
Phylogenetic Characterization and Genome Sequence Analysis of Burkholderia glumae Strains Isolated in Thailand as the Causal Agent of Rice Bacterial Panicle Blight

 , and
, and
Abstract
:1. Introduction
2. Results
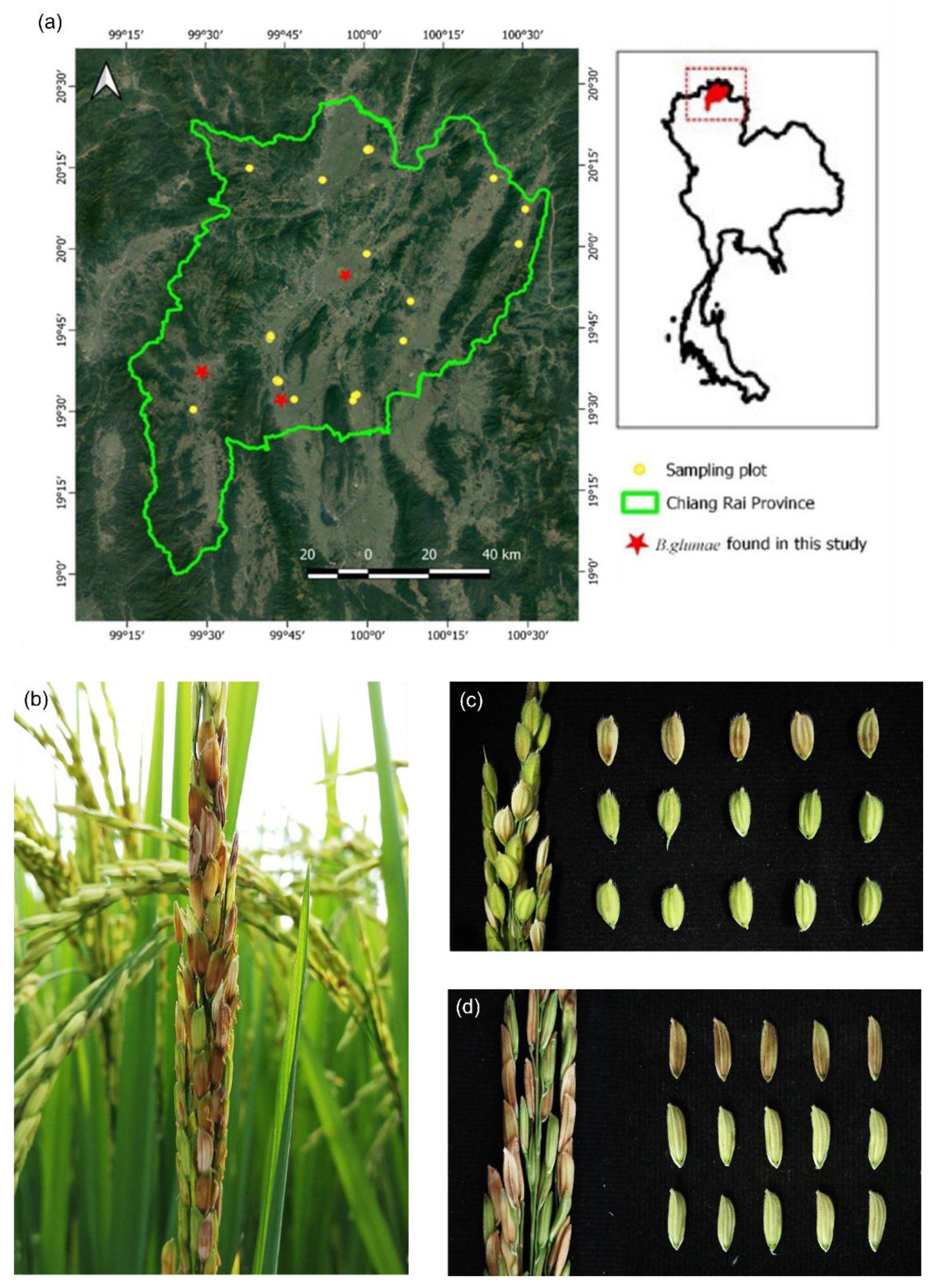
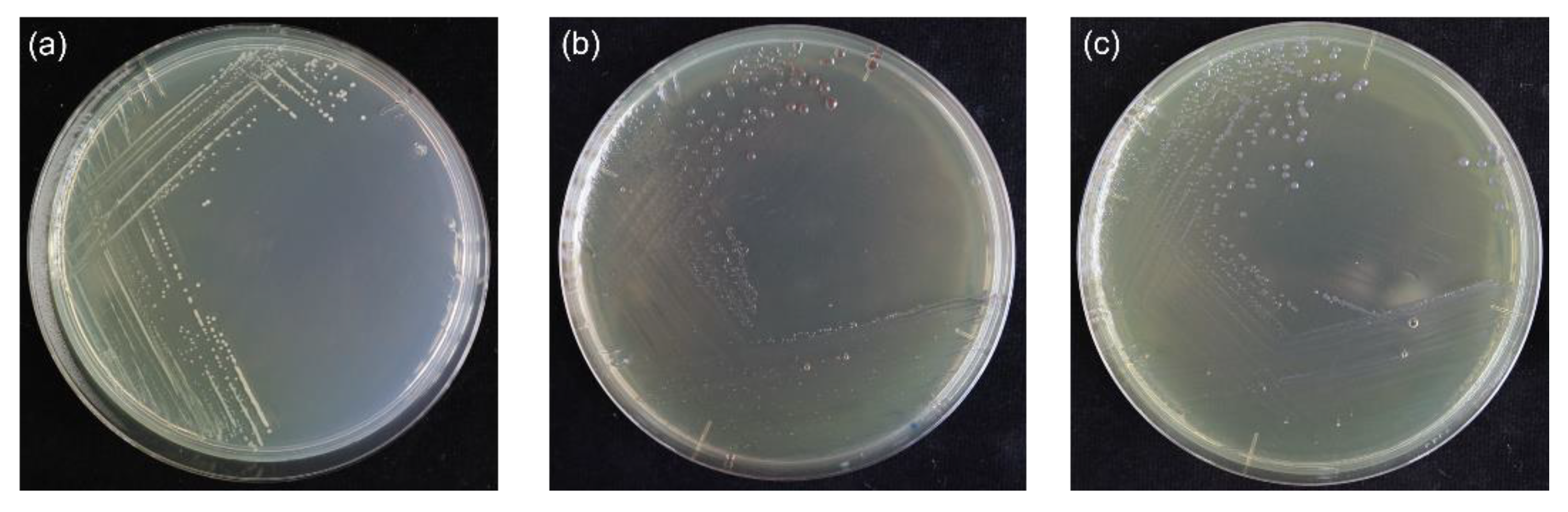
2.1. Isolation and PCR-Based Identification of Burkholderia glumae
2.2. Pathogenic and Biochemical Characterization of Six Selected Burkholderia glumae Strains
2.3. Genome Features of Six Selected Burkholderia glumae Strains
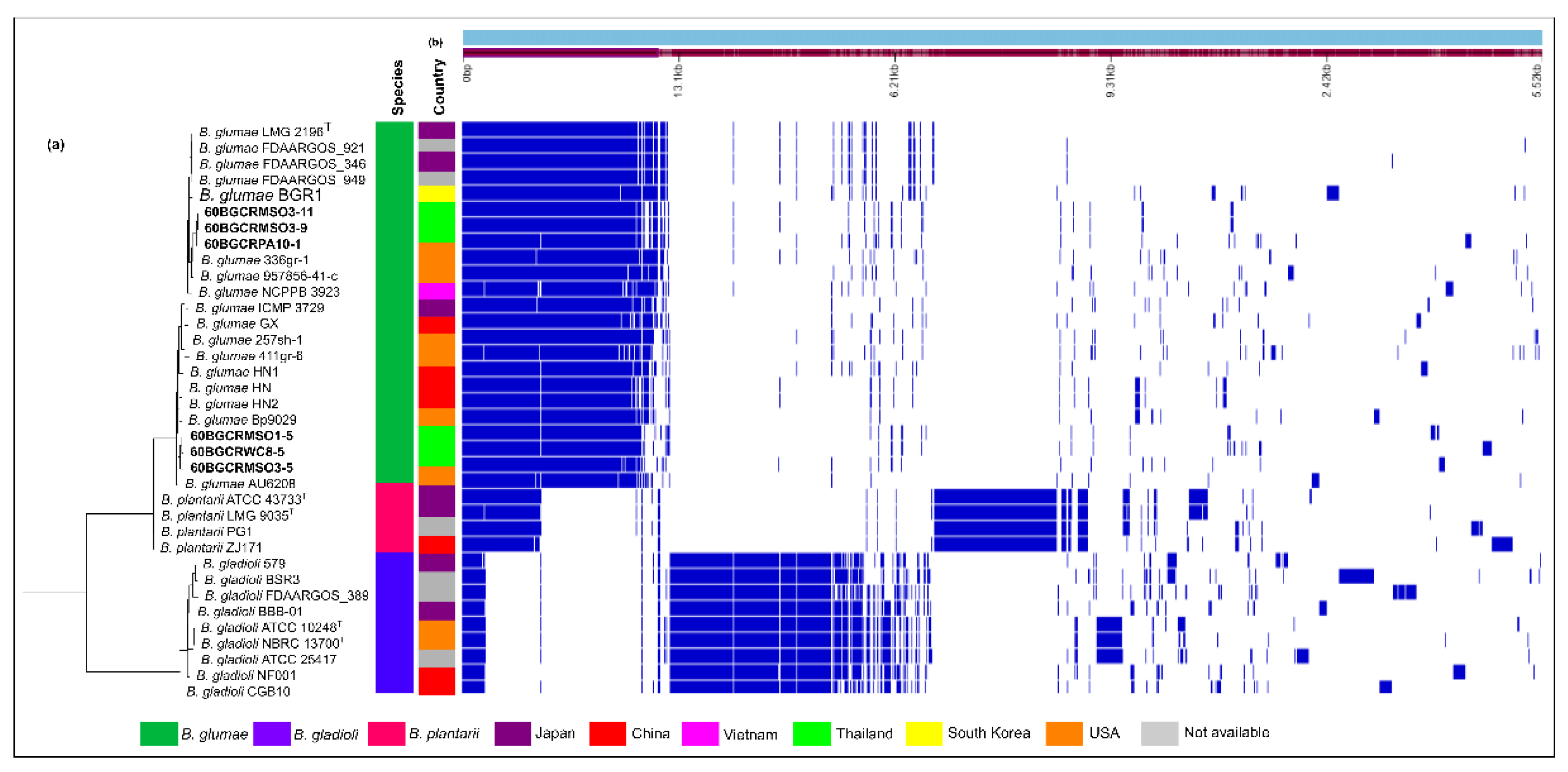
2.4. Burkholderia glumae Strains from Thailand and Worldwide Harbor Specific Prophage Regions
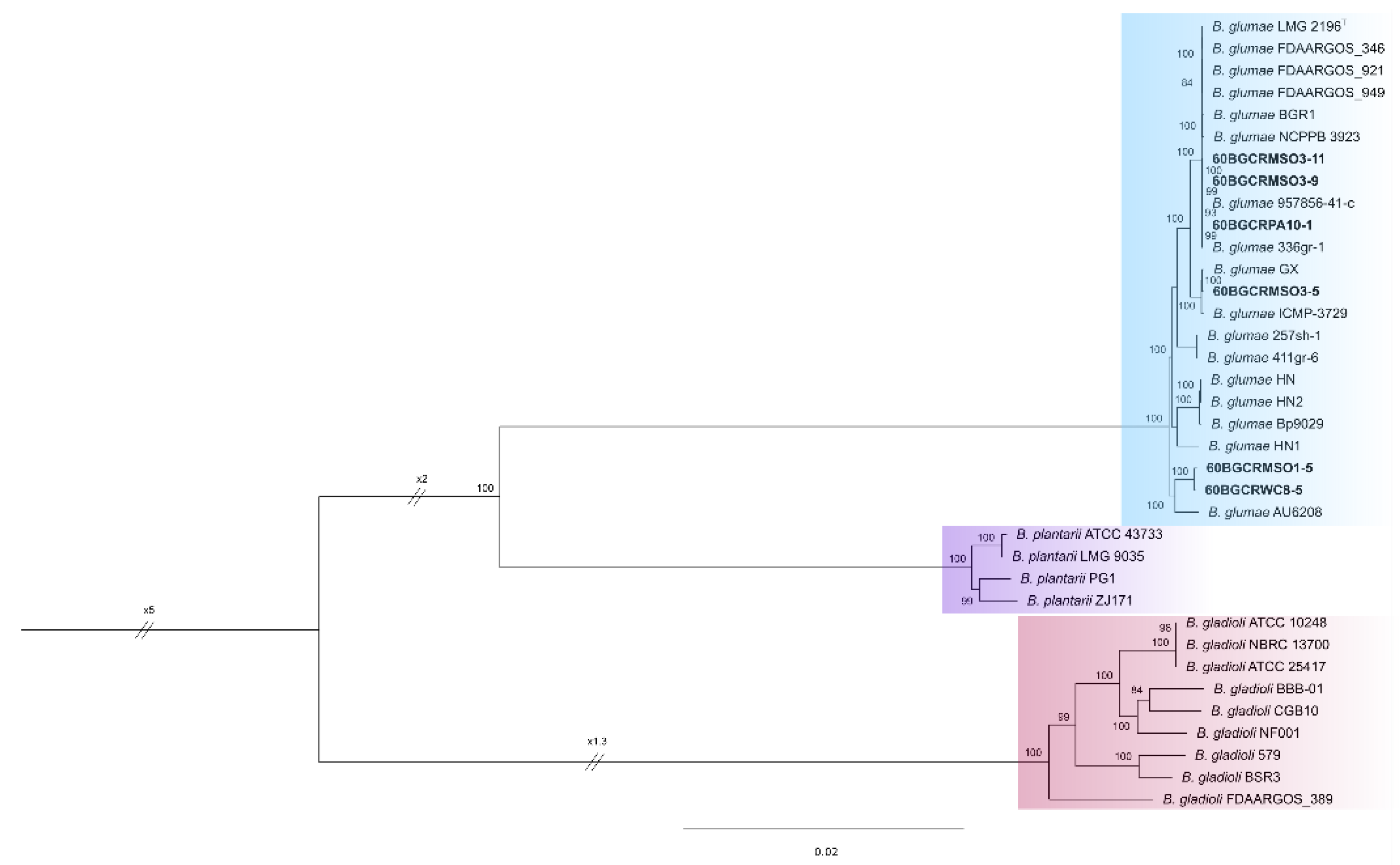
2.5. Phylogenetic and Phylogenomic Analysis of Six Selected Burkholderia glumae Strains

2.6. Taxogenomic analysis of Six Selected Burkholderia glumae Strains
2.7. Core Genome Characterization of Six Selected Burkholderia glumae Strains
3. Discussion
4. Materials and Methods
4.1. Sample Collection, Isolation, and PCR-Based Identification
4.2. Hypersensitive Response and Pathogenicity Test
4.3. Phenotypic and Biochemical Characterization
4.4. Whole-Genome Sequencing, Assembly, and Annotation
4.5. Phylogenetic and Phylogenomic Analysis
4.6. Taxogenomic Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Trung, H.M.; Van, N.V.; Vien, N.V.; Lam, D.T.; Lien, M. Occurrence of rice grain rot disease in Vietnam. Int. Rice Res. Notes 1993, 18, 30. [Google Scholar]
- Cui, Z.-Q.; Zhu, B.; Xie, G.-L.; Lin, B.; Huang, S.-W. Research status and prospect of Burkholderia glumae, the pathogen causing bacterial panicle blight. Rice Sci. 2016, 23, 111–118. [Google Scholar]
- Goto, K.; Ohata, K. New bacterial diseases of rice (brown stripe and grain rot). Jpn. J. Phytopathol. 1956, 21, 46–47. [Google Scholar]
