parasitology 2018, 145, 1723–1726. [Google Scholar] [CrossRef]
- Angora, E.K.; Allienne, J.F.; Rey, O.; Menan, H.; Toure, A.O.; Coulibaly, J.T.; Raso, G.; Yavo, W.; N’Goran, E.K.; Utzinger, J.; et al. High prevalence of Schistosoma haematobium × Schistosoma bovis hybrids in schoolchildren in Cote d’Ivoire. Parasitology 2020, 147, 287–294. [Google Scholar] [CrossRef]
- Wright, C.A.; Ross, G.C. Hybrids between Schistosoma haematobium and S. mattheei and their identification by isoelectric focusing of enzymes. Trans. R Soc. Trop Med. Hyg. 1980, 74, 326–332. [Google Scholar] [CrossRef]
- Kruger, F.J.; Evans, A.C. Do all human urinary infections with Schistosoma mattheei represent hybridization between S. haematobium and S. mattheei? J. Helminthol. 1990, 64, 330–332. [Google Scholar] [CrossRef]
- Webster, B.L.; Diaw, O.T.; Seye, M.M.; Webster, J.P.; Rollinson, D. Introgressive hybridization of Schistosoma haematobium group species in Senegal: Species barrier break down between ruminant and human schistosomes. PLoS Negl. Trop Dis. 2013, 7, e2110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huyse, T.; Van den Broeck, F.; Hellemans, B.; Volckaert, F.A.; Polman, K. Hybridisation between the two major African schistosome species of humans. Int. J. Parasitol. 2013, 43, 687–689. [Google Scholar] [CrossRef]
- Le Govic, Y.; Kincaid-Smith, J.; Allienne, J.F.; Rey, O.; de Gentile, L.; Boissier, J. Schistosoma haematobium-Schistosoma mansoni Hybrid Parasite in Migrant Boy, France, 2017. Emerg. Infect. Dis. 2019, 25, 365–367. [Google Scholar] [CrossRef] [Green Version]
- Depaquit, J.; Akhoundi, M.; Haouchine, D.; Mantelet, S.; Izri, A. No limit in interspecific hybridization in schistosomes: Observation from a case report. Parasite 2019, 26, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brémond, P.; Sellin, B.; Sellin, E.; Naméoua, B.; Labbo, R.; Théron, A.; Combes, C. Arguments en faveur d’une modification du génome (introgression) du parasite humain Schistosoma haematobium par des gènes de S. bovis, au Niger. Comptes-Rendus L’académie Des. Sci. 1993, 316, 667–670. [Google Scholar]
- Mone, H.; Holtfreter, M.C.; Allienne, J.F.; Mintsa-Nguema, R.; Ibikounle, M.; Boissier, J.; Berry, A.; Mitta, G.; Richter, J.; Mouahid, G. Introgressive hybridizations of Schistosoma haematobium by Schistosoma bovis at the origin of the first case report of schistosomiasis in Corsica (France, Europe). Parasitol. Res. 2015, 114, 4127–4133. [Google Scholar] [CrossRef] [PubMed]
- Boissier, J.; Grech-Angelini, S.; Webster, B.L.; Allienne, J.F.; Huyse, T.; Mas-Coma, S.; Toulza, E.; Barré-Cardi, H.; Rollinson, D.; Kincaid-Smith, J.; et al. Outbreak of urogenital schistosomiasis in Corsica (France): An epidemiological case study. Lancet Infect. Dis. 2016, 16, 971–979. [Google Scholar] [CrossRef]
