
Oncolytic Rodent Protoparvoviruses Evade a TLR- and RLR-Independent Antiviral Response in Transformed Cells

 ,
,  ,
,  and
and 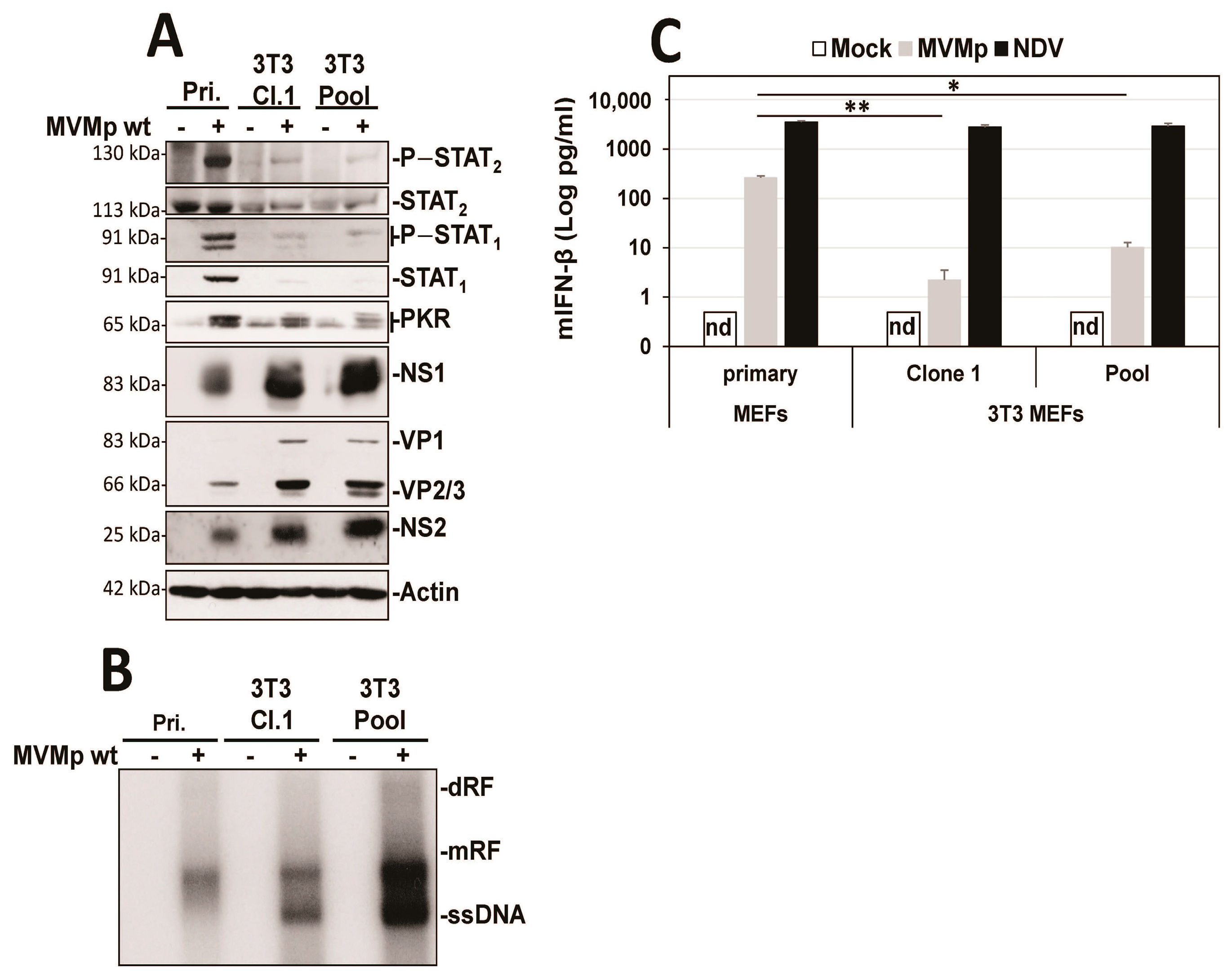{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Antibodies, Kits and Compounds
2.2. Cell Culture
2.3. Production and Titration of Virus Stocks
2.4. Cell Infection
2.5. Cell Transfection
2.6. Viral DNA Extraction and Southern Blot Analysis
2.7. SDS-PAGE and Western Blot Analysis
2.8. Fluorescence Microscopy
2.9. Detection of Type-I IFN Production
2.10. Statistical Analysis
3. Results
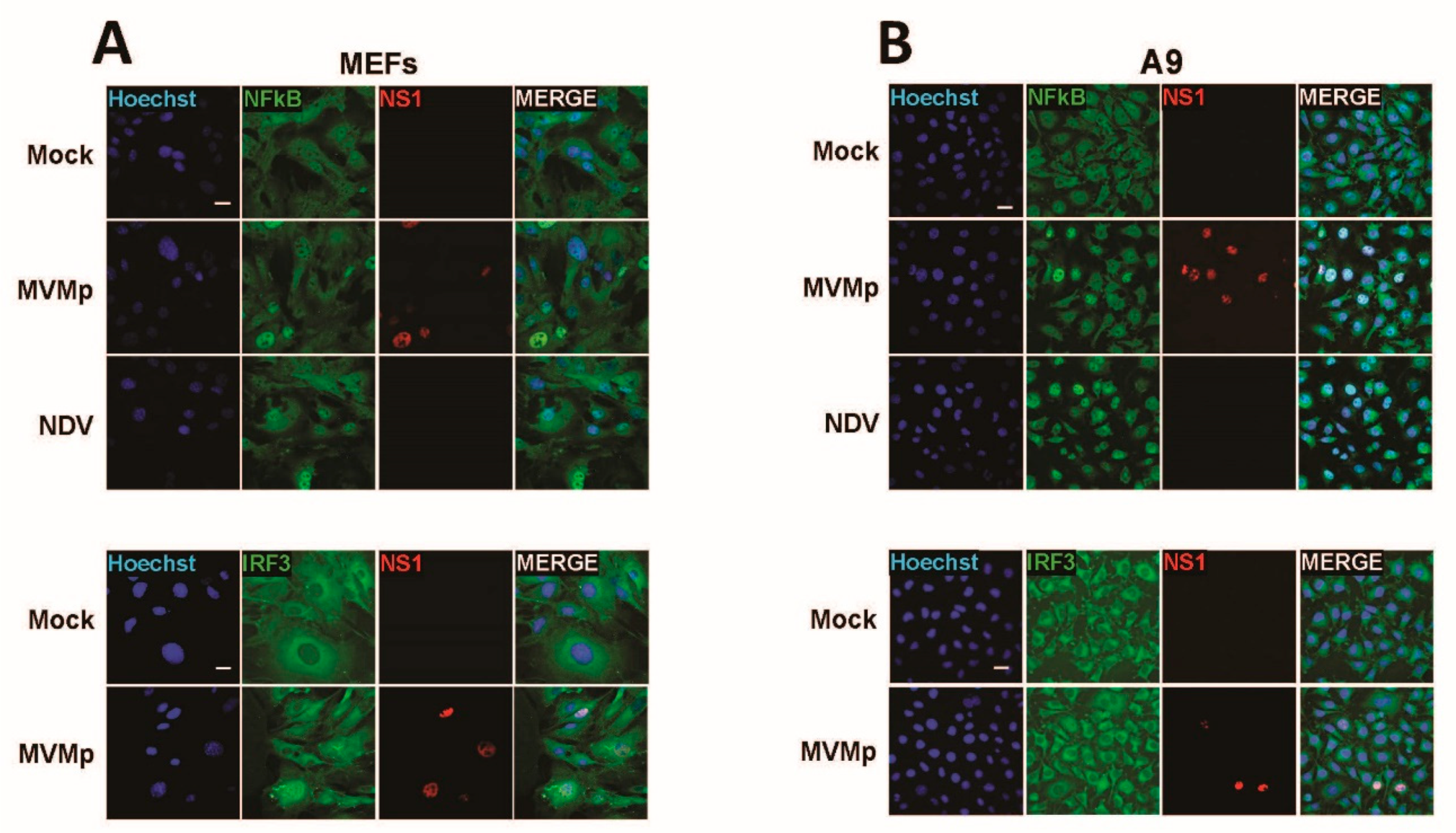
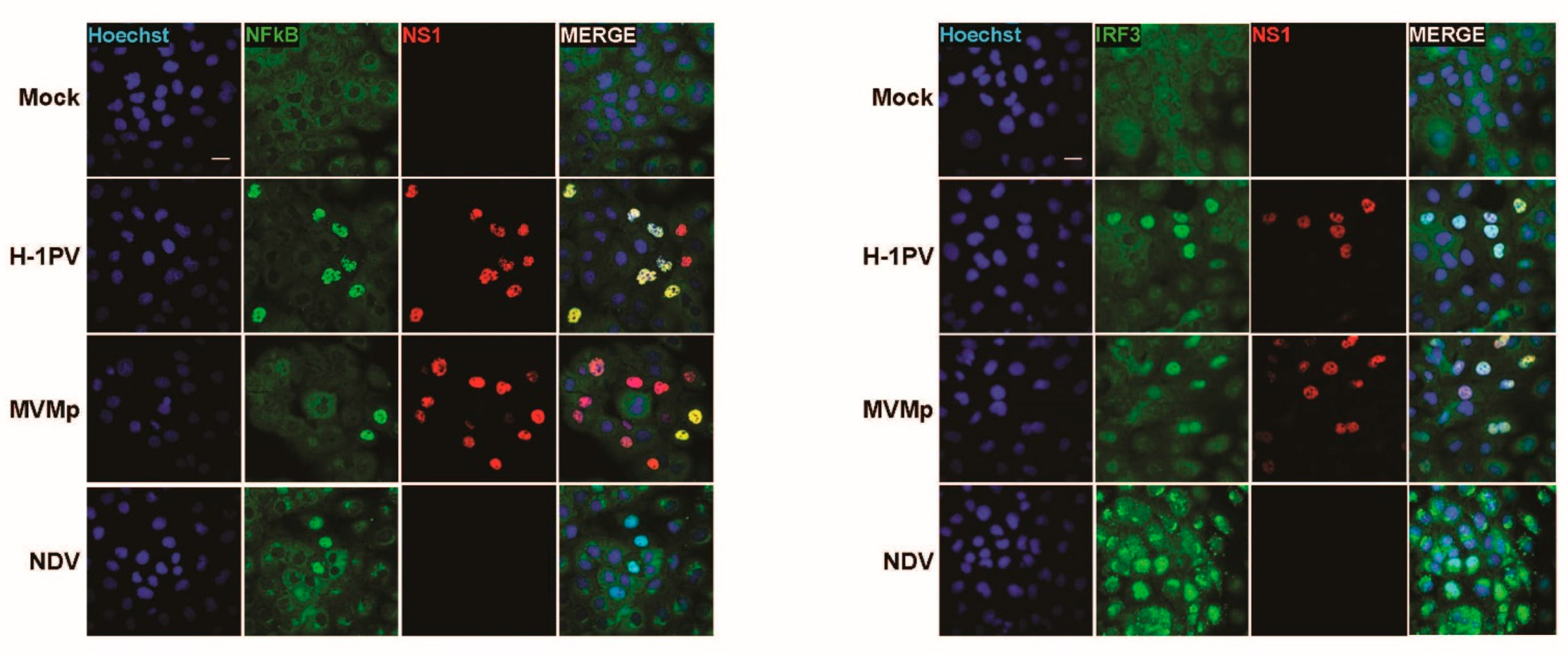
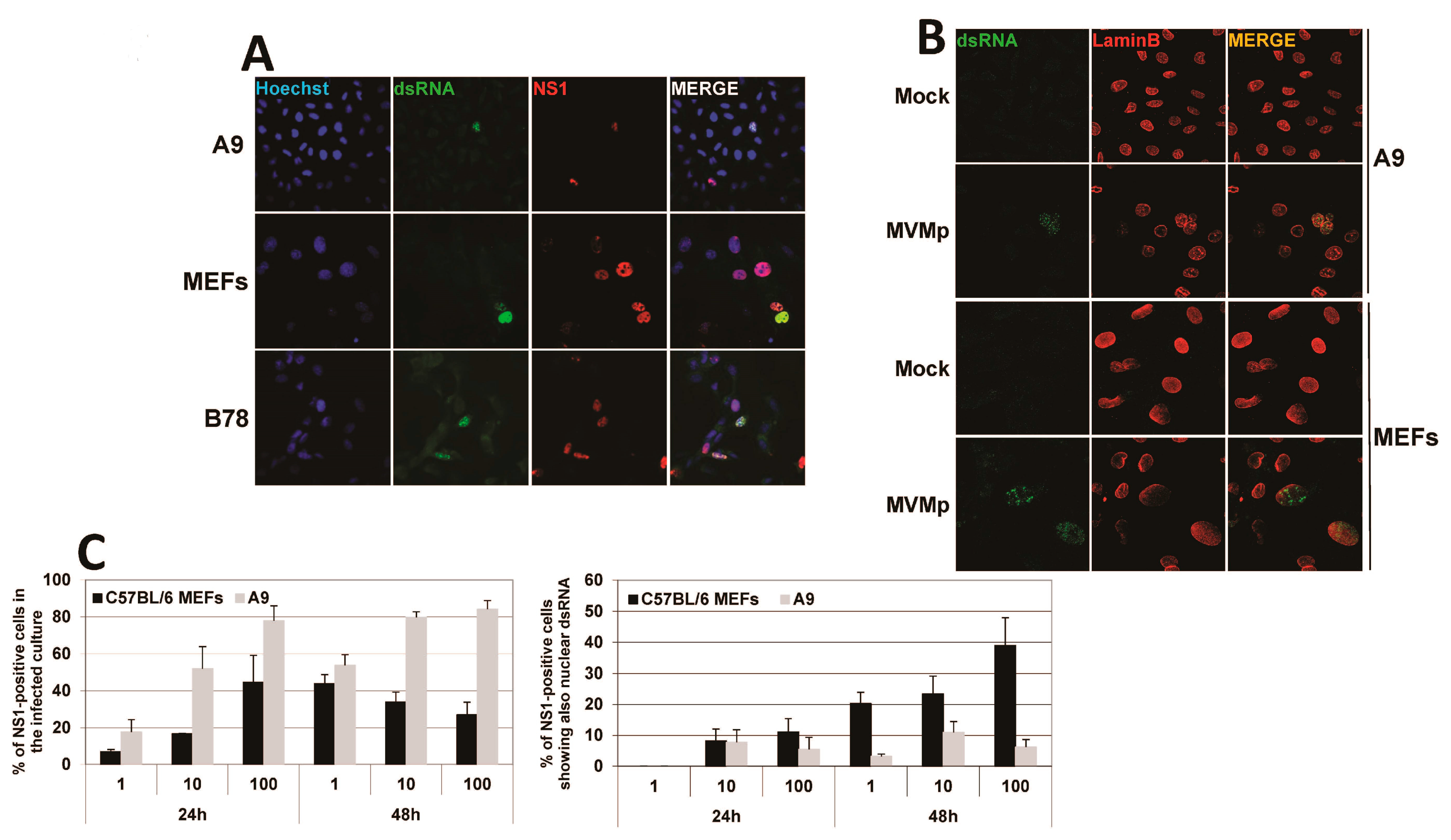
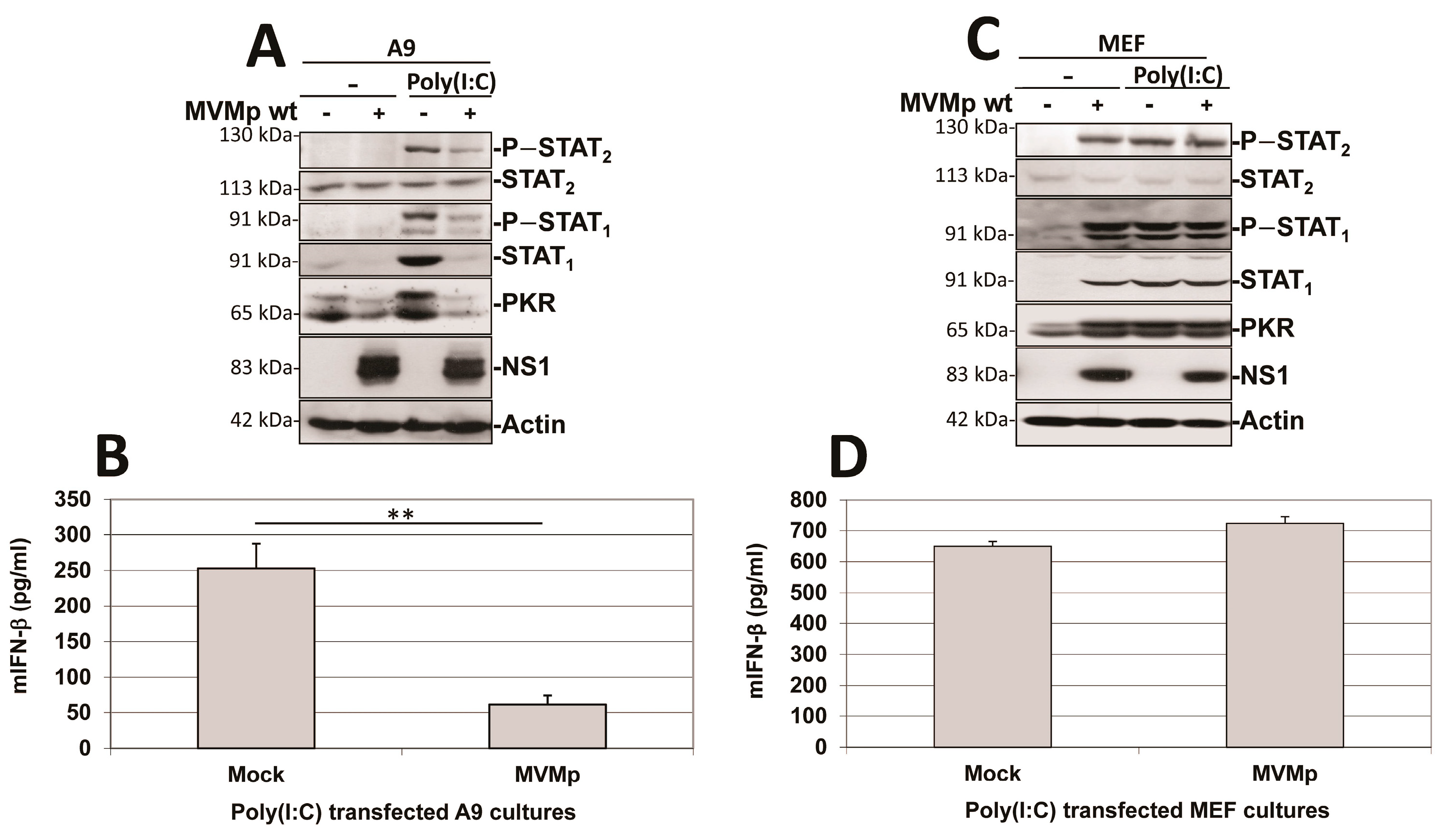
3.1. Parvovirus Infection Is Sensed by Both Normal and Transformed Cells but Results in Type-I IFN Production Only in the Former Cells
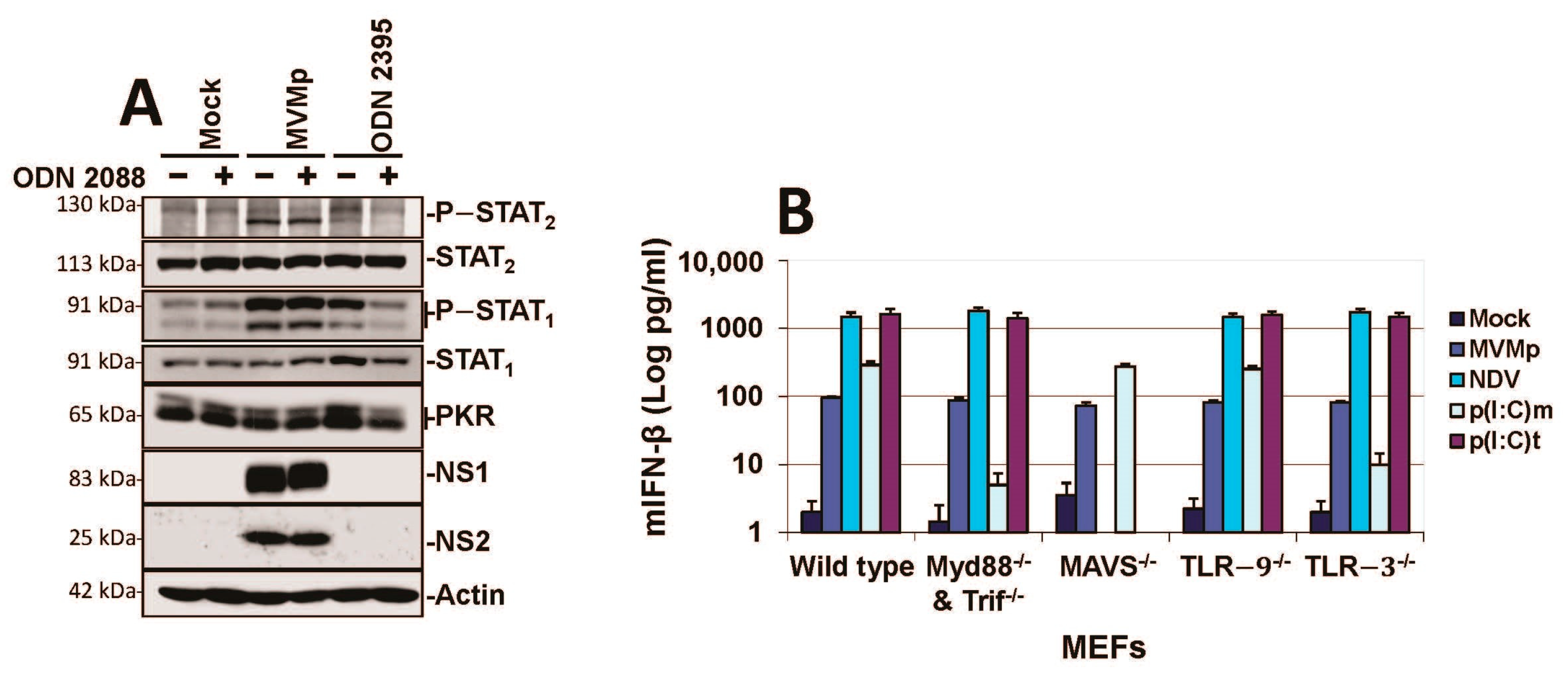
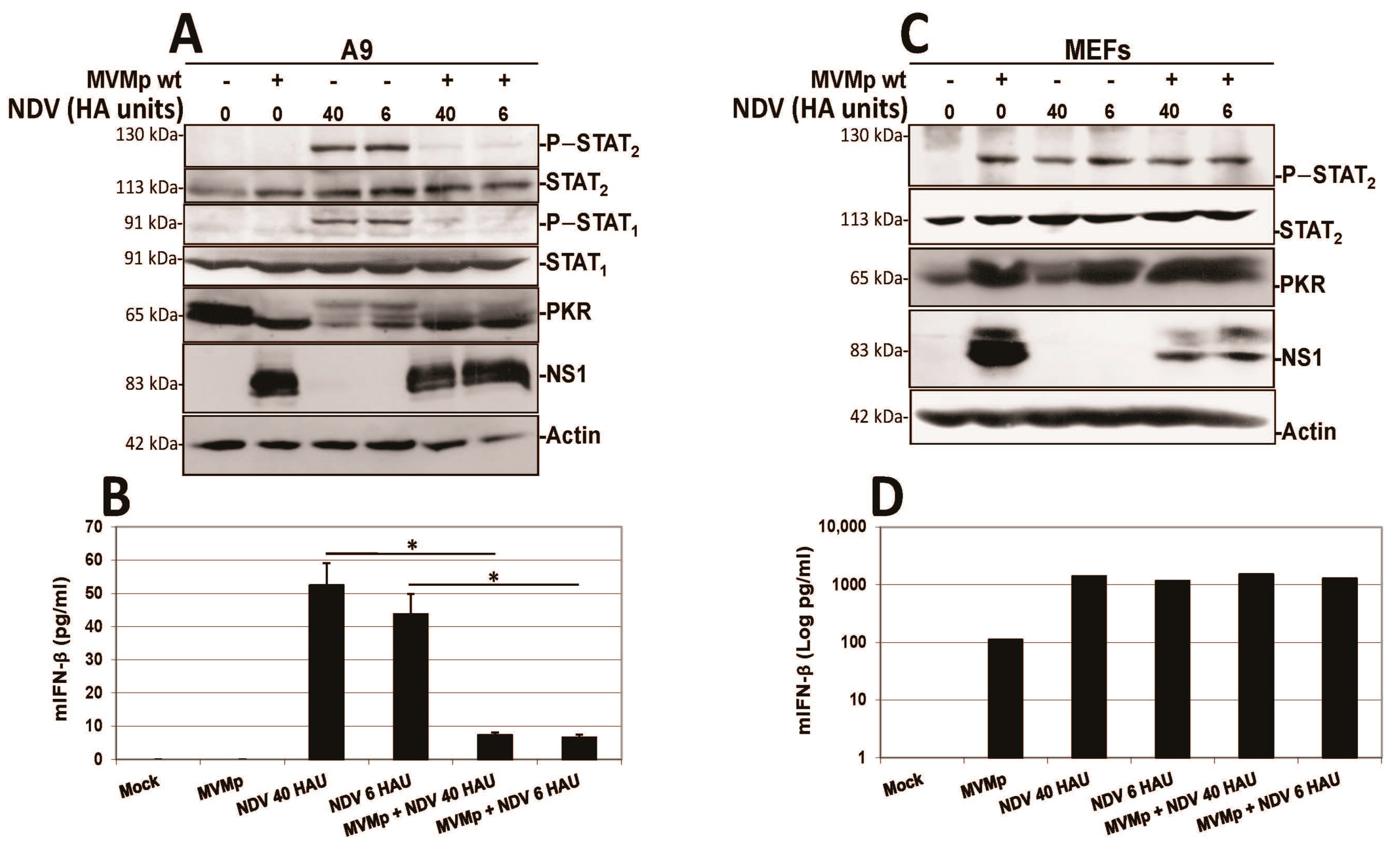
3.2. Parvovirus Sensing Can Take Place Independently of Conventional TLR and RLR Receptors and Requires Some Extent of Parvovirus Replication
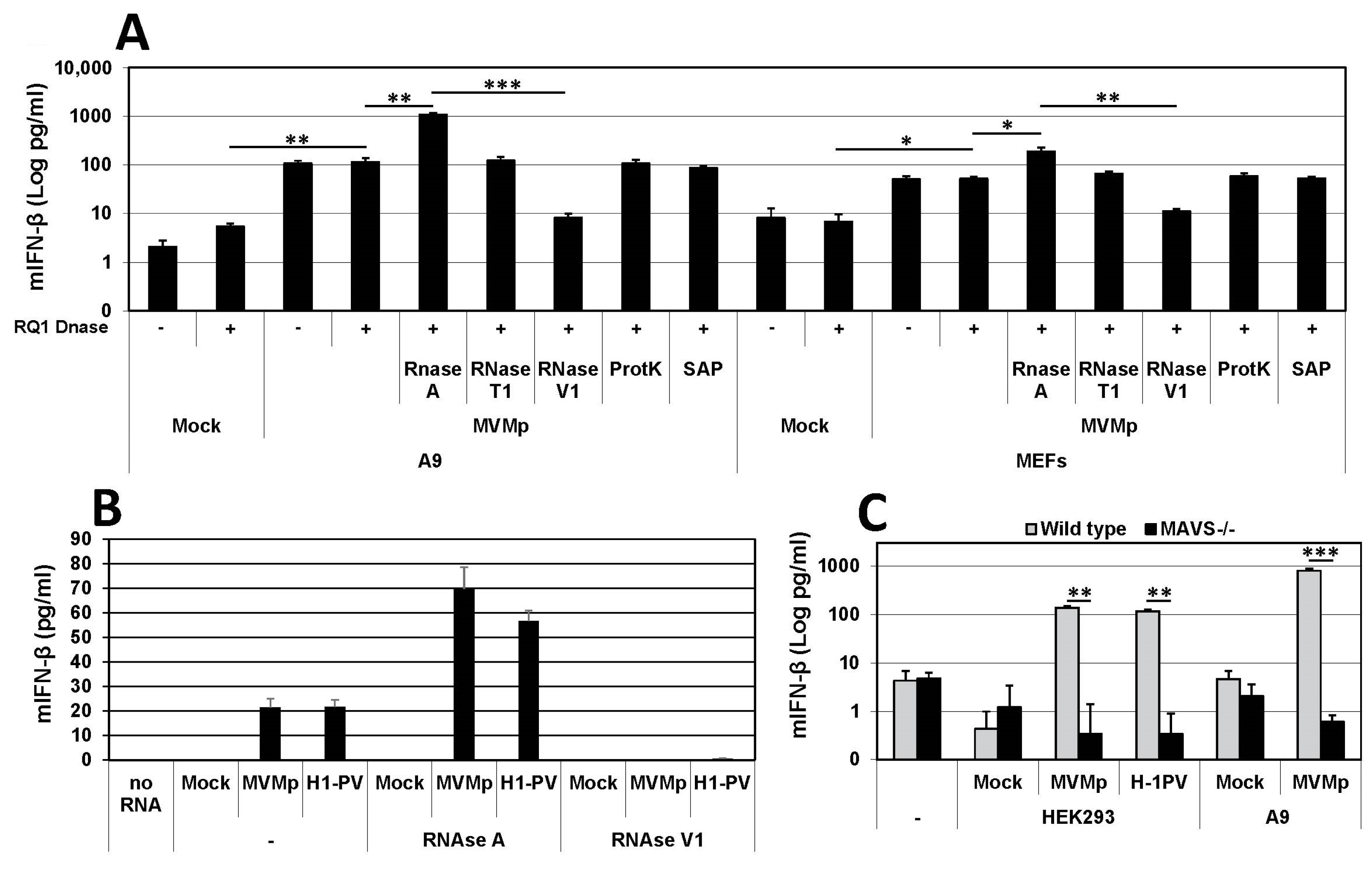
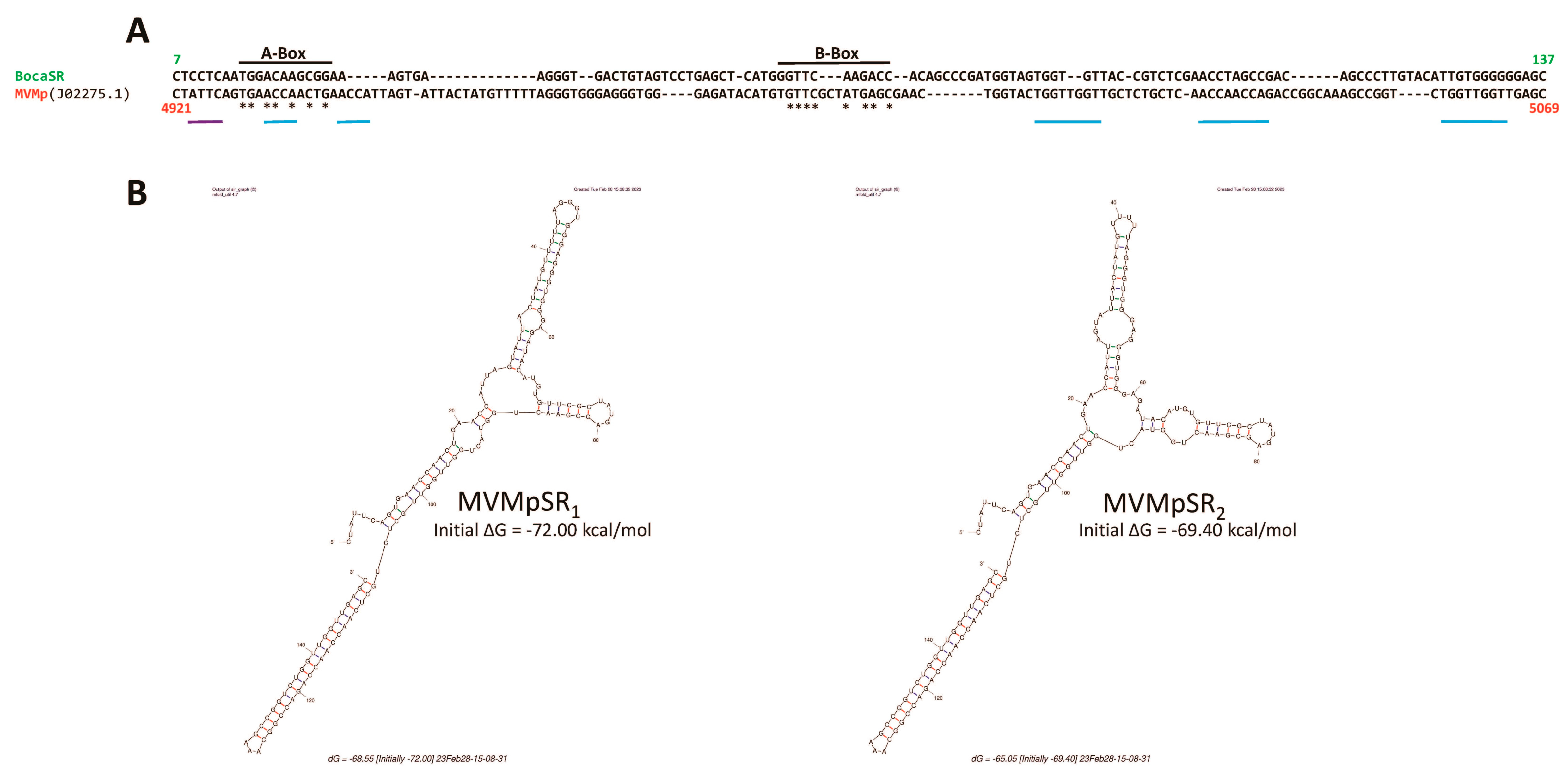
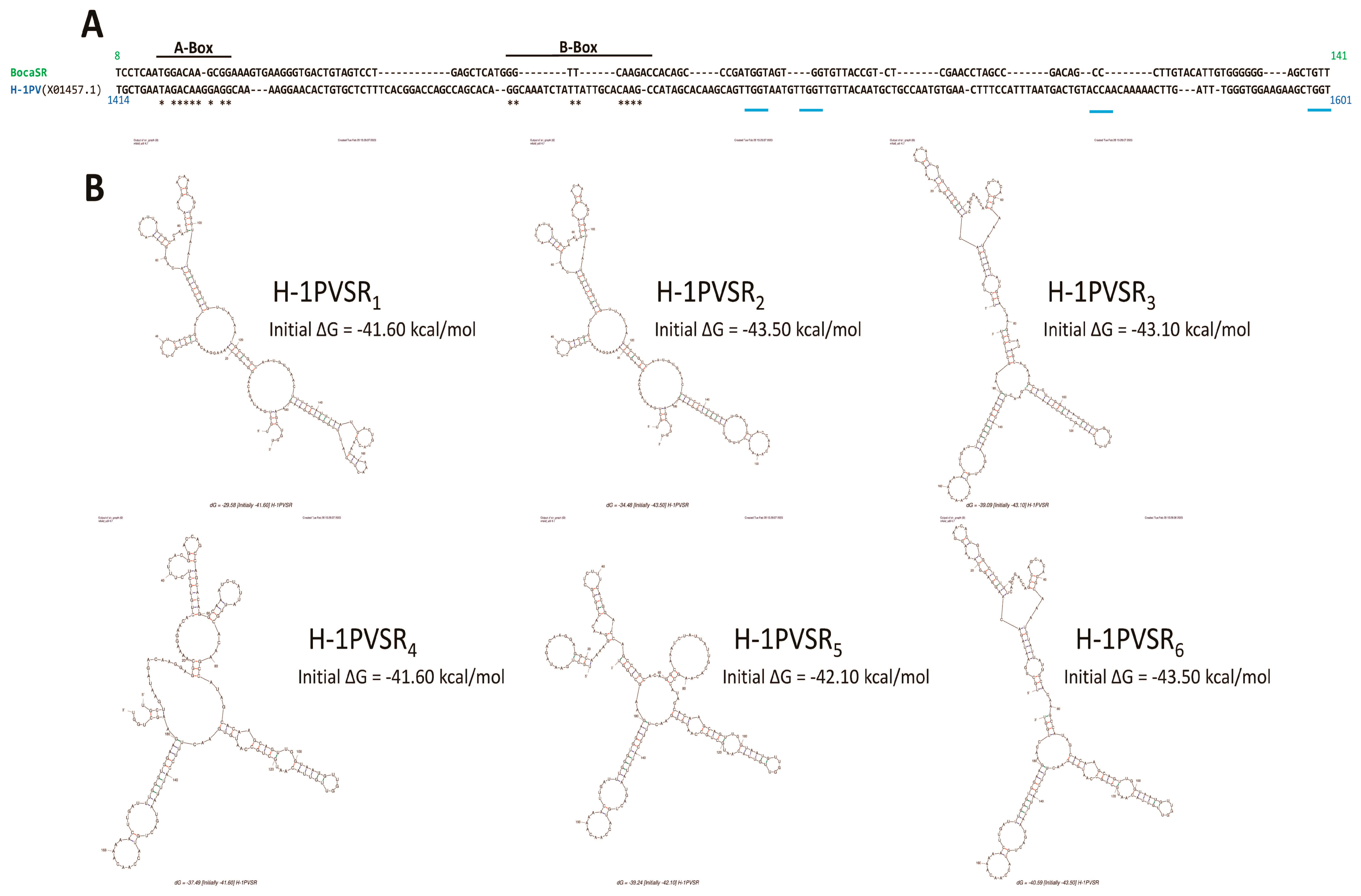
3.3. Parvovirus-Induced dsRNA Is a PAMP Candidate for PV Sensing
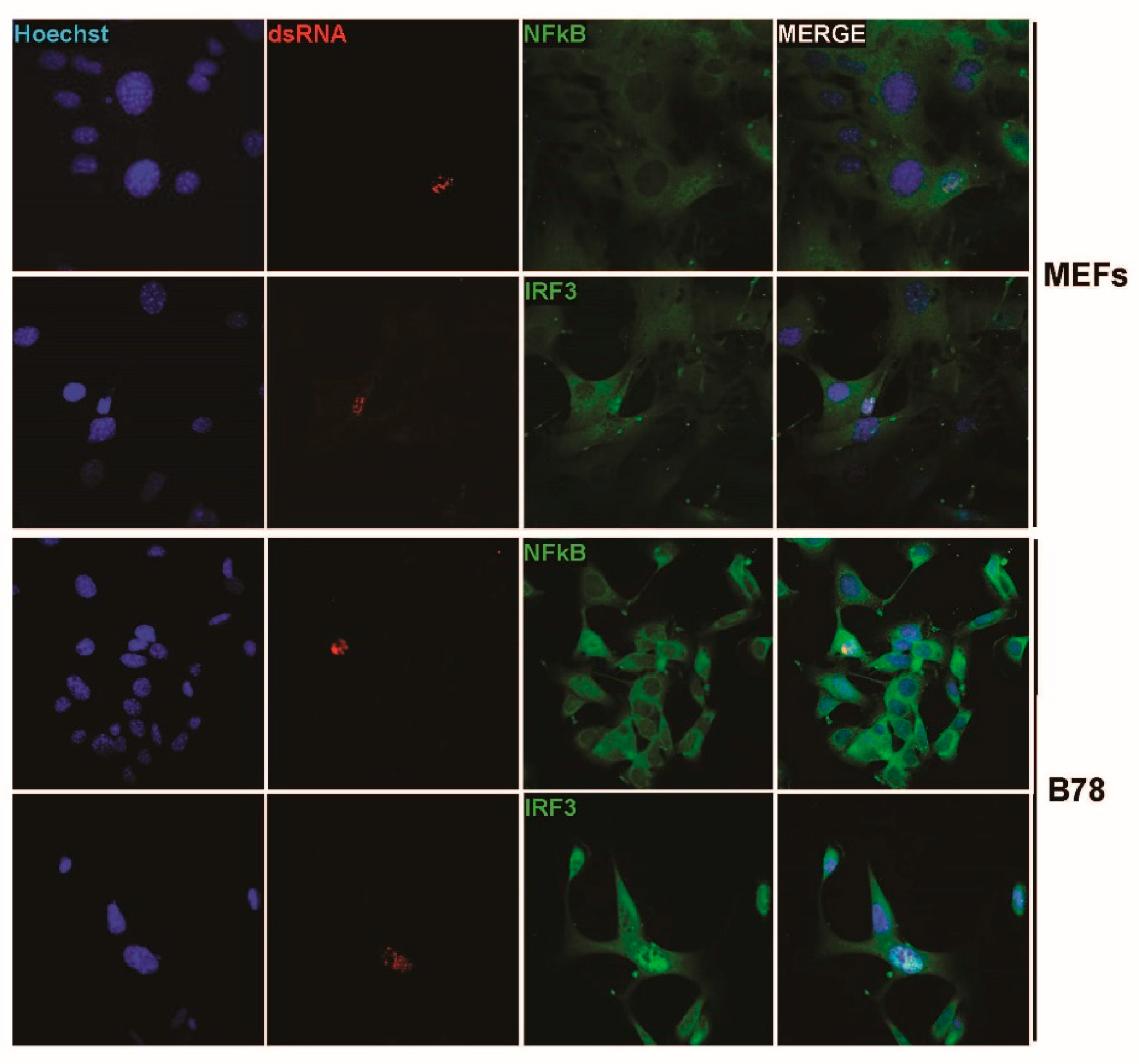
3.4. IFN Induction Aborts in Transformed PV-Infected Cells That Evade the Antiviral Response
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marchini, A.; Bonifati, S.; Scott, E.M.; Angelova, A.L.; Rommelaere, J. Oncolytic parvoviruses: From basic virology to clinical applications. Virol. J. 2015, 12, 6. [Google Scholar] [CrossRef] [PubMed]
- Boagni, D.A.; Ravirala, D.; Zhang, S.X. Current strategies in engaging oncolytic viruses with antitumor immunity. Mol. Ther. Oncolytics 2021, 22, 98–113. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.M.; McFadden, G. Oncolytic Viruses: Newest Frontier for Cancer Immunotherapy. Cancers 2021, 13, 5452. [Google Scholar] [CrossRef]
- Bommareddy, P.K.; Shettigar, M.; Kaufman, H.L. Integrating oncolytic viruses in combination cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 498–513. [Google Scholar] [CrossRef]
- Ahmed, A.; Tait, S.W.G. Targeting immunogenic cell death in cancer. Mol. Oncol. 2020, 14, 2994–3006. [Google Scholar] [CrossRef] [PubMed]
- Fucikova, J.; Kepp, O.; Kasikova, L.; Petroni, G.; Yamazaki, T.; Liu, P.; Zhao, L.; Spisek, R.; Kroemer, G.; Galluzzi, L. Detection of immunogenic cell death and its relevance for cancer therapy. Cell Death Dis. 2020, 11, 1013. [Google Scholar] [CrossRef]
- Carty, M.; Guy, C.; Bowie, A.G. Detection of Viral Infections by Innate Immunity. Biochem. Pharmacol. 2020, 183, 114316. [Google Scholar] [CrossRef]
