

Low Genetic Diversity of the Only Clade of the Tick Rhipicephalus microplus in the Neotropics

 , , ,
, , ,  , ,
, ,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Collection of Ticks and Preparation of Samples
2.2. DNA Extraction, PCR, and Sequencing
2.3. Alignment and Phylogenetic Analysis
2.4. Haplotype Networks and Genetic Diversity of R. microplus
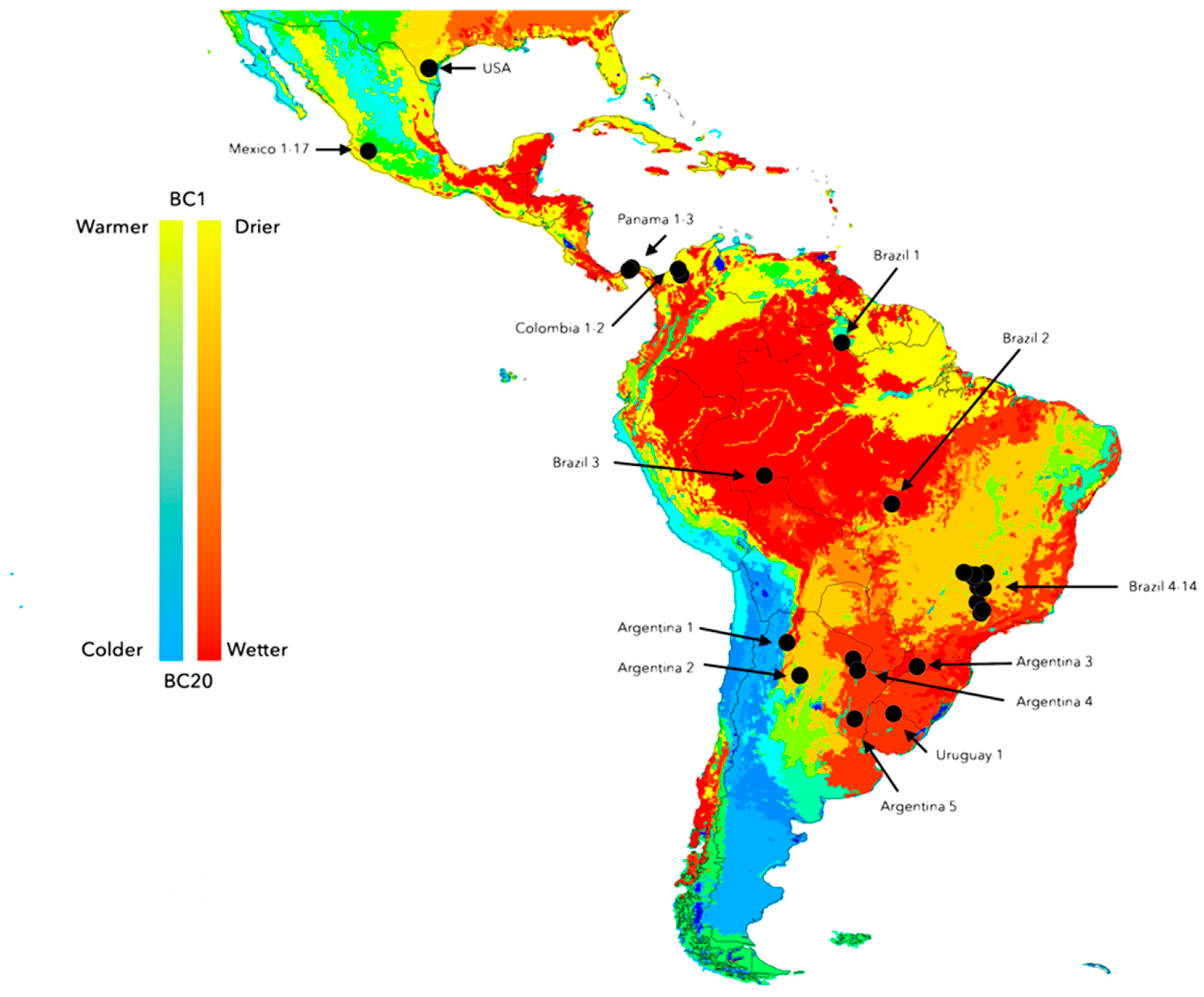
2.5. Climate and Genetic Variability among Populations of Neotropical R. microplus
2.6. Geographic Distance among Populations as Possible Driver of Genetic Variability of R. microplus
3. Results
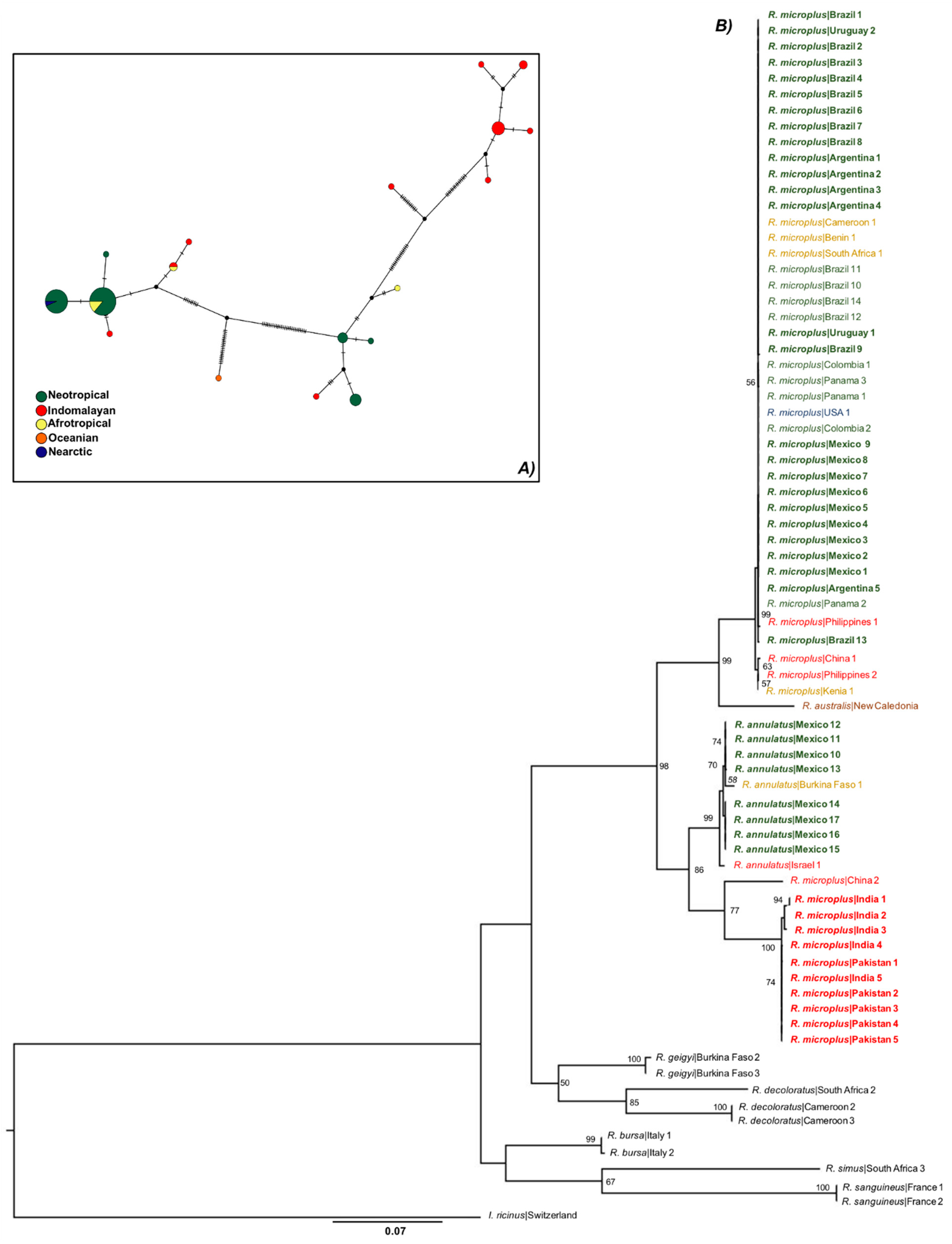
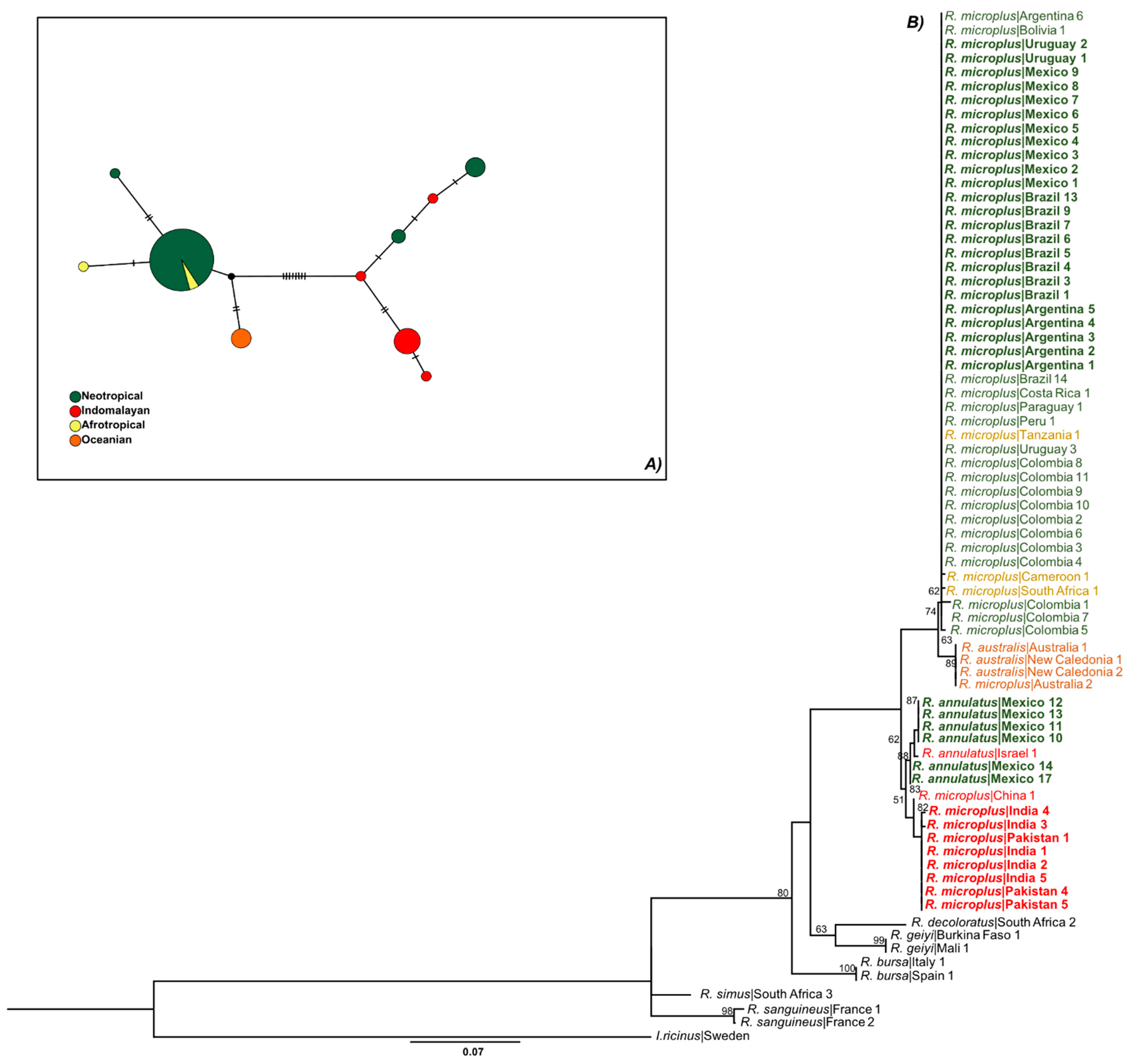
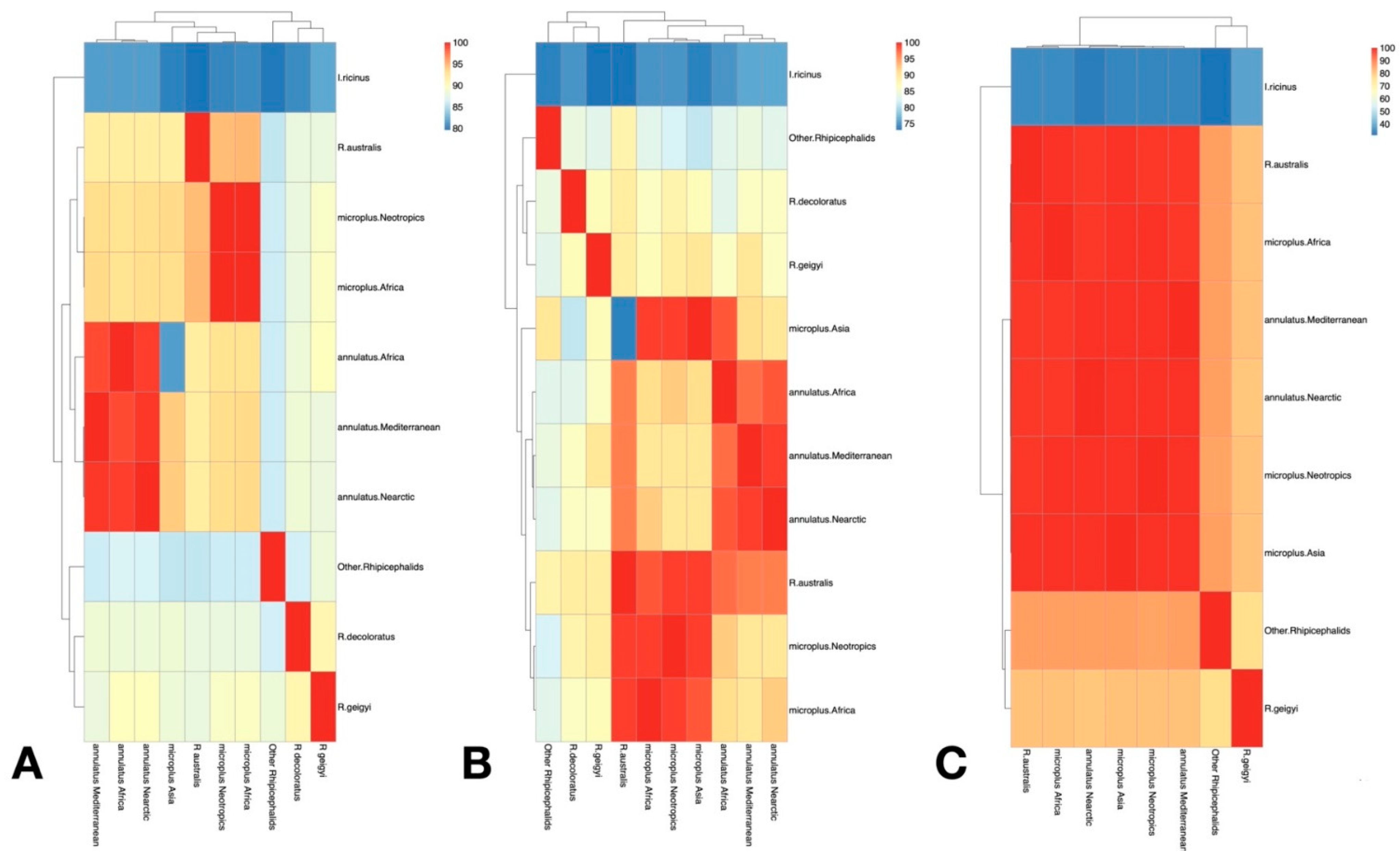
3.1. Only R. microplus s.s. (Clade A) Exists in the Nearctic–Neotropics
3.2. Climate Traits Are Not Driving the Mutation Rates of Three Genes of R. microplus
3.3. There Is a Random Pattern of Genetic Variability of R. microplus According to Distance in Neotropics
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Roy, B.C.; Estrada-Peña, A.; Krücken, J.; Rehman, A.; Nijhof, A.M. Morphological and phylogenetic analyses of Rhipicephalus microplus ticks from Bangladesh, Pakistan and Myanmar. Ticks Tick-Borne Dis. 2018, 9, 1069–1079. [Google Scholar] [CrossRef] [PubMed]
- Madder, M.; Thys, E.; Achi, L.; Touré, A.; De Deken, R. Rhipicephalus (Boophilus) microplus: A most successful invasive tick species in West-Africa. Exp. Appl. Acarol. 2011, 53, 139–145. [Google Scholar] [CrossRef]
- Sutherst, R.W.; Maywald, G.F.; Kerr, J.D.; Stegeman, D.A. The effect of cattle tick (Boophilus microplus) on the growth of Bos indicus × B. taurus steers. Aust. J. Agric. Res. 1983, 34, 317–327. [Google Scholar] [CrossRef]