- Catalano, S.; Sene, M.; Diouf, N.D.; Fall, C.B.; Borlase, A.; Leger, E.; Ba, K.; Webster, J.P. Rodents as Natural Hosts of Zoonotic Schistosoma Species and Hybrids: An Epidemiological and Evolutionary Perspective From West Africa. J. Infect. Dis. 2018, 218, 429–433. [Google Scholar] [CrossRef] [Green Version]
- Savassi, B.; Dobigny, G.; Etougbetche, J.R.; Avocegan, T.T.; Quinsou, F.T.; Gauthier, P.; Ibikounle, M.; Mone, H.; Mouahid, G. Mastomys natalensis (Smith, 1834) as a natural host for Schistosoma haematobium (Bilharz, 1852) Weinland, 1858 × Schistosoma bovis Sonsino, 1876 introgressive hybrids. Parasitol. Res. 2021, 120, 1755–1770. [Google Scholar] [CrossRef]
- Savassi, B.; Mouahid, G.; Lasica, C.; Mahaman, S.K.; Garcia, A.; Courtin, D.; Allienne, J.F.; Ibikounle, M.; Mone, H. Cattle as natural host for Schistosoma haematobium (Bilharz, 1852) Weinland, 1858 × Schistosoma bovis Sonsino, 1876 interactions, with new cercarial emergence and genetic patterns. Parasitol. Res. 2020, 119, 2189–2205. [Google Scholar] [CrossRef]
- Southgate, V.R.; Rollinson, D.; Ross, G.C.; Knowles, R.J.; Vercruysse, J. On Schistosoma curassoni, S. haematobium and S. bovis from Senegal: Development in Mesocricetus auratus, compatibility with species of Bulinus and their enzymes. J. Nat. Hist. 1985, 19, 1249–1267. [Google Scholar] [CrossRef]
- Vercruysse, J.; Southgate, V.R.; Rollinson, D.; De Clercq, D.; Sacko, M.; De Bont, J.; Mungomba, L.M. Studies on transmission and schistosome interactions in Senegal, Mali and Zambia. Trop Geogr. Med. 1994, 46, 220–226. [Google Scholar] [PubMed]
- Webster, B.L.; Tchuem Tchuente, L.A.; Jourdane, J.; Southgate, V.R. The interaction of Schistosoma haematobium and S. guineensis in Cameroon. J. Helminthol. 2005, 79, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Mone, H.; Minguez, S.; Ibikounle, M.; Allienne, J.F.; Massougbodji, A.; Mouahid, G. Natural Interactions between S. haematobium and S. guineensis in the Republic of Benin. Sci. World J. 2012, 2012, 793420. [Google Scholar] [CrossRef] [Green Version]
- De Bont, J.; Vercruysse, J.; Southgate, V.R.; Rollinson, D.; Kaukas, A. Cattle schistosomiasis in Zambia. J. Helminthol. 1994, 68, 295–299. [Google Scholar] [CrossRef]
- Rey, O.; Webster, B.L.; Huyse, T.; Rollinson, D.; Van den Broeck, F.; Kincaid-Smith, J.; Onyekwere, A.; Boissier, J. Population genetics of African Schistosoma species. Infect. Genet. Evol. 2021, 89, 104727. [Google Scholar] [CrossRef]