- Sun, H.; Hu, W.; Yan, Y.; Zhang, Z.; Chen, Y.; Yao, X.; Teng, L.; Wang, X.; Chai, D.; Zheng, J.; et al. Using PAMPs and DAMPs as adjuvants in cancer vaccines. Hum. Vaccines Immunother. 2021, 17, 5546–5557. [Google Scholar] [CrossRef]
- Hennessy, C.; McKernan, D.P. Anti-Viral Pattern Recognition Receptors as Therapeutic Targets. Cells 2021, 10, 2258. [Google Scholar] [CrossRef]
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2020, 20, 95–112. [Google Scholar] [CrossRef]
- Olive, C. Pattern recognition receptors: Sentinels in innate immunity and targets of new vaccine adjuvants. Expert Rev. Vaccines 2012, 11, 237–256. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Chiu, Y.-H.; Chen, Z.J. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol. Cell 2014, 54, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Velloso, F.J.; Trombetta-Lima, M.; Anschau, V.; Sogayar, M.C.; Correa, R.G. NOD-like receptors: Major players (and targets) in the interface between innate immunity and cancer. Biosci. Rep. 2019, 39, BSR20181709. [Google Scholar] [CrossRef] [PubMed]
- Dolasia, K.; Bisht, M.K.; Pradhan, G.; Udgata, A.; Mukhopadhyay, S. TLRs/NLRs: Shaping the landscape of host immunity. Int. Rev. Immunol. 2018, 37, 3–19. [Google Scholar] [CrossRef]
- Gern, O.L.; Mulenge, F.; Pavlou, A.; Ghita, L.; Steffen, I.; Stangel, M.; Kalinke, U. Toll-like Receptors in Viral Encephalitis. Viruses 2021, 13, 2065. [Google Scholar] [CrossRef]
- Wu, Y.; Song, K.; Hao, W.; Li, J.; Wang, L.; Li, S. Nuclear soluble cGAS senses double-stranded DNA virus infection. Commun. Biol. 2022, 5, 433. [Google Scholar] [CrossRef]
- Cui, S.; Yu, Q.; Chu, L.; Cui, Y.; Ding, M.; Wang, Q.; Wang, H.; Chen, Y.; Liu, X.; Wang, C. Nuclear cGAS Functions Non-canonically to Enhance Antiviral Immunity via Recruiting Methyltransferase Prmt5. Cell Rep. 2020, 33, 108490. [Google Scholar] [CrossRef]
- Li, D.; Xie, L.; Qiao, Z.; Zhu, J.; Yao, H.; Qin, Y.; Yan, Y.; Chen, Z.; Ma, F. IFI16 Isoforms with Cytoplasmic and Nuclear Locations Play Differential Roles in Recognizing Invaded DNA Viruses. J. Immunol. 2021, 207, 2699–2709. [Google Scholar] [CrossRef]
- Huérfano, S.; Šroller, V.; Bruštíková, K.; Horníková, L.; Forstová, J. The Interplay between Viruses and Host DNA Sensors. Viruses 2022, 14, 666. [Google Scholar] [CrossRef]
- Musella, M.; Galassi, C.; Manduca, N.; Sistigu, A. The Yin and Yang of Type I IFNs in Cancer Promotion and Immune Activation. Biology 2021, 10, 856. [Google Scholar] [CrossRef]
- Zanin, N.; Viaris de Lesegno, C.; Lamaze, C.; Blouin, C.M. Interferon Receptor Trafficking and Signaling: Journey to the Cross Roads. Front. Immunol. 2020, 11, 615603. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W. Interferon-Stimulated Genes: What Do They All Do? Annu. Rev. Virol. 2019, 6, 567–584. [Google Scholar] [CrossRef] [PubMed]