- Sayler, R.J.; Cartwright, R.D.; Yang, Y. Genetic characterization and real-time PCR detection of Burkholderia glumae, a newly emerging bacterial pathogen of rice in the United States. Plant Dis. 2006, 90, 603–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karki, H.; Shrestha, B.; Han, J.; Groth, D.; Barphagha, I.; Rush, M.C.; Melanson, R.A.; Kim, B.S.; Ham, J.H. Diversities in virulence, antifungal activity, pigmentation and dna fingerprint among strains of Burkholderia glumae. PLoS ONE 2012, 7, e45376. [Google Scholar] [CrossRef] [PubMed]
- Ham, J.H.; Melanson, R.A.; Rush, M.C. Burkholderia glumae: Next major pathogen of rice? Mol. Plant Pathol. 2011, 12, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Schaad, N.W.; Jones, J.B.; Chun, W. Laboratory Guide for Identification of Plant Pathogenic Bacteria, 2nd ed.; APS Press: St. Paul, MN, USA, 2001; pp. 1–373. [Google Scholar]
- Tsushima, S.; Wakimoto, S.; Mogi, S. Selective medium for detecting Pseudomonas glumae Kurita et Tabei, the causal bacterium of grain rot of rice. Jpn. J. Phytopathol. 1986, 52, 253–259. [Google Scholar] [CrossRef]
- Takeuchi, T.; Sawada, H.; Suzuki, F.; Matsuda, I. Specific detection of Burkholderia plantarii and B. glumae by PCR using primers selected from the 16S-23S rDNA spacer regions. Jpn. J. Phytopathol. 1997, 63, 455–462. [Google Scholar] [CrossRef] [Green Version]
- Pet-amphai, W.; Watcharachaiyakup, J.; Patarapuwadol, S.; Kositratana, W. Identification of bacterial pathogens causing panicle blight and dirty panicle of rice by multilocus sequence analysis. J. Agric. Sci. 2017, 48, 297–311. [Google Scholar]