- Barré, N.; Garris, G.; Camus, E. Propagation of the tick Amblyomma variegatum in the Caribbean. Rev. Sci. Tech. 1995, 14, 841. [Google Scholar] [CrossRef] [PubMed]
- Labruna, M.B.; Naranjo, V.; Mangold, A.J.; Thompson, C.; Estrada-Peña, A.; Guglielmone, A.A.; de La Fuente, J. Allopatric speciation in ticks: Genetic and reproductive divergence between geographic strains of Rhipicephalus (Boophilus) microplus. BMC Evol. Biol. 2009, 9, 46. [Google Scholar] [CrossRef]
- Burger, T.D.; Shao, R.; Barker, S.C. Phylogenetic analysis of mitochondrial genome sequences indicates that the cattle tick, Rhipicephalus (Boophilus) microplus, contains a cryptic species. Mol. Phylogenetics Evol. 2014, 76, 241. [Google Scholar] [CrossRef]
- Low, V.L.; Tay, S.T.; Kho, K.L.; Koh, F.X.; Tan, T.K.; Lim, Y.A.L.; Ong, L.B.; Panchadcharam, C.; Norma-Rashid, Y.; Sofian-Azirum, M. Molecular characterisation of the tick Rhipicephalus microplus in Malaysia: New insights into the cryptic diversity and distinct genetic assemblages throughout the world. Parasites Vectors 2015, 8, 341. [Google Scholar] [CrossRef]
- Baron, S.; van der Merwe, N.A.; Maritz-Olivier, C. The genetic relationship between R. microplus and R. decoloratus ticks in South Africa and their population structure. Mol. Phylogenetics Evol. 2018, 129, 60–69. [Google Scholar] [CrossRef]
- Makenov, M.T.; Toure, A.H.; Korneev, M.G.; Sacko, N.; Porshakov, A.M.; Yakovlev, S.A.; Radyuk, E.V.; Zakharov, K.S.; Shipovalov, A.V.; Boumbaly, S.; et al. Rhipicephalus microplus and its vector-borne haemoparasites in Guinea: Further species expansion in West Africa. Parasitol. Res. 2021, 120, 1563–1570. [Google Scholar] [CrossRef]