- Beachboard, D.C.; Horner, S.M. Innate immune evasion strategies of DNA and RNA viruses. Curr. Opin. Microbiol. 2016, 32, 113–119. [Google Scholar] [CrossRef]
- Zou, S.-S.; Qiao, Y.; Zhu, S.; Gao, B.; Yang, N.; Liu, Y.-J.; Chen, J. Intrinsic strategies for the evasion of cGAS-STING signaling-mediated immune surveillance in human cancer: How therapy can overcome them. Pharmacol. Res. 2021, 166, 105514. [Google Scholar] [CrossRef]
- Znaidia, M.; Demeret, C.; van der Werf, S.; Komarova, A.V. Characterization of SARS-CoV-2 Evasion: Interferon Pathway and Therapeutic Options. Viruses 2022, 14, 1247. [Google Scholar] [CrossRef]
- Lin, Y.; Zheng, C. A Tug of War: DNA-Sensing Antiviral Innate Immunity and Herpes Simplex Virus Type I Infection. Front. Microbiol. 2019, 10, 2627. [Google Scholar] [CrossRef]
- Coldbeck-Shackley, R.C.; Eyre, N.S.; Beard, M.R. The Molecular Interactions of ZIKV and DENV with the Type-I IFN Response. Vaccines 2020, 8, 530. [Google Scholar] [CrossRef]
- Humeau, J.; Le Naour, J.; Galluzzi, L.; Kroemer, G.; Pol, J.G. Trial watch: Intratumoral immunotherapy. Oncoimmunology 2021, 10, 1984677. [Google Scholar] [CrossRef]
- Feola, S.; Russo, S.; Ylösmäki, E.; Cerullo, V. Oncolytic ImmunoViroTherapy: A long history of crosstalk between viruses and immune system for cancer treatment. Pharmacol. Ther. 2021, 236, 108103. [Google Scholar] [CrossRef]
- Vitiello, G.A.F.; Ferreira, W.A.S.; Cordeiro de Lima, V.C.; Medina, T. da S. Antiviral Responses in Cancer: Boosting Antitumor Immunity Through Activation of Interferon Pathway in the Tumor Microenvironment. Front. Immunol. 2021, 12, 782852. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.J.; Kroemer, G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 2015, 15, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Zhu, B.; Chen, D. Type I interferon-mediated tumor immunity and its role in immunotherapy. Cell. Mol. Life Sci. CMLS 2022, 79, 191. [Google Scholar] [CrossRef]
- Rommelaere, J.; Geletneky, K.; Angelova, A.L.; Daeffler, L.; Dinsart, C.; Kiprianova, I.; Schlehofer, J.R.; Raykov, Z. Oncolytic parvoviruses as cancer therapeutics. Cytokine Growth Factor Rev. 2010, 21, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Angelova, A.L.; Geletneky, K.; Nüesch, J.P.F.; Rommelaere, J. Tumor Selectivity of Oncolytic Parvoviruses: From in vitro and Animal Models to Cancer Patients. Front. Bioeng. Biotechnol. 2015, 3, 55. [Google Scholar] [CrossRef]
- Geletneky, K.; Nüesch, J.P.; Angelova, A.; Kiprianova, I.; Rommelaere, J. Double-faceted mechanism of parvoviral oncosuppression. Curr. Opin. Virol. 2015, 13, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Geletneky, K.; Hajda, J.; Angelova, A.L.; Leuchs, B.; Capper, D.; Bartsch, A.J.; Neumann, J.-O.; Schöning, T.; Hüsing, J.; Beelte, B.; et al. Oncolytic H-1 Parvovirus Shows Safety and Signs of Immunogenic Activity in a First Phase I/IIa Glioblastoma Trial. Mol. Ther. J. Am. Soc. Gene Ther. 2017, 25, 2620–2634. [Google Scholar] [CrossRef]