- Prakash, O.; Verma, M.; Sharma, P.; Kumar, M.; Kumari, K.; Singh, A.; Kumari, H.; Jit, S.; Gupta, S.K.; Khanna, M.; et al. Polyphasic approach of bacterial classification—An overview of recent advances. Indian J. Microbiol. 2007, 47, 98–108. [Google Scholar] [CrossRef] [Green Version]
- Nandakumar, R.; Shahjahan, A.; Yuan, X.; Dickstein, E.; Groth, D.; Clark, C.A.; Cartwright, R.D.; Rush, M.C. Burkholderia glumae and B. gladioli cause bacterial panicle blight in rice in the southern united states. Plant Dis. 2009, 93, 896–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, D.; Vishunavat, K. Identification of a seed-borne rice bacterium, Burkholderia glumae using cultural, morphological and biochemical methods. J. Appl. Nat. Sci. 2015, 7, 562–566. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.-P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier-Kolthoff, J.P.; Sardà Carbasse, J.; Peinado-Olarte, R.L.; Göker, M. TYGS and LPSN: A database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acid Res. 2022, 50, D801–D807. [Google Scholar] [CrossRef]
- Parks, D.H.; Chuvochina, M.; Chaumeil, P.A.; Rinke, C.; Mussig, A.J.; Hugenholtz, P. A complete domain-to-species taxonomy for bacteria and archaea. Nat. Biotechnol. 2020, 38, 1079–1086. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Y.; Zhou, J.; Zhou, J.; Hu, M.; Zhang, Q.; Kong, N.; Ren, H.; Liang, L.; Yue, J. Genome-based classification of Burkholderia cepacia complex provides new insight into its taxonomic status. Biol. Direct. 2020, 15, 6. [Google Scholar] [CrossRef]
- Bach, E.; Passaglia, L.M.P.; Jiao, J.; Gross, H. Burkholderia in the genomic era: From taxonomy to the discovery of new antimicrobial secondary metabolites. Crit. Rev. Microbiol. 2022, 48, 121–160. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Lee, T.H.; Nahm, B.H.; Choi, Y.D.; Kim, M.; Hwang, I. Complete genome sequence of Burkholderia glumae BGR1. J. Bacteriol. 2009, 191, 3758–3759. [Google Scholar] [CrossRef] [Green Version]