- Boon, N.A.M.; Mbow, M.; Paredis, L.; Moris, P.; Sy, I.; Maes, T.; Webster, B.L.; Sacko, M.; Volckaert, F.A.M.; Polman, K.; et al. No barrier breakdown between human and cattle schistosome species in the Senegal River Basin in the face of hybridisation. Int J. Parasitol. 2019, 49, 1039–1048. [Google Scholar] [CrossRef]
- Boon, N.A.M.; Van Den Broeck, F.; Faye, D.; Volckaert, F.A.M.; Mboup, S.; Polman, K.; Huyse, T. Barcoding hybrids: Heterogeneous distribution of Schistosoma haematobium × Schistosoma bovis hybrids across the Senegal River Basin. Parasitology 2018, 145, 634–645. [Google Scholar] [CrossRef] [Green Version]
- Leger, E.; Borlase, A.; Fall, C.B.; Diouf, N.D.; Diop, S.D.; Yasenev, L.; Catalano, S.; Thiam, C.T.; Ndiaye, A.; Emery, A.; et al. Prevalence and distribution of schistosomiasis in human, livestock, and snail populations in northern Senegal: A One Health epidemiological study of a multi-host system. Lancet Planet Health 2020, 4, e330–e342. [Google Scholar] [CrossRef]
- Onyekwere, A.M.; Rey, O.; Nwanchor, M.C.; Alo, M.; Angora, E.K.; Allienne, J.F.; Boissier, J. Prevalence and Risk Factors Associated with Urogenital Schistosomiasis among Primary School Pupils in Nigeria. Parasite Epidemiol. Control 2021, Submitted. [Google Scholar]
- Angora, E.K.; Boissier, J.; Menan, H.; Rey, O.; Tuo, K.; Toure, A.O.; Coulibaly, J.T.; Meite, A.; Raso, G.; N’Goran, E.K.; et al. Prevalence and Risk Factors for Schistosomiasis among Schoolchildren in two Settings of Cote d’Ivoire. Trop Med. Infect. Dis. 2019, 4, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sang, T.; Crawford, D.J.; Stuessy, T.F. Documentation of reticulate evolution in peonies (Paeonia) using internal transcribed spacer sequences of nuclear ribosomal DNA: Implications for biogeography and concerted evolution. Proc. Natl. Acad. Sci. USA 1995, 92, 6813–6817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kincaid-Smith, J.; Mathieu-Begne, E.; Chaparro, C.; Reguera-Gomez, M.; Mulero, S.; Allienne, J.F.; Toulza, E.; Boissier, J. No pre-zygotic isolation mechanisms between Schistosoma haematobium and Schistosoma bovis parasites: From mating interactions to differential gene expression. PLoS Negl. Trop Dis. 2021, 15, e0009363. [Google Scholar] [CrossRef]
- Platt, R.N.; McDew-White, M.; Le Clec’h, W.; Chevalier, F.D.; Allan, F.; Emery, A.M.; Garba, A.; Hamidou, A.A.; Ame, S.M.; Webster, J.P.; et al. Ancient Hybridization and Adaptive Introgression of an Invadolysin Gene in Schistosome Parasites. Mol. Biol. Evol. 2019, 36, 2127–2142. [Google Scholar] [CrossRef] [Green Version]