- Hajda, J.; Leuchs, B.; Angelova, A.L.; Frehtman, V.; Rommelaere, J.; Mertens, M.; Pilz, M.; Kieser, M.; Krebs, O.; Huber, B.; et al. Phase 2 trial of oncolytic H-1 parvovirus therapy shows safety and immune cell activity in patients with metastatic pancreatic ductal adenocarcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 5546–5556. [Google Scholar] [CrossRef]
- Angelova, A.; Ferreira, T.; Bretscher, C.; Rommelaere, J.; Marchini, A. Parvovirus-Based Combinatorial Immunotherapy: A Reinforced Therapeutic Strategy against Poor-Prognosis Solid Cancers. Cancers 2021, 13, 342. [Google Scholar] [CrossRef]
- Grekova, S.; Zawatzky, R.; Hörlein, R.; Cziepluch, C.; Mincberg, M.; Davis, C.; Rommelaere, J.; Daeffler, L. Activation of an antiviral response in normal but not transformed mouse cells: A new determinant of minute virus of mice oncotropism. J. Virol. 2010, 84, 516–531. [Google Scholar] [CrossRef]
- Raykov, Z.; Grekova, S.P.; Hörlein, R.; Leuchs, B.; Giese, T.; Giese, N.A.; Rommelaere, J.; Zawatzky, R.; Daeffler, L. TLR-9 contributes to the antiviral innate immune sensing of rodent parvoviruses MVMp and H-1PV by normal human immune cells. PLoS ONE 2013, 8, 55086. [Google Scholar] [CrossRef] [PubMed]
- Raykov, Z.; Grekova, S.; Leuchs, B.; Aprahamian, M.; Rommelaere, J. Arming parvoviruses with CpG motifs to improve their oncosuppressive capacity. Int. J. Cancer 2008, 122, 2880–2884. [Google Scholar] [CrossRef] [PubMed]
- Bodendorf, U.; Cziepluch, C.; Jauniaux, J.C.; Rommelaere, J.; Salomé, N. Nuclear export factor CRM1 interacts with nonstructural proteins NS2 from parvovirus minute virus of mice. J. Virol. 1999, 73, 7769–7779. [Google Scholar] [CrossRef] [PubMed]
- López-Bueno, A.; Mateu, M.G.; Almendral, J.M. High mutant frequency in populations of a DNA virus allows evasion from antibody therapy in an immunodeficient host. J. Virol. 2003, 77, 2701–2708. [Google Scholar] [CrossRef]
- Ghita, L.; Breitkopf, V.; Mulenge, F.; Pavlou, A.; Gern, O.L.; Durán, V.; Prajeeth, C.K.; Kohls, M.; Jung, K.; Stangel, M.; et al. Sequential MAVS and MyD88/TRIF signaling triggers anti-viral responses of tick-borne encephalitis virus-infected murine astrocytes. J. Neurosci. Res. 2021, 99, 2478–2492. [Google Scholar] [CrossRef]
- Nilausen, K.; Green, H. Reversible arrest of growth in G1 of an established fibroblast line (3T3). Exp. Cell Res. 1965, 40, 166–168. [Google Scholar] [CrossRef]
