
Mechanism of Immune Evasion in Mosquito-Borne Diseases

 and
and 
Abstract
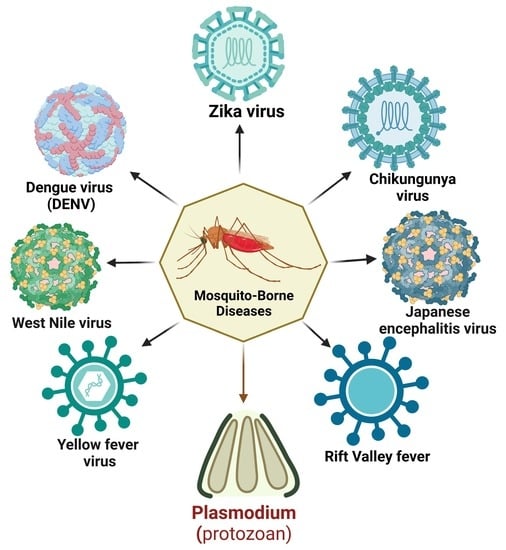
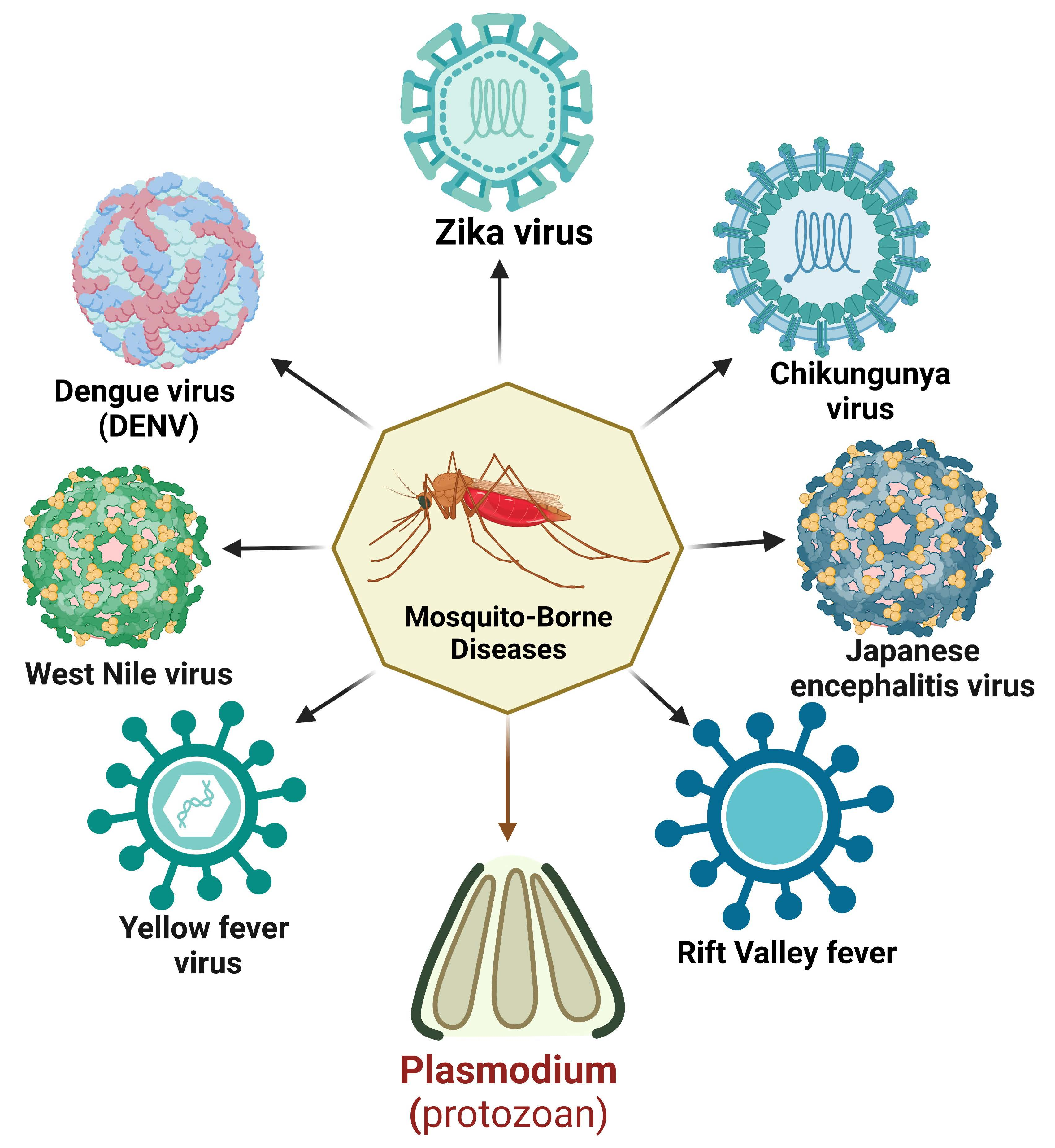
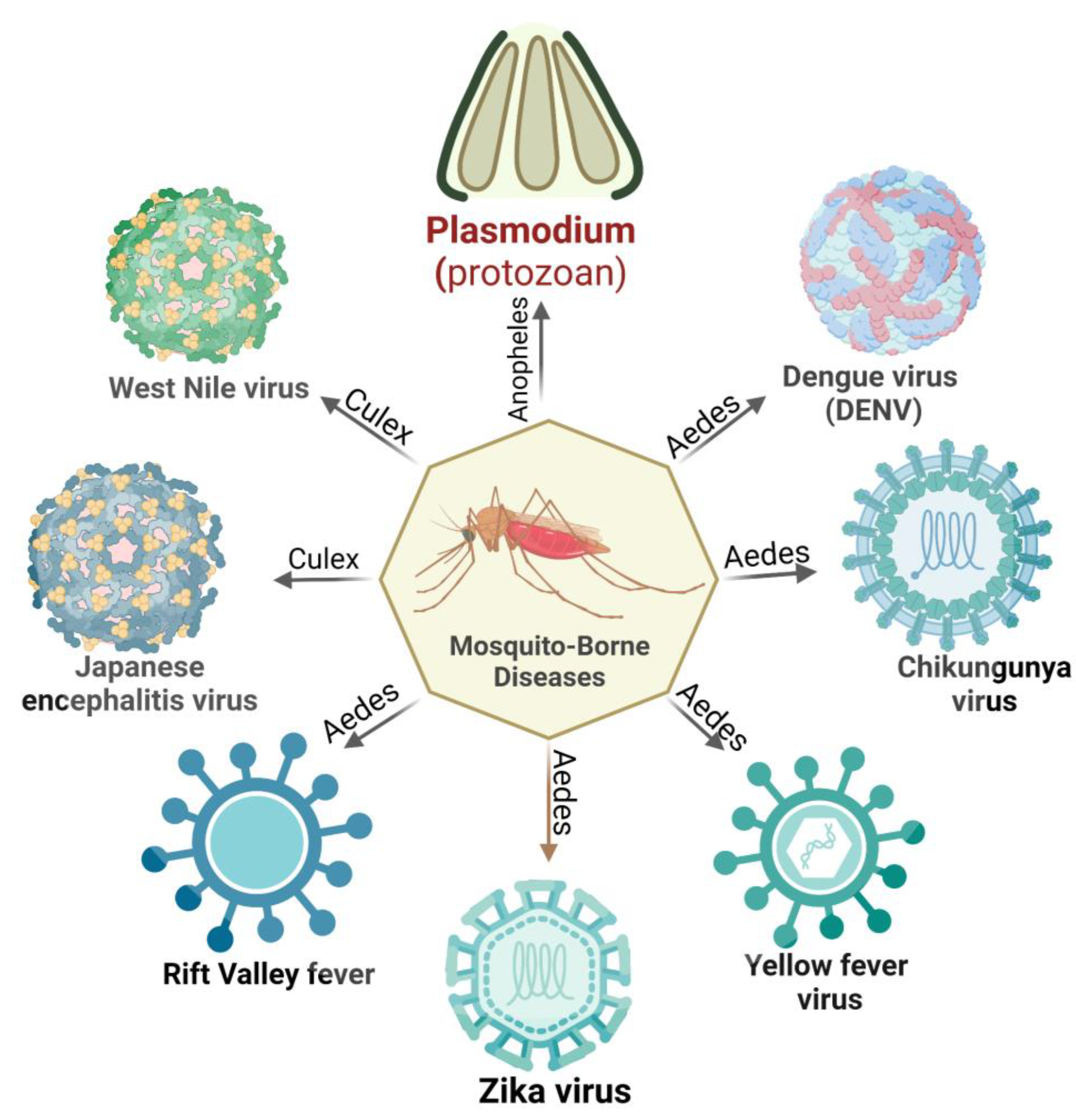
:
1. Introduction