- Amrutha, B.M.; Kumar, K.G.A.; Kurbet, P.S.; Varghese, A.; Deepa, C.K.; Pradeep, R.K.; Nimisha, M.; Asaf, M.; Juliet, S.; Ravindran, R.; et al. Morphological and molecular characterization of Rhipicephalus microplus and Rhipicephalus annulatus from selected states of southern India. Ticks Tick-Borne Dis. 2023, 14, 102086. [Google Scholar] [CrossRef]
- Paulauskas, A.; Galdikas, M.; Galdikaitė-Brazienė, E.; Stanko, M.; Kahl, O.; Karbowiak, G.; Radzijevskaja, J. Microsatellite-based genetic diversity of Dermacentor reticulatus in Europe. Inf. Gen. Evol. 2018, 66, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Lado, P.; Luan, B.; Allerdice, M.E.; Paddock, C.D.; Karpathy, S.E.; Klompen, H. Integrating population genetic structure, microbiome, and pathogens presence data in Dermacentor variabilis. Peer J. 2020, 8, e9367. [Google Scholar] [CrossRef] [PubMed]
- Beati, L.; Nava, S.; Burkman, E.J.; Barros-Battesti, D.M.; Labruna, M.B.; Guglielmone, A.A.; Faccini, J.L. Amblyomma cajennense (Fabricius, 1787) (Acari: Ixodidae), the Cayenne tick: Phylogeography and evidence for allopatric speciation. BMC Evol. Biol. 2013, 13, 267. [Google Scholar] [CrossRef] [PubMed]
- Gulia-Nuss, M.; Nuss, A.B.; Meyer, J.M.; Sonenshine, D.E.; Roe, R.M.; Waterhouse, R.M.; Hill, C.A. Genomic insights into the Ixodes scapularis tick vector of Lyme disease. Nat. Comm. 2016, 9, 10507–10514. [Google Scholar] [CrossRef] [PubMed]