- Zolotukhin, S.; Byrne, B.J.; Mason, E.; Zolotukhin, I.; Potter, M.; Chesnut, K.; Summerford, C.; Samulski, R.J.; Muzyczka, N. Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther. 1999, 6, 973–985. [Google Scholar] [CrossRef]
- Wilden, H.; Fournier, P.; Zawatzky, R.; Schirrmacher, V. Expression of RIG-I, IRF3, IFN-beta and IRF7 determines resistance or susceptibility of cells to infection by Newcastle Disease Virus. Int. J. Oncol. 2009, 34, 971–982. [Google Scholar] [CrossRef]
- Daeffler, L.; Hörlein, R.; Rommelaere, J.; Nüesch, J.P.F. Modulation of minute virus of mice cytotoxic activities through site-directed mutagenesis within the NS coding region. J. Virol. 2003, 77, 12466–12478. [Google Scholar] [CrossRef]
- Schirrmacher, V.; van Gool, S.; Stuecker, W. Breaking Therapy Resistance: An Update on Oncolytic Newcastle Disease Virus for Improvements of Cancer Therapy. Biomedicines 2019, 7, 66. [Google Scholar] [CrossRef]
- Schirrmacher, V. Molecular Mechanisms of Anti-Neoplastic and Immune Stimulatory Properties of Oncolytic Newcastle Disease Virus. Biomedicines 2022, 10, 562. [Google Scholar] [CrossRef] [PubMed]
- Kowalinski, E.; Lunardi, T.; McCarthy, A.A.; Louber, J.; Brunel, J.; Grigorov, B.; Gerlier, D.; Cusack, S. Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell 2011, 147, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Xu, H.; Ranjith-Kumar, C.T.; Brooks, M.T.; Hou, T.Y.; Hu, F.; Herr, A.B.; Strong, R.K.; Kao, C.C.; Li, P. The structural basis of 5’ triphosphate double-stranded RNA recognition by RIG-I C-terminal domain. Structure 2010, 18, 1032–1043. [Google Scholar] [CrossRef] [PubMed]
- Mattei, L.M.; Cotmore, S.F.; Tattersall, P.; Iwasaki, A. Parvovirus evades interferon-dependent viral control in primary mouse embryonic fibroblasts. Virology 2013, 442, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Cotmore, S.F.; Agbandje-McKenna, M.; Canuti, M.; Chiorini, J.A.; Eis-Hubinger, A.-M.; Hughes, J.; Mietzsch, M.; Modha, S.; Ogliastro, M.; Pénzes, J.J.; et al. ICTV Virus Taxonomy Profile: Parvoviridae. J. Gen. Virol. 2019, 100, 367–368. [Google Scholar] [CrossRef]
- Wang, Z.; Shen, W.; Cheng, F.; Deng, X.; Engelhardt, J.F.; Yan, Z.; Qiu, J. Parvovirus Expresses a Small Noncoding RNA That Plays an Essential Role in Virus Replication. J. Virol. 2017, 91, e02375-16. [Google Scholar] [CrossRef]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Buijs, P.; van Nieuwkoop, S.; Vaes, V.; Fouchier, R.; van Eijck, C.; van den Hoogen, B. Recombinant Immunomodulating Lentogenic or Mesogenic Oncolytic Newcastle Disease Virus for Treatment of Pancreatic Adenocarcinoma. Viruses 2015, 7, 2980–2998. [Google Scholar] [CrossRef]
- Mattei, L.M.; Cotmore, S.F.; Li, L.; Tattersall, P.; Iwasaki, A. Toll-like receptor 9 in plasmacytoid dendritic cells fails to detect parvoviruses. J. Virol. 2013, 87, 3605–3608. [Google Scholar] [CrossRef]