- Francis, F.; Kim, J.; Ramaraj, T.; Farmer, A.; Rush, M.C.; Ham, J.H. Comparative genomic analysis of two Burkholderia glumae strains from different geographic origins reveals a high degree of plasticity in genome structure associated with genomic islands. Mol. Genet. Genom. 2013, 288, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, T.D.; Flett, K.B.; Yelin, I.; Martin, T.R.; McAdam, A.J.; Priebe, G.P.; Kishony, R. Genetic variation of a bacterial pathogen within individuals with cystic fibrosis provides a record of selective pressures. Nat. Genet. 2014, 46, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Chewapreecha, C.; Holden, M.T.; Vehkala, M.; Välimäki, N.; Yang, Z.; Harris, S.R.; Mather, A.E.; Tuanyok, A.; De Smet, B.; Le Hello, S.; et al. Global and regional dissemination and evolution of Burkholderia pseudomallei. Nat. Microbiol. 2017, 2, 16263. [Google Scholar] [CrossRef]
- Hassan, A.A.; Dos Santos, S.C.; Cooper, V.S.; Sá-Correia, I. Comparative evolutionary patterns of Burkholderia cenocepacia and B. multivorans during chronic co-infection of a cystic fibrosis patient lung. Front. Microbiol. 2020, 11, 574626. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Ren, H.; Hu, M.; Zhou, J.; Li, B.; Kong, N.; Zhang, Q.; Jin, Y.; Liang, L.; Yue, J. Characterization of Burkholderia cepacia complex core genome and the underlying recombination and positive selection. Front. Genet. 2020, 11, 506. [Google Scholar] [CrossRef]
- Kim, J.; Kim, J.G.; Kang, Y.; Jang, J.Y.; Jog, G.J.; Lim, J.Y.; Kim, S.; Suga, H.; Nagamatsu, T.; Hwang, I. Quorum sensing and the LysR-type transcriptional activator ToxR regulate toxoflavin biosynthesis and transport in Burkholderia glumae. Mol. Microbiol. 2004, 54, 921–934. [Google Scholar] [CrossRef]