- Kovalev, S.Y.; Golovljova, I.V.; Mukhacheva, T.A. Natural hybridization between Ixodes ricinus and Ixodes persulcatus ticks evidenced by molecular genetics methods. Ticks Tick-Borne Dis. 2016, 7, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Leo, S.S.T.; Davis, C.S.; Sperling, F.A.H. Characterization of 14 microsatellite loci developed for Dermacentor albipictus and cross-species amplification in D. andersoni and D. variabilis (Acari: Ixodidae). Conserv. Gen. Res. 2014, 4, 379–382. [Google Scholar] [CrossRef]
- Martins, T.F.; Barbieri, A.R.M.; Costa, F.B.; Terassini, F.A.; Carmargo, C.L.; Peterga, R.M.; Labruna, M.B. Geographical distribution of Amblyomma cajennense (sensu lato) ticks (Parasitiformes: Ixodidae) in Brazil, with description of the nymph of A. cajennense (sensu stricto). Parasites Vectors 2016, 9, 186–189. [Google Scholar] [CrossRef]
- Hutchinson, G. Cold Spring harbor symposium on quantitative biology. Concluding remarks. Symp. Quant. Biol. 1957, 22, 415–427. [Google Scholar] [CrossRef]
- Wiens, J.J.; Graham, C.H. Niche conservatism: Integrating evolution, ecology, and conservation biology. Ann. Rev. Ecol. Evol. Syst. 2005, 36, 519–539. [Google Scholar] [CrossRef]
- Kozak, K.H.; Wiens, J.J. Accelerated rates of climate-niche evolution underlie rapid species diversification. Ecol. Lett. 2010, 13, 1378–1389. [Google Scholar] [CrossRef]
- Nee, S.; Mooers, A.O.; Harvey, P.H. Tempo and mode of evolution revealed from molecular phylogenies. Proc. Natl. Acad. Sci. USA 1992, 89, 8322–8326. [Google Scholar] [CrossRef] [PubMed]
- Donoghue, M.J. A phylogenetic perspective on the distribution of plant diversity. Proc. Natl. Acad. Sci. USA 2008, 105, 11549–11551. [Google Scholar] [CrossRef] [PubMed]
- Olalla-Tárraga, M.A.; McInnes, L.; Bini, L.M.; Diniz-Filho, J.A.; Fritz, S.A.; Hawkins, B.A.; Hortal, J.; Orme, C.D.L.; Rahbek, C.; Rodríguez, M.A.; et al. Climatic niche conservatism and the evolutionary dynamics in species range boundaries: Global congruence across mammals and amphibians. J. Biogeogr. 2011, 38, 2237–2247. [Google Scholar] [CrossRef]