- Rey, O.; Toulza, E.; Chaparro, C.; Allienne, J.F.; Kincaid-Smith, J.; Mathieu-Begne, E.; Allan, F.; Rollinson, D.; Webster, B.L.; Boissier, J. Diverging patterns of introgression from Schistosoma bovis across S. haematobium African lineages. PLoS Pathog. 2021, 17, e1009313. [Google Scholar]
- Gower, C.M.; Gabrielli, A.F.; Sacko, M.; Dembele, R.; Golan, R.; Emery, A.M.; Rollinson, D.; Webster, J.P. Population genetics of Schistosoma haematobium: Development of novel microsatellite markers and their application to schistosomiasis control in Mali. Parasitology 2011, 138, 978–994. [Google Scholar] [CrossRef] [PubMed]
- Agola, L.E.; Mburu, D.N.; DeJong, R.J.; Mungai, B.N.; Muluvi, G.M.; Njagi, E.N.; Loker, E.S.; Mkoji, G.M. Microsatellite typing reveals strong genetic structure of Schistosoma mansoni from localities in Kenya. Infect Genet. Evol. 2006, 6, 484–490. [Google Scholar] [CrossRef]
- Aemero, M.; Boissier, J.; Climent, D.; Mone, H.; Mouahid, G.; Berhe, N.; Erko, B. Genetic diversity, multiplicity of infection and population structure of Schistosoma mansoni isolates from human hosts in Ethiopia. BMC Genet. 2015, 16, 137. [Google Scholar] [CrossRef] [Green Version]
- Beltran, S.; Galinier, R.; Allienne, J.F.; Boissier, J. Cheap, rapid and efficient DNA extraction method to perform multilocus microsatellite genotyping on all Schistosoma mansoni stages. Mem. Inst. Oswaldo Cruz 2008, 103, 501–503. [Google Scholar] [CrossRef] [Green Version]
- Webster, B.L.; Rollinson, D.; Stothard, J.R.; Huyse, T. Rapid diagnostic multiplex PCR (RD-PCR) to discriminate Schistosoma haematobium and S. bovis. J. Helminthol. 2010, 84, 107–114. [Google Scholar] [CrossRef] [Green Version]
- Webster, B.L.; Emery, A.M.; Webster, J.P.; Gouvras, A.; Garba, A.; Diaw, O.; Seye, M.M.; Tchuente, L.A.; Simoonga, C.; Mwanga, J.; et al. Genetic diversity within Schistosoma haematobium: DNA barcoding reveals two distinct groups. PLoS Negl. Trop Dis. 2012, 6, e1882. [Google Scholar] [CrossRef] [PubMed]
- Webster, B.L.; Rabone, M.; Pennance, T.; Emery, A.M.; Allan, F.; Gouvras, A.; Knopp, S.; Garba, A.; Hamidou, A.A.; Mohammed, K.A.; et al. Development of novel multiplex microsatellite polymerase chain reactions to enable high-throughput population genetic studies of Schistosoma haematobium. Parasit Vectors 2015, 8, 432, Erratum Parasit Vectors 2015, 8, 519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goudet, J.; Perrin, N.; Waser, P. Tests for sex-biased dispersal using bi-parentally inherited genetic markers. Mol. Ecol. 2002, 11, 1103–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weir, B.S.; Cockerham, C.C. Estimating F-Statistics for the Analysis of Population Structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [PubMed]