- Suzuki, F.; Sawada, H.; Azegami, K.; Tsuchiya, K. Molecular characterization of the tox operon involved in toxoflavin biosynthesis of Burkholderia glumae. J. Gen. Plant Pathol. 2004, 70, 97–107. [Google Scholar] [CrossRef]
- Chen, R.; Barphagha, I.K.; Karki, H.S.; Ham, J.H. Dissection of quorum-sensing genes in Burkholderia glumae reveals non-canonical regulation and the new regulatory gene tofM for toxoflavin production. PLoS ONE 2012, 7, e52150. [Google Scholar] [CrossRef]
- Chen, R.; Barphagha, I.K.; Ham, J.H. Identification of potential genetic components involved in the deviant quorum-sensing signaling pathways of Burkholderia glumae through a functional genomics approach. Front. Cell. Infect. Microbiol. 2015, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Furuya, N.; Iiyama, K.; Ueda, Y.; Matsuyama, N. Reaction of tobacco and rice leaf tissue infiltrated with Burkholderia glumae or B. gladioli. J. Fac. Agric. Kyushu Univ. 1997, 42, 43–51. [Google Scholar] [CrossRef]
- Urakami, T.; Ito-yoshida, C.; Araki, H.; Kijima, T.; Suzuki, K.; Komagata, K. Transfer of Pseudomonas plantarii and Pseudomonas glumae to Burkholderia as Burkholderia spp.; description of Burkholderia vandii sp. Int. J. Syst. Bacteriol. 1994, 44, 235–245. [Google Scholar] [CrossRef]
- Hussain, A.; Shahbaz, M.; Tariq, M.; Ibrahim, M.; Hong, X.; Naeem, F.; Khalid, Z.; Raza, H.M.Z.; Bo, Z.; Bin, L. Genome re-sequence and analysis of Burkholderia glumae strain AU6208 and evidence of toxoflavin: A potential bacterial toxin. Comput. Biol. Chem. 2020, 86, 107245. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.H.; Lelis, T.; Ontoy, J.; Bruno, J.; Ham, J.H.; Seo, Y.S. Complete genome sequence data of four Burkholderia glumae strains isolated from rice fields in the United States. Mol. Plant-Microbe Interact. 2021, 34, 1324–1327. [Google Scholar] [CrossRef] [PubMed]