- Losos, J.B. Phylogenetic niche conservatism, phylogenetic signal and the relationship between phylogenetic relatedness and ecological similarity among species. Ecol. Lett. 2008, 11, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Kellermann, V.; Loeschcke, V.; Hoffmann, A.A.; Kristensen, T.N.; Fløjgaard, C.; David, J.R.; Overgaard, J. Phylogenetic constraints in key functional traits behind species’ climate niches: Patterns of desiccation and cold resistance across 95 Drosophila species. Int. J. Org. Evol. 2012, 66, 3377–3389. [Google Scholar] [CrossRef]
- Nava, S.; Beati, L.; Labruna, M.B.; Cáceres, A.G.; Mangold, A.J.; Guglielmone, A.A. Reassessment of the taxonomic status of Amblyomma cajennense (F.) with the description of three new species, Amblyomma tonelliae n. sp.; Amblyomma interandinum n. sp. and Amblyomma patinoi n. sp.; and reinstatement of Amblyomma mixtum, and Amblyomma sculptum (Ixodida: Ixodidae). Ticks Tick-Borne Dis. 2014, 5, 252–276. [Google Scholar]
- Cuervo, P.F.; Flores, F.S.; Venzal, J.M.; Nava, S. Niche divergence among closely related taxa provides insight on evolutionary patterns of ticks. J. Biogeogr. 2021, 48, 2865. [Google Scholar] [CrossRef]
- Murrell, A.; Campbell, N.J.H.; Barker, S.C. Phylogenetic analyses of the rhipicephaline ticks indicate that the genus Rhipicephalus is paraphyletic. Mol. Phylogenetics Evol. 2000, 16, 1–7. [Google Scholar] [CrossRef]