- Paglino, J.C.; Andres, W.; van den Pol, A.N. Autonomous parvoviruses neither stimulate nor are inhibited by the type I interferon response in human normal or cancer cells. J. Virol. 2014, 88, 4932–4942. [Google Scholar] [CrossRef]
- Rostovsky, I.; Davis, C. Induction of an embryonic mouse innate immune response following inoculation in utero with minute virus of mice. J. Virol. 2015, 89, 2182–2191. [Google Scholar] [CrossRef] [PubMed]
- Loew, L.; Goonawardane, N.; Ratcliff, J.; Nguyen, D.; Simmonds, P. Use of a small DNA virus model to investigate mechanisms of CpG dinucleotide-induced attenuation of virus replication. J. Gen. Virol. 2020, 101, 1202–1218. [Google Scholar] [CrossRef] [PubMed]
- Panne, D. The enhanceosome. Curr. Opin. Struct. Biol. 2008, 18, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Liu, D.; Tian, J.; Hu, X.; Zhang, X.; Yin, H.; Wu, H.; Liu, C.; Guo, D.; Li, Z.; et al. Feline Panleucopenia Virus NS2 Suppresses the Host IFN-β Induction by Disrupting the Interaction between TBK1 and STING. Viruses 2017, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- Runde, A.P.; Mack, R.; Breslin S.J., P.; Zhang, J. The role of TBK1 in cancer pathogenesis and anticancer immunity. J. Exp. Clin. Cancer Res. CR 2022, 41, 135. [Google Scholar] [CrossRef]
- Langer, S.; Hammer, C.; Hopfensperger, K.; Klein, L.; Hotter, D.; De Jesus, P.D.; Herbert, K.M.; Pache, L.; Smith, N.; van der Merwe, J.A.; et al. HIV-1 Vpu is a potent transcriptional suppressor of NF-κB-elicited antiviral immune responses. eLife 2019, 8, e41930. [Google Scholar] [CrossRef]
- Irie, T.; Nagata, N.; Igarashi, T.; Okamoto, I.; Sakaguchi, T. Conserved charged amino acids within Sendai virus C protein play multiple roles in the evasion of innate immune responses. PLoS ONE 2010, 5, 10719. [Google Scholar] [CrossRef]
- Zandi, M.; Shafaati, M.; Kalantar-Neyestanaki, D.; Pourghadamyari, H.; Fani, M.; Soltani, S.; Kaleji, H.; Abbasi, S. The role of SARS-CoV-2 accessory proteins in immune evasion. Biomed. Pharmacother. Biomed. Pharmacother. 2022, 156, 113889. [Google Scholar] [CrossRef]
- Tal, S.; Mincberg, M.; Rostovsky, I.; Rommelaere, J.; Salome, N.; Davis, C. The Minute Virus of Mice NS2 proteins are not essential for productive infection of embryonic murine cells in utero. Virology 2014, 468–470, 631–636. [Google Scholar] [CrossRef]
- Mihaylov, I.S.; Cotmore, S.F.; Tattersall, P. Complementation for an essential ancillary non-structural protein function across parvovirus genera. Virology 2014, 468–470, 226–237. [Google Scholar] [CrossRef]