2. Immune Evasion in Mosquitoes
3. Immune Evasion in the Vertebrate Host
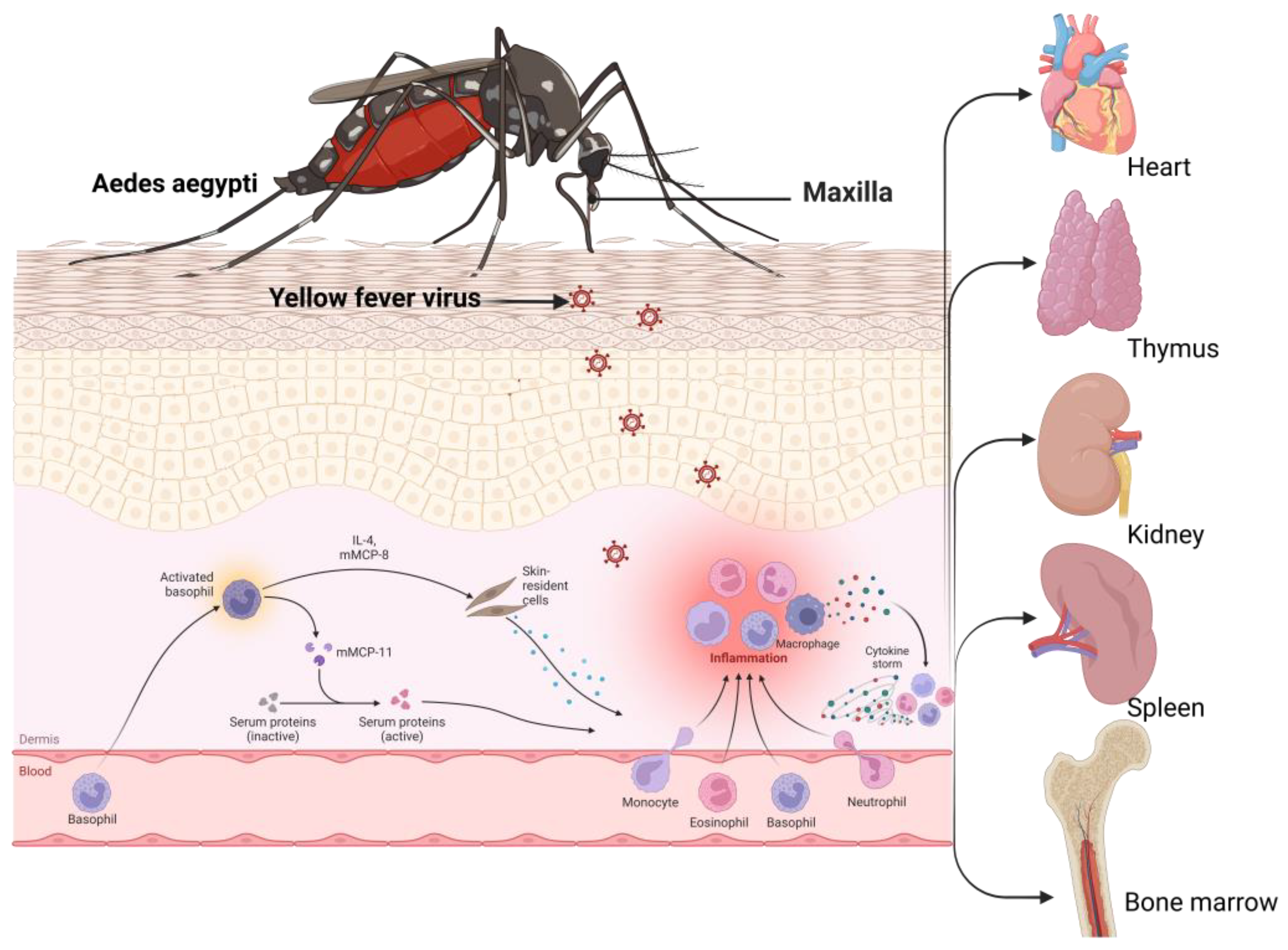
3.1. Immune Response at the Skin Barrier
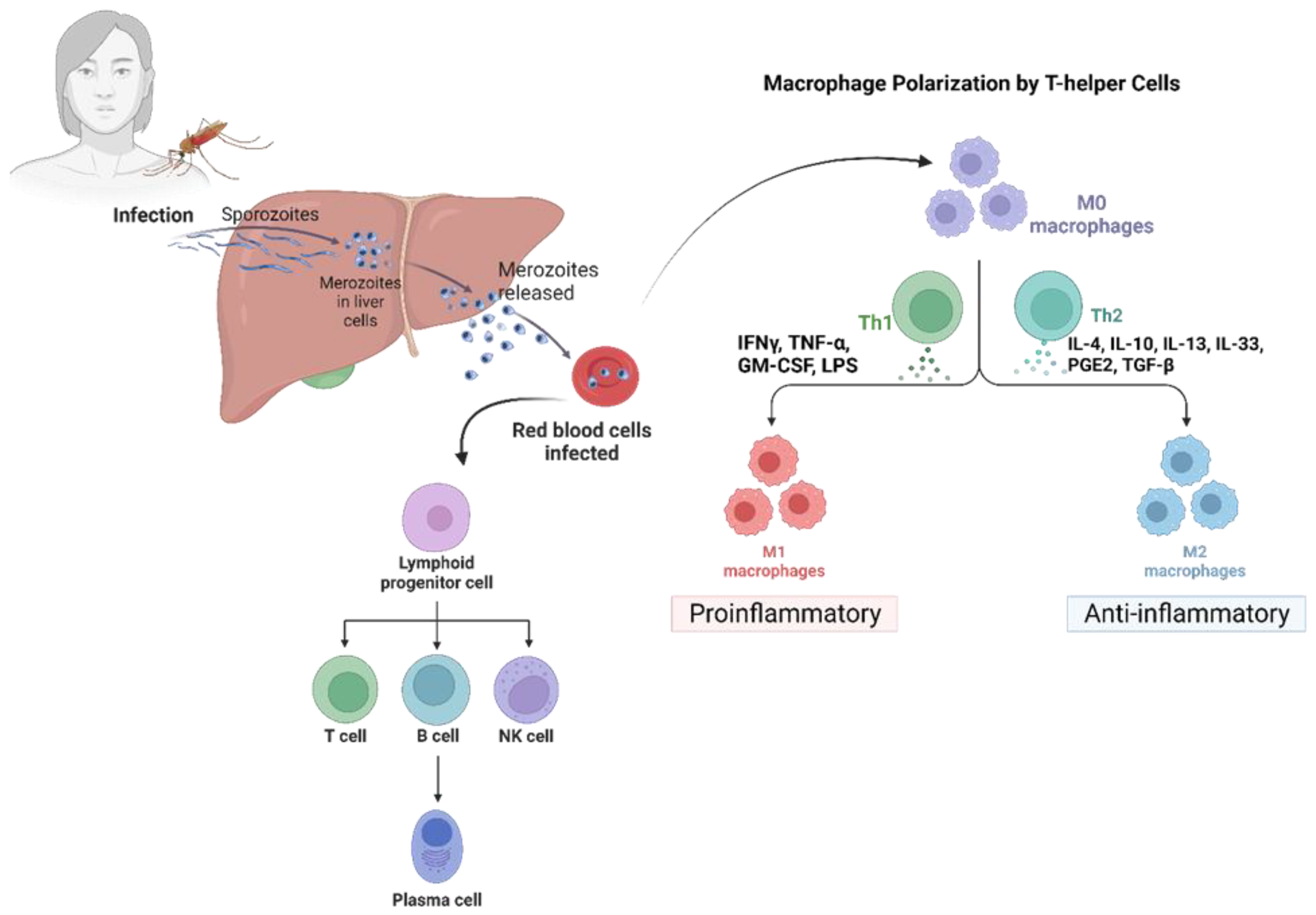
3.2. Immune Evasion Strategies by Plasmodium Parasites
3.2.1. Immune Evasion Strategies at the Liver Stage
3.2.2. Immune Evasion in the Hepatocyte Stage
3.2.3. Immune Evasion Strategies at the Pre-Erythrocytic Stage
3.2.4. Immune Evasion Strategies at the Erythrocytic Stage
3.2.5. Antigenic Variation on the Surface of iRBCs
3.2.6. Cytoadherence and Sequestration as an Immune Evasion Strategy
3.2.7. Rosetting as an Immune Evasion Strategy
3.2.8. Malaria and Its Association with Other Disease Outcomes