- Varani, A.M.; Monteiro-Vitorello, C.B.; Nakaya, H.I.; Van Sluys, M.A. The role of prophage in plant-pathogenic bacteria. Annu. Rev. Phytopathol. 2013, 51, 429–451. [Google Scholar] [CrossRef]
- Dorgai, L.; Oberto, J.; Weisberg, R.A. Xis and Fis proteins prevent site-specific DNA inversion in lysogens of phage HK022. J. Bacteriol. 1993, 175, 693–700. [Google Scholar] [CrossRef] [Green Version]
- Brüssow, H.; Canchaya, C.; Hardt, W.D. Phages and the evolution of bacterial pathogens: From genomic rearrangements to lysogenic conversion. Microbiol. Mol. Biol. Rev. 2004, 68, 560–602. [Google Scholar] [CrossRef] [Green Version]
- Adachi, N.; Tsukamoto, S.; Inoue, Y.; Azegami, K. Control of bacterial seedling rot and seedling blight of rice by bacteriophage. Plant Dis. 2012, 96, 1033–1036. [Google Scholar] [CrossRef] [Green Version]
- Jungkhun, N.; Farias, A.R.; Barphagha, I.; Patarapuwadol, S.; Ham, J.H. Isolation and characterization of bacteriophages infecting Burkholderia glumae, the major causal agent of bacterial panicle blight in rice. Plant Dis. 2021, 105, 2551–2559. [Google Scholar] [CrossRef]
- Spilker, T.; Baldwin, A.; Bumford, A.; Dowson, C.G.; Mahenthiralingam, E.; LiPuma, J.J. Expanded multilocus sequence typing for Burkholderia species. J. Clin. Microbiol. 2009, 47, 2607–2610. [Google Scholar] [CrossRef] [Green Version]
- Choi, O.; Kim, S.; Kang, B.; Lee, Y.; Bae, J.; Kim, J. Genetic diversity and distribution of Korean isolates of Burkholderia glumae. Plant Dis. 2021, 105, 1398–1407. [Google Scholar] [CrossRef]