- Belkhir, K.; Borsa, P.; Chikhi, L.; Raufaste, N.; Bonhomme, F. GENETIX 4.05, Logiciel Sous Windows TM Pour la Génétique des Populations. Laboratoire Génome, Populations, Interactions, CNRS UMR 5000, Université de Montpellier II, Montpellier (France) 1996. Available online: http://www.genetix.univ-montp2.fr/genetix/constr.htm (accessed on 30 March 2022).
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [Green Version]
- Francis, R.M. pophelper: An R package and web app to analyse and visualize population structure. Mol. Ecol. Resour. 2017, 17, 27–32. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, N.A. DISTRUCT: A program for the graphical display of population structure. Mol. Ecol. Notes 2004, 4, 137–138. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Sampling Site | No. of Children Tested | No. Miracidia Genotyped | No. of Miracidia with Cox1 S. bovis | No. of Miracidia with Cox1 S. haematobium | Min. % of Hybrids (S. bovis Cox1) |
|---|---|---|---|---|---|
| 1 | 10 | 90 | 90 | 0 | 100% |
| 2 | 20 | 156 | 156 | 0 | 100% |
| 3 | 18 | 152 | 28 | 124 | 18% |
| 4 | 11 | 74 | 46 | 28 | 62% |
| 5 | 3 | 30 | 30 | 0 | 100% |
| 6 | 12 | 66 | 66 | 0 | 100% |
| 7 | 12 | 84 | 84 | 0 | 100% |
| 8 | 12 | 103 | 103 | 0 | 100% |
| 9 | 17 | 164 | 164 | 0 | 100% |
| 10 | 12 | 90 | 90 | 0 | 100% |
| 11 | 20 | 193 | 193 | 0 | 100% |
| 12 | 18 | 162 | 162 | 0 | 100% |
| Total | 165 | 1364 | 1212 | 152 | 89% |
| Genotype | Site | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | Total Alleles (%) | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sb ITS2 | Sh ITS2 | ||||||||||||||||
| COX1 | ITS2 | ||||||||||||||||
| Sb | SbSb | 3 | 1 | 0 | 0 | 0 | 0 | 2 | 0 | 4 | 4 | 5 | 4 | 23 (39.0) | 46 (39.0) | 0 (0) | |
| Sb | ShSh | 2 | 0 | 4 | 2 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 9 (15.3) | 0 (0) | 18 (15.3) | |
| Sb | SbSh | 2 | 6 | 2 | 1 | 0 | 2 | 0 | 1 | 3 | 2 | 1 | 2 | 22 (37.2) | 22 (18.6) | 22 (18.6) | |
| Sh | SbSb | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 (0) | 0 (0) | 0 (0) | |
| Sh | ShSh | 0 | 0 | 2 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 3 (5.1) | 0 (0) | 6 (5.1) | |
| Sh | SbSh | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 (3.4) | 2 (1.7) | 2 (1.7) | |
| Total | 7 | 7 | 10 | 4 | 0 | 2 | 2 | 1 | 7 | 7 | 6 | 6 | 59 (100) | 70 (59.3) | 48 (40.7) | ||
| Locus | Sh9 | Sh3 | C102 | Sh1 | Sh14 | Sh6 | C111 | Sh7 | Sh13 | Sh11 | Sh2 | Sh5 | Sh10 | Sh12 | Mean | SE |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Site 1 n = 74 | ||||||||||||||||
| He | 0.633 | 0.824 | 0.000 | 0.704 | 0.875 | 0.365 | 0.608 | 0.560 | 0.696 | 0.454 | 0.877 | 0.841 | 0.536 | 0.335 | 0.593 | 0.247 |