- Zhuandi, G.; Zhaofang, Y.; Dianyu, L.; Mengyuan, P.; Suocheng, W. Immune escape of bovine parvovirus by VP1 inhibiting IFN-β production through the RIG-I-like receptor pathway. Int. Microbiol. Off. J. Span. Soc. Microbiol. 2023, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Yuan, W.; Davis, I.; Parrish, C.R. Nonstructural protein-2 and the replication of canine parvovirus. Virology 1998, 240, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Hoelzer, K.; Shackelton, L.A.; Holmes, E.C.; Parrish, C.R. Within-Host Genetic Diversity of Endemic and Emerging Parvoviruses of Dogs and Cats. J. Virol. 2008, 82, 11096–11105. [Google Scholar] [CrossRef] [PubMed]
- Gentili, M.; Lahaye, X.; Nadalin, F.; Nader, G.F.P.; Puig Lombardi, E.; Herve, S.; De Silva, N.S.; Rookhuizen, D.C.; Zueva, E.; Goudot, C.; et al. The N-Terminal Domain of cGAS Determines Preferential Association with Centromeric DNA and Innate Immune Activation in the Nucleus. Cell Rep. 2019, 26, 2377–2393. [Google Scholar] [CrossRef]
- Ventoso, I.; Berlanga, J.J.; Almendral, J.M. Translation control by protein kinase R restricts minute virus of mice infection: Role in parvovirus oncolysis. J. Virol. 2010, 84, 5043–5051. [Google Scholar] [CrossRef]
- Gil-Ranedo, J.; Gallego-García, C.; Almendral, J.M. Viral targeting of glioblastoma stem cells with patient-specific genetic and post-translational p53 deregulations. Cell Rep. 2021, 36, 109673. [Google Scholar] [CrossRef]
- Son, K.-N.; Liang, Z.; Lipton, H.L. Double-Stranded RNA Is Detected by Immunofluorescence Analysis in RNA and DNA Virus Infections, Including Those by Negative-Stranded RNA Viruses. J. Virol. 2015, 89, 9383–9392. [Google Scholar] [CrossRef]
- Dzananovic, E.; Patel, T.R.; Chojnowski, G.; Boniecki, M.J.; Deo, S.; McEleney, K.; Harding, S.E.; Bujnicki, J.M.; McKenna, S.A. Solution conformation of adenovirus virus associated RNA-I and its interaction with PKR. J. Struct. Biol. 2014, 185, 48–57. [Google Scholar] [CrossRef]
- Cesaro, T.; Michiels, T. Inhibition of PKR by Viruses. Front. Microbiol. 2021, 12, 757238. [Google Scholar] [CrossRef]
- Hur, S. Double-Stranded RNA Sensors and Modulators in Innate Immunity. Annu. Rev. Immunol. 2019, 37, 349–375. [Google Scholar] [CrossRef]
- Hall, A.E.; Turnbull, C.; Dalmay, T. Y RNAs: Recent developments. Biomol. Concepts 2013, 4, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Vabret, N.; Najburg, V.; Solovyov, A.; Gopal, R.; McClain, C.; Šulc, P.; Balan, S.; Rahou, Y.; Beauclair, G.; Chazal, M.; et al. Y RNAs are conserved endogenous RIG-I ligands across RNA virus infection and are targeted by HIV-1. iScience 2022, 25, 104599. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Liu, S.; Li, Y.; Yang, G.; Luo, Y.; Li, S.; Du, H.; Zhao, Y.; Wang, D.; Chen, J.; et al. The Nuclear Matrix Protein SAFA Surveils Viral RNA and Facilitates Immunity by Activating Antiviral Enhancers and Super-enhancers. Cell Host Microbe 2019, 26, 369–384. [Google Scholar] [CrossRef] [PubMed]