- Weinberg, J.B.; Alexander, B.D.; Majure, J.M.; Williams, L.W.; Kim, J.Y.; Vandamme, P.; LiPuma, J.J. Burkholderia glumae infection in an infant with chronic granulomatous disease. J. Clin. Microbiol. 2007, 45, 662–665. [Google Scholar] [CrossRef] [Green Version]
- Cui, Z.; Wang, S.; Kakar, K.U.; Xie, G.; Li, B.; Chen, G.; Zhu, B. Genome sequence and adaptation analysis of the human and rice-pathogenic strain Burkholderia glumae AU6208. Pathogens 2021, 10, 87. [Google Scholar] [CrossRef] [PubMed]
- Glandorf, B.; Brand, I.; Bakker, P.; Schippers, B. Stability of rifampicin as a marker for root colonization studies of Pseudomonas putida in the field. Plant Soil 1992, 147, 135–142. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 10 January 2022).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [Green Version]
- Bosi, E.; Donati, B.; Galardini, M.; Brunetti, S.; Sagot, M.F.; Lió, P.; Crescenzi, P.; Fani, R.; Fondi, M. MeDuSa: A multi-draft based scaffolder. Bioinformatics 2015, 31, 2443–2451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Zdobnov, E.M. BUSCO: Assessing genomic data quality and beyond. Curr. Protoc. 2021, 1, e323. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [Green Version]
- The UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, A.; Mahenthiralingam, E.; Drevinek, P.; Pope, C.; Waine, D.J.; Henry, D.A.; Speert, D.P.; Carter, P.; Vandamme, P.; LiPuma, J.J.; et al. Elucidating global epidemiology of Burkholderia multivorans in cases of cystic fibrosis by multilocus sequence typing. J. Clin. Microbiol. 2008, 46, 290–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. Mrbayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A. FigTree v1.4.4. 2010. Available online: https://github.com/rambaut/figtree/releases/tag/v1.4.4 (accessed on 7 February 2022).