| A | 5 | 10 | 1 | 6 | 11 | 5 | 5 | 4 | 8 | 5 | 10 | 9 | 7 | 4 | 6.429 | 2.848 |
| Ar | 4.590 | 9.603 | 1.000 | 5.550 | 10.934 | 4.947 | 4.645 | 3.636 | 7.330 | 4.980 | 9.574 | 8.710 | 6.014 | 3.956 | 6.105 | 2.774 |
| Fis | 0.569 | 0.241 | NA | 0.177 | 0.082 | 0.273 | 0.088 | 0.377 | −0.026 | 0.416 | 0.411 | 0.415 | 0.501 | 0.048 | 0.275 | 0.196 |
| Site 2 n = 206 | ||||||||||||||||
| He | 0.624 | 0.860 | 0.025 | 0.659 | 0.883 | 0.335 | 0.636 | 0.677 | 0.710 | 0.480 | 0.821 | 0.821 | 0.365 | 0.473 | 0.598 | 0.241 |
| A | 11 | 11 | 2 | 6 | 12 | 6 | 6 | 4 | 10 | 5 | 12 | 13 | 8 | 5 | 7.929 | 3.518 |
| Ar | 7.111 | 9.906 | 1.694 | 5.502 | 11.469 | 4.337 | 4.960 | 4.000 | 7.839 | 4.465 | 10.851 | 10.045 | 5.225 | 4.212 | 6.544 | 3.017 |
| Fis | 0.414 | 0.116 | −0.010 | 0.013 | 0.063 | 0.090 | 0.039 | 0.468 | −0.010 | 0.411 | 0.264 | 0.423 | 0.219 | −0.086 | 0.172 | 0.191 |
| Site 3 n = 219 | ||||||||||||||||
| He | 0.698 | 0.801 | 0.331 | 0.624 | 0.767 | 0.360 | 0.630 | 0.656 | 0.656 | 0.231 | 0.833 | 0.875 | 0.326 | 0.136 | 0.566 | 0.241 |
| A | 9 | 14 | 7 | 12 | 10 | 5 | 6 | 5 | 11 | 5 | 12 | 13 | 6 | 5 | 8.571 | 3.368 |
| Ar | 6.925 | 10.399 | 5.671 | 8.790 | 8.091 | 3.698 | 5.003 | 4.623 | 9.302 | 3.715 | 10.252 | 12.187 | 4.802 | 3.541 | 6.929 | 2.890 |
| Fis | 0.363 | 0.107 | 0.130 | −0.006 | 0.151 | 0.344 | 0.108 | 0.094 | 0.103 | 0.562 | 0.335 | 0.202 | 0.399 | 0.158 | 0.218 | 0.157 |
| Site 4 n = 76 | ||||||||||||||||
| He | 0.583 | 0.885 | 0.039 | 0.675 | 0.865 | 0.419 | 0.602 | 0.579 | 0.652 | 0.580 | 0.796 | 0.821 | 0.308 | 0.409 | 0.587 | 0.235 |
| A | 5 | 11 | 3 | 6 | 12 | 5 | 6 | 4 | 8 | 5 | 11 | 9 | 5 | 4 | 6.714 | 2.946 |
| Ar | 4.683 | 10.865 | 2.354 | 5.831 | 11.624 | 4.757 | 5.418 | 3.863 | 7.602 | 4.675 | 10.836 | 8.517 | 4.322 | 3.934 | 6.377 | 2.986 |
| Fis | 0.216 | 0.135 | −0.009 | −0.015 | 0.183 | 0.040 | 0.145 | 0.330 | −0.062 | 0.655 | 0.392 | 0.513 | 0.333 | −0.033 | 0.202 | 0.219 |
| Site 7 n = 77 | ||||||||||||||||
| He | 0.585 | 0.781 | 0.208 | 0.569 | 0.731 | 0.000 | 0.659 | 0.534 | 0.822 | 0.194 | 0.750 | 0.758 | 0.666 | 0.703 | 0.569 | 0.253 |
| A | 8 | 9 | 2 | 5 | 7 | 1 | 6 | 3 | 11 | 2 | 7 | 7 | 7 | 4 | 5.643 | 2.925 |
| Ar | 6.624 | 8.526 | 2.000 | 4.714 | 6.506 | 1.000 | 5.133 | 2.998 | 9.906 | 2.000 | 5.969 | 6.680 | 6.897 | 4.000 | 5.211 | 2.591 |
| Fis | 0.399 | 0.179 | 0.126 | 0.214 | 0.088 | NA | 0.262 | 1.000 | 0.083 | 0.525 | −0.167 | 0.330 | 0.675 | 0.196 | 0.301 | 0.298 |
| Site8 n = 90 | ||||||||||||||||
| He | 0.732 | 0.802 | 0.163 | 0.525 | 0.673 | 0.000 | 0.629 | 0.509 | 0.736 | 0.229 | 0.711 | 0.671 | 0.677 | 0.664 | 0.552 | 0.245 |