- Harrington, K.; Freeman, D.J.; Kelly, B.; Harper, J.; Soria, J.-C. Optimizing oncolytic virotherapy in cancer treatment. Nat. Rev. Drug Discov. 2019, 18, 689–706. [Google Scholar] [CrossRef]
- Jamieson, T.R.; Poutou, J.; Ilkow, C.S. Redirecting oncolytic viruses: Engineering opportunists to take control of the tumour microenvironment. Cytokine Growth Factor Rev. 2020, 56, 102–114. [Google Scholar] [CrossRef]
- Sprooten, J.; Agostinis, P.; Garg, A.D. Type I interferons and dendritic cells in cancer immunotherapy. Int. Rev. Cell Mol. Biol. 2019, 348, 217–262. [Google Scholar] [CrossRef]
- Liang, Y.; Hannan, R.; Fu, Y.-X. Type I IFN Activating Type I Dendritic Cells for Antitumor Immunity. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 3818–3824. [Google Scholar] [CrossRef]
- Vitale, G.; van Eijck, C.H.J.; van Koetsveld Ing, P.M.; Erdmann, J.I.; Speel, E.J.M.; van der Wansem Ing, K.; Mooij, D.M.; Colao, A.; Lombardi, G.; Croze, E.; et al. Type I interferons in the treatment of pancreatic cancer: Mechanisms of action and role of related receptors. Ann. Surg. 2007, 246, 259–268. [Google Scholar] [CrossRef]
- Zhang, F.; Manna, S.; Pop, L.M.; Chen, Z.J.; Fu, Y.-X.; Hannan, R. Type I Interferon Response in Radiation-Induced Anti-Tumor Immunity. Semin. Radiat. Oncol. 2020, 30, 129–138. [Google Scholar] [CrossRef]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angelova, A.; Pierrard, K.; Detje, C.N.; Santiago, E.; Grewenig, A.; Nüesch, J.P.F.; Kalinke, U.; Ungerechts, G.; Rommelaere, J.; Daeffler, L. Oncolytic Rodent Protoparvoviruses Evade a TLR- and RLR-Independent Antiviral Response in Transformed Cells. Pathogens 2023, 12, 607. https://doi.org/10.3390/pathogens12040607
Angelova A, Pierrard K, Detje CN, Santiago E, Grewenig A, Nüesch JPF, Kalinke U, Ungerechts G, Rommelaere J, Daeffler L. Oncolytic Rodent Protoparvoviruses Evade a TLR- and RLR-Independent Antiviral Response in Transformed Cells. Pathogens. 2023; 12(4):607. https://doi.org/10.3390/pathogens12040607
Chicago/Turabian StyleAngelova, Assia, Kristina Pierrard, Claudia N. Detje, Estelle Santiago, Annabel Grewenig, Jürg P. F. Nüesch, Ulrich Kalinke, Guy Ungerechts, Jean Rommelaere, and Laurent Daeffler. 2023. "Oncolytic Rodent Protoparvoviruses Evade a TLR- and RLR-Independent Antiviral Response in Transformed Cells" Pathogens 12, no. 4: 607. https://doi.org/10.3390/pathogens12040607
APA StyleAngelova, A., Pierrard, K., Detje, C. N., Santiago, E., Grewenig, A., Nüesch, J. P. F., Kalinke, U., Ungerechts, G., Rommelaere, J., & Daeffler, L. (2023). Oncolytic Rodent Protoparvoviruses Evade a TLR- and RLR-Independent Antiviral Response in Transformed Cells. Pathogens, 12(4), 607. https://doi.org/10.3390/pathogens12040607