- Nava, S.; Beati, L.; Venzal, J.M.; Labruna, M.B.; Szabo, M.P.; Petney, T.; Saracho-Bottero, M.N.; Tarragona, E.L.; Dantas-Torres, F.; Silva, M.M.; et al. Rhipicephalus sanguineus (Latreille, 1806): Neotype designation, morphological re-description of all parasitic stages and molecular characterization. Ticks Tick-Borne Dis. 2018, 9, 1573–1585. [Google Scholar] [CrossRef]
- Silatsa, B.A.; Kuiate, J.R.; Njiokou, F.; Simo, G.; Feussom, J.M.K.; Tunrayo, A.; Pelle, R. A countrywide molecular survey leads to a seminal identification of the invasive cattle tick Rhipicephalus (Boophilus) microplus in Cameroon, a decade after it was reported in Cote d’Ivoire. Ticks Tick-Borne Dis. 2019, 10, 585–593. [Google Scholar] [CrossRef]
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 1994, 3, 294–299. [Google Scholar] [PubMed]
- Black, W.C.; Piesman, J. Phylogeny of hard- and soft-tick taxa (Acari:Ixodida) based on mitochondrial 16S rDNA sequences. Proc. Natl. Acad. Sci. USA 1994, 91, 10034–10038. [Google Scholar] [CrossRef]
- Barker, S.C. Distinguishing species and populations of rhipicephaline ticks with its ribosomal RNA. J. Parasitol. 1998, 84, 887–892. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772–779. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.P.; Ronquist, F. MrBayes: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef]
- Sugiura, N. Further analysts of the data by Akaike’s information criterion and the finite corrections. Commun. Stat.-Theory Methods 1978, 7, 13–26. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Rambaut, A. FigTree v1. 4.2. A Graphical Viewer of Phylogenetic Trees. Institute of Evolutionary Biology University of Edinburgh. 2014. Available online: https://evomics.org/resources/software/molecular-evolution-software/figtree/ (accessed on 16 August 2021).