| A | 9 | 8 | 2 | 5 | 7 | 1 | 4 | 4 | 5 | 4 | 7 | 8 | 9 | 7 | 5.714 | 2.525 |
| Ar | 8.112 | 7.737 | 2.000 | 4.448 | 6.387 | 1.000 | 3.859 | 3.669 | 4.999 | 3.424 | 6.643 | 7.285 | 7.628 | 6.179 | 5.241 | 2.242 |
| Fis | 0.700 | 0.213 | −0.092 | 0.102 | −0.008 | NA | 0.043 | 0.690 | 0.233 | 0.630 | −0.102 | 0.109 | 0.636 | 0.089 | 0.249 | 0.300 |
| Site 9 n = 87 | ||||||||||||||||
| He | 0.755 | 0.778 | 0.269 | 0.457 | 0.702 | 0.000 | 0.578 | 0.400 | 0.771 | 0.052 | 0.737 | 0.714 | 0.494 | 0.667 | 0.527 | 0.263 |
| A | 8 | 8 | 2 | 5 | 6 | 1 | 4 | 2 | 6 | 2 | 7 | 8 | 7 | 6 | 5.143 | 2.507 |
| Ar | 7.445 | 7.121 | 2.000 | 3.973 | 5.971 | 1.000 | 3.488 | 2.000 | 5.518 | 1.983 | 6.718 | 6.826 | 6.302 | 5.709 | 4.718 | 2.245 |
| Fis | 0.470 | 0.044 | 0.081 | −0.080 | −0.043 | NA | 0.376 | 0.826 | 0.408 | −0.018 | −0.192 | 0.159 | 0.349 | 0.104 | 0.191 | 0.275 |
| Site 10 n = 77 | ||||||||||||||||
| He | 0.761 | 0.762 | 0.232 | 0.534 | 0.677 | 0.000 | 0.567 | 0.490 | 0.766 | 0.262 | 0.787 | 0.727 | 0.702 | 0.704 | 0.569 | 0.244 |
| A | 8 | 8 | 3 | 3 | 8 | 1 | 5 | 2 | 7 | 3 | 8 | 6 | 8 | 6 | 5.429 | 2.563 |
| Ar | 7.334 | 7.561 | 2.553 | 3.000 | 7.071 | 1.000 | 4.120 | 2.000 | 6.151 | 2.667 | 7.614 | 5.856 | 7.568 | 5.791 | 5.020 | 2.379 |
| Fis | 0.371 | 0.075 | 0.092 | 0.241 | −0.113 | NA | 0.201 | 0.819 | 0.231 | 0.697 | −0.059 | 0.089 | 0.719 | 0.105 | 0.267 | 0.298 |
| Site 11 n = 61 | ||||||||||||||||
| He | 0.747 | 0.768 | 0.242 | 0.692 | 0.677 | 0.075 | 0.557 | 0.551 | 0.763 | 0.406 | 0.776 | 0.723 | 0.593 | 0.696 | 0.590 | 0.213 |
| A | 9 | 9 | 2 | 4 | 6 | 4 | 4 | 3 | 9 | 2 | 8 | 9 | 8 | 5 | 5.857 | 2.742 |
| Ar | 8.542 | 8.910 | 2.000 | 4.000 | 5.962 | 3.543 | 3.700 | 3.000 | 10.162 | 2.000 | 7.439 | 8.736 | 7.466 | 4.999 | 5.747 | 2.779 |
| Fis | 0.596 | 0.030 | 0.256 | 0.261 | 0.091 | 0.494 | 0.281 | 0.637 | −0.063 | 0.707 | −0.013 | 0.202 | 0.603 | 0.116 | 0.300 | 0.262 |
| Site 12 n = 169 | ||||||||||||||||
| He | 0.760 | 0.752 | 0.200 | 0.619 | 0.725 | 0.045 | 0.608 | 0.420 | 0.816 | 0.123 | 0.738 | 0.712 | 0.468 | 0.728 | 0.551 | 0.259 |
| A | 10 | 11 | 4 | 6 | 7 | 2 | 5 | 4 | 11 | 3 | 8 | 9 | 8 | 7 | 6.786 | 2.914 |
| Ar | 7.487 | 8.692 | 3.555 | 4.973 | 6.448 | 1.895 | 4.220 | 3.188 | 9.908 | 2.340 | 6.648 | 7.411 | 6.542 | 4.797 | 5.579 | 2.401 |
| Fis | 0.513 | 0.167 | 0.038 | 0.134 | −0.025 | 0.855 | 0.147 | 0.836 | 0.020 | 0.471 | −0.034 | 0.174 | 0.665 | −0.000 | 0.283 | 0.321 |
| Total (n = 1136) | ||||||||||||||||
| Population Number | 1 | 2 | 3 | 4 | 7 | 8 | 9 | 10 | 11 | 12 |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | -- | 0.0104 | 0.0681 * | 0.0441 * | 0.1493 * | 0.1454 * | 0.1544 * | 0.1295 * | 0.1424 * | 0.1579 * |