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan-genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minh, B.Q.; Nguyen, M.A.T.; von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pritchard, L.; Glover, R.H.; Humphris, S.; Elphinstone, J.G.; Toth, I.K. Genomics and taxonomy in diagnostics for food security: Soft-rotting enterobacterial plant pathogens. Anal. Methods. 2016, 8, 12–24. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| District | Surveyed Fields | Fields with Symptomatic Samples | Panicle Samples | Bacteria Isolates | Positive PCR Result |
|---|---|---|---|---|---|
| Mae Suai | 30 | 12 | 123 | 12 | 7 |
| Wiang Chai | 15 | 4 | 40 | 13 | 11 |
| Pa Daet | 25 | 3 | 30 | 5 | 4 |
| Mae Chan | 18 | 2 | 20 | 9 | 9 |
| Chiang Khong | 16 | 16 | 160 | 4 | 2 |
| Chiang Saen | 17 | 4 | 31 | 1 | 1 |
| Wiang Kaen | 3 | 3 | 23 | 7 | 5 |
| Phan | 44 | 16 | 142 | 2 | 2 |
| Thoeng | 17 | 7 | 59 | 1 | 1 |
| Mae Lao | 6 | 6 | 38 | 3 | 2 |
| Phaya Mengrai | 14 | 2 | 20 | 1 | 0 |
| Wiang Chiang Rung | 14 | 5 | 37 | 1 | 0 |
| Mae Fa Luang | 4 | 4 | 23 | 2 | 0 |
| Muang Chiang Rai | 16 | 6 | 56 | 0 | 0 |
| Mae Sai | 33 | 3 | 22 | 0 | 0 |
| Wiang Pa Pao | 7 | 5 | 30 | 0 | 0 |
| Kun Tan | 7 | 4 | 41 | 0 | 0 |
| Doi Luang | 3 | 3 | 30 | 0 | 0 |
| Total | 289 | 105 | 925 | 61 | 44 |
| Feature | 60BGCRMSO31-5 | 60BGCRMSO3-5 | 60BGCRMSO3-9 | 60BGCRMSO3-11 | 60BGCRPA10-1 | 60BGCRWC8-5 | Average |
|---|---|---|---|---|---|---|---|
| No. contigs | 191 | 162 | 173 | 174 | 183 | 186 | 178 |
| Size (Mb) | 6.45 | 6.30 | 6.56 | 6.56 | 6.59 | 6.59 | 6.51 |
| GC content (%) | 68.54 | 68.76 | 68.39 | 68.39 | 68.39 | 68.36 | 68.42 |
| N50 (bp) | 113,987 | 110,811 | 161,337 | 161,338 | 109,662 | 132,818 | 131,659 |
| No. scaffolds | 27 | 12 | 10 | 11 | 13 | 14 | 14.5 |
| Size (Mb) | 6.46 | 6.31 | 6.57 | 6.57 | 6.59 | 6.59 | 6.52 |
| Completeness (%) | 99.90 | 99.70 | 99.90 | 99.90 | 99.90 | 100.00 | 99.89 |
| No. coding sequences | 5594 | 5433 | 5587 | 5580 | 5688 | 5752 | 5606 |
| No. tRNA | 67 | 62 | 69 | 69 | 69 | 64 | 67 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jungkhun, N.; Gomes de Farias, A.R.; Watcharachaiyakup, J.; Kositcharoenkul, N.; Ham, J.H.; Patarapuwadol, S. Phylogenetic Characterization and Genome Sequence Analysis of Burkholderia glumae Strains Isolated in Thailand as the Causal Agent of Rice Bacterial Panicle Blight. Pathogens 2022, 11, 676. https://doi.org/10.3390/pathogens11060676
Jungkhun N, Gomes de Farias AR, Watcharachaiyakup J, Kositcharoenkul N, Ham JH, Patarapuwadol S. Phylogenetic Characterization and Genome Sequence Analysis of Burkholderia glumae Strains Isolated in Thailand as the Causal Agent of Rice Bacterial Panicle Blight. Pathogens. 2022; 11(6):676. https://doi.org/10.3390/pathogens11060676
Chicago/Turabian StyleJungkhun, Nootjarin, Antonio Roberto Gomes de Farias, Jutatape Watcharachaiyakup, Nuttima Kositcharoenkul, Jong Hyun Ham, and Sujin Patarapuwadol. 2022. "Phylogenetic Characterization and Genome Sequence Analysis of Burkholderia glumae Strains Isolated in Thailand as the Causal Agent of Rice Bacterial Panicle Blight" Pathogens 11, no. 6: 676. https://doi.org/10.3390/pathogens11060676
APA StyleJungkhun, N., Gomes de Farias, A. R., Watcharachaiyakup, J., Kositcharoenkul, N., Ham, J. H., & Patarapuwadol, S. (2022). Phylogenetic Characterization and Genome Sequence Analysis of Burkholderia glumae Strains Isolated in Thailand as the Causal Agent of Rice Bacterial Panicle Blight. Pathogens, 11(6), 676. https://doi.org/10.3390/pathogens11060676