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP v6. DNA Sequence polymorphisms analysis of large datasets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Leigh, J.W.; Bryant, D. POPART. Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Caliński, T.; Harabasz, J. A dendrite method for cluster analysis. Commun. Stat.-Theory Methods 1974, 3, 1–27. [Google Scholar] [CrossRef]
- Warren, D.; Dinnage, R. ENMTools: Analysis of Niche Evolution Using Niche and Distribution Models. R Package Version 1.0.2. 2020. Available online: https://CRAN.R-project.org/package=ENMTools (accessed on 3 September 2023).
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020; Available online: https://www.R-project.org/ (accessed on 3 September 2023).
- Blomberg, S.P.; Garland, T., Jr.; Ives, A.R. Testing for phylogenetic signal in comparative data: Behavioral traits are more labile. Evolution 2003, 57, 717–745. [Google Scholar] [PubMed]
- Pagel, M. Inferring the historical patterns of biological evolution. Nature 1999, 401, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Revell, L. Phytools: An R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 2012, 3, 217–223. [Google Scholar] [CrossRef]
- Padgham, M.; Sumner, M.D. Geodist: Fast, Dependency-Free Geodesic Distance Calculations. R Package Version 0.0.6. 2020. Available online: https://CRAN.R-project.org/package=geodist (accessed on 3 September 2023).
- Hebert, P.D.N.; Cywinska, A.; Ball, S.L.; de Waard, J.R. Biological identifications through DNA barcodes. Proc. R. Soc. Lond. Ser. B Biol. Sci. 2003, 270, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Hebert, P.D.N.; Ratnasingham, S.; de Waard, J.R. Barcoding animal life: Cytochrome c oxidase subunit 1 divergences among closely related species. Proc. R. Soc. Lond. Ser. B Biol. Sci. 2003, 270, S96–S99. [Google Scholar] [CrossRef]
- Coimbra-Dores, M.J.; Maia-Silva, M.; Marques, W.; Oliveira, A.C.; Rosa, F.; Dias, D. Phylogenetic insights on Mediterranean and Afrotropical Rhipicephalus species (Acari: Ixodida) based on mitochondrial DNA. Exp. Appl. Acarol. 2018, 75, 107–128. [Google Scholar] [CrossRef]
- Mohamed, W.M.A.; Moustafa, M.A.M.; Kelava, S.; Barker, D.; Matsuno, K.; Nonaka, N.; Nakao, R. Reconstruction of mitochondrial genomes from raw sequencing data provides insights on the phylogeny of Ixodes ticks but suggests the caution for species misidentification. Ticks Tick-Borne Dis. 2021, 13, 101832. [Google Scholar] [CrossRef]
- Estrada-Peña, A.; Venzal, J.M.; Nava, S.; Mangold, A.J.; Guglielmone, A.A.; Labruna, M.B.; de la Fuente, J. Reinstatement of Rhipicephalus (Boophilus) australis (Acari: Ixodidae) with redescription of the adult and larval stages. J. Med. Entomol. 2012, 49, 794–802. [Google Scholar] [CrossRef]
- Kanduma, E.G.; Emery, D.; Githaka, N.W.; Nguu, E.K.; Bishop, R.P.; Šlapeta, J. Molecular evidence confirms occurrence of Rhipicephalus microplus Clade A in Kenya and sub-Saharan Africa. Parasites Vectors 2020, 13, 432. [Google Scholar] [CrossRef]
- Berry, C.M. Resolution of the Taxonomic Status of Rhipicephalus (Boophilus) microplus. Ph.D. Dissertation, University of Glasgow, Glasgow, UK, 2017. [Google Scholar]
- Thomson, G.R.; Tambi, E.N.; Hargreaves, S.K.; Leyland, T.J.; Catley, A.P.; Van‘T Klooster, G.G.M.; Penrith, M.L. International trade in livestock and livestock products: The need for a commodity-based approach. Vet. Rec. 2004, 155, 429–433. [Google Scholar] [PubMed]