| 2 | 5.6 | -- | 0.0546 * | 0.0187 * | 0.1206 * | 0.1286 * | 0.1323 * | 0.1157 * | 0.1188 * | 0.1348 * |
| 3 | 72.7 | 68.2 | -- | 0.0445 * | 0.1195 * | 0.1497 * | 0.1387 * | 0.1346 * | 0.1179 * | 0.1332 * |
| 4 | 97.6 | 92.8 | 25.5 | -- | 0.1274 * | 0.1683 * | 0.1688 * | 0.1521 * | 0.1181 * | 0.1487 * |
| 7 | 372.3 | 377.9 | 436.3 | 461.5 | -- | 0.0358 * | 0.0371 * | 0.0318 * | 0.0112 | 0.0209 |
| 8 | 311.7 | 317.3 | 376.4 | 401.6 | 24.3 | -- | 0.0122 | 0.0052 | 0.0356 * | 0.0241 * |
| 9 | 298.9 | 304.5 | 363.3 | 388.5 | 36.1 | 13.2 | -- | 0.0194 | 0.0485 * | 0.0261 * |
| 10 | 225.1 | 230.5 | 295.4 | 320.9 | 113.8 | 105.4 | 96.2 | -- | 0.0244 * | 0.0286 * |
| 11 | 365.8 | 371.2 | 414.7 | 437.4 | 153.3 | 138.0 | 138.0 | 221.5 | -- | 0.0220 * |
| 12 | 497.9 | 501.3 | 506.5 | 518.5 | 439.3 | 419.8 | 415.4 | 471.3 | 291.0 | -- |
| Site No | Sampling Site | No. of Children | No. of Miracidia Collected | No. of Miracidia Genotyped with Cox1 | No. of Miracidia Genotyped with Microsatellites |
|---|---|---|---|---|---|
| 1 | Ipogun | 10 | 268 | 95 | 74 |
| 2 | Ilara-Mokin | 20 | 560 | 156 | 206 |
| 3 | Alie Ilie | 18 | 405 | 152 | 219 |
| 4 | Lie Twon | 11 | 279 | 74 | 76 |
| 5 | Ikwo | 3 | 82 | 30 | 0 |
| 6 | Ohaozara | 12 | 279 | 66 | 0 |
| 7 | Onicha | 12 | 277 | 84 | 77 |
| 8 | Ishielu | 12 | 278 | 103 | 90 |
| 9 | Nkanu east | 17 | 418 | 164 | 87 |
| 10 | Anambra west | 12 | 279 | 90 | 77 |
| 11 | Gwer east | 20 | 465 | 193 | 61 |
| 12 | Jos north | 18 | 417 | 162 | 169 |
| Total | 165 | 4007 | 1364 | 1136 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Onyekwere, A.M.; Rey, O.; Allienne, J.-F.; Nwanchor, M.C.; Alo, M.; Uwa, C.; Boissier, J. Population Genetic Structure and Hybridization of Schistosoma haematobium in Nigeria. Pathogens 2022, 11, 425. https://doi.org/10.3390/pathogens11040425
Onyekwere AM, Rey O, Allienne J-F, Nwanchor MC, Alo M, Uwa C, Boissier J. Population Genetic Structure and Hybridization of Schistosoma haematobium in Nigeria. Pathogens. 2022; 11(4):425. https://doi.org/10.3390/pathogens11040425
Chicago/Turabian StyleOnyekwere, Amos Mathias, Olivier Rey, Jean-François Allienne, Monday Chukwu Nwanchor, Moses Alo, Clementina Uwa, and Jerome Boissier. 2022. "Population Genetic Structure and Hybridization of Schistosoma haematobium in Nigeria" Pathogens 11, no. 4: 425. https://doi.org/10.3390/pathogens11040425
APA StyleOnyekwere, A. M., Rey, O., Allienne, J. -F., Nwanchor, M. C., Alo, M., Uwa, C., & Boissier, J. (2022). Population Genetic Structure and Hybridization of Schistosoma haematobium in Nigeria. Pathogens, 11(4), 425. https://doi.org/10.3390/pathogens11040425