- Beerli, P.; Palczewski, M. Unified framework to evaluate panmixia and migration direction among multiple sampling locations. Genetics 2010, 185, 313. [Google Scholar] [CrossRef] [PubMed]
- Busch, J.D.; Stone, N.E.; Nottingham, R.; Araya-Anchetta, A.; Lewis, J.; Hochhalter, C.; Wagner, D.M. Widespread movement of invasive cattle fever ticks (Rhipicephalus microplus) in southern Texas leads to shared local infestations on cattle and deer. Parasites Vectors 2014, 7, 188. [Google Scholar] [CrossRef] [PubMed]
- Estrada-Peña, A.; Bouattour, A.; Camicas, J.-L.; Walker, A. Ticks of Domestic Animals in the Mediterranean Region. A Guide to Identification of Species; University of Zaragoza: Zaragoza, Spain, 2004; ISBN 8.4862141-84. [Google Scholar]
- Kroepelin, S. Revisiting the age of the Sahara Desert. Science 2006, 312, 1138–1139. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target (Gene) | Primers | Sequence (5′→3′) | PCR Program | Reference Primers |
|---|---|---|---|---|
| COI | LCO1490 | GGTCAACAAATCATAAAGATATTGG | Initial denaturation 94 °C, 1 min Pre-amplification: 5 cycles Denature 94 °C, 1 min Anneal 45 °C, 1.5 min Extend 72 °C, 1.5 min Amplification: 35 cycles Denature 94 °C, 1 min Anneal 57 °C, 1.5 min Extend 72 °C, 1 min Final elongation 72 °C, 1 min | [31] |
| HC02198 | TAAACTTCAGGGTGACCAAAA AATCA | |||
| 16S | 16 + 1 | CTGCTCAATGATTTTTTAAATTGCTGTGG | Initial denaturation: 94 °C, 4 min Amplification: 30 cycles Denature 94 °C, 1 min Anneal 54 °C, 30 s Extend 72 °C, 30 s Final elongation: 72 °C, 10 min | [32] |
| 16 – 1 | CCGGTCTGAACTCAGATCAAGT | |||
| ITS2 | 3SA | CTAAGC GGTGGATCACTCGG | Initial denaturation: 94 °C, 4 min Amplification: 30 cycles Denature 94 °C, 1 min Anneal 55 °C, 30 s Extend 72 °C, 30 s Final elongation: 72 °C, 10 min | [33] |
| JB9A | GCACTATCAAGCAACACGACTC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Díaz-Sánchez, S.; Hernández-Triana, L.M.; Labruna, M.B.; Merino, O.; Mosqueda, J.; Nava, S.; Szabó, M.; Tarragona, E.; Venzal, J.M.; de la Fuente, J.; et al. Low Genetic Diversity of the Only Clade of the Tick Rhipicephalus microplus in the Neotropics. Pathogens 2023, 12, 1344. https://doi.org/10.3390/pathogens12111344
Díaz-Sánchez S, Hernández-Triana LM, Labruna MB, Merino O, Mosqueda J, Nava S, Szabó M, Tarragona E, Venzal JM, de la Fuente J, et al. Low Genetic Diversity of the Only Clade of the Tick Rhipicephalus microplus in the Neotropics. Pathogens. 2023; 12(11):1344. https://doi.org/10.3390/pathogens12111344
Chicago/Turabian StyleDíaz-Sánchez, Sandra, Luis M. Hernández-Triana, Marcelo B. Labruna, Octavio Merino, Juan Mosqueda, Santiago Nava, Matias Szabó, Evelina Tarragona, José M. Venzal, José de la Fuente, and et al. 2023. "Low Genetic Diversity of the Only Clade of the Tick Rhipicephalus microplus in the Neotropics" Pathogens 12, no. 11: 1344. https://doi.org/10.3390/pathogens12111344
APA StyleDíaz-Sánchez, S., Hernández-Triana, L. M., Labruna, M. B., Merino, O., Mosqueda, J., Nava, S., Szabó, M., Tarragona, E., Venzal, J. M., de la Fuente, J., & Estrada-Peña, A. (2023). Low Genetic Diversity of the Only Clade of the Tick Rhipicephalus microplus in the Neotropics. Pathogens, 12(11), 1344. https://doi.org/10.3390/pathogens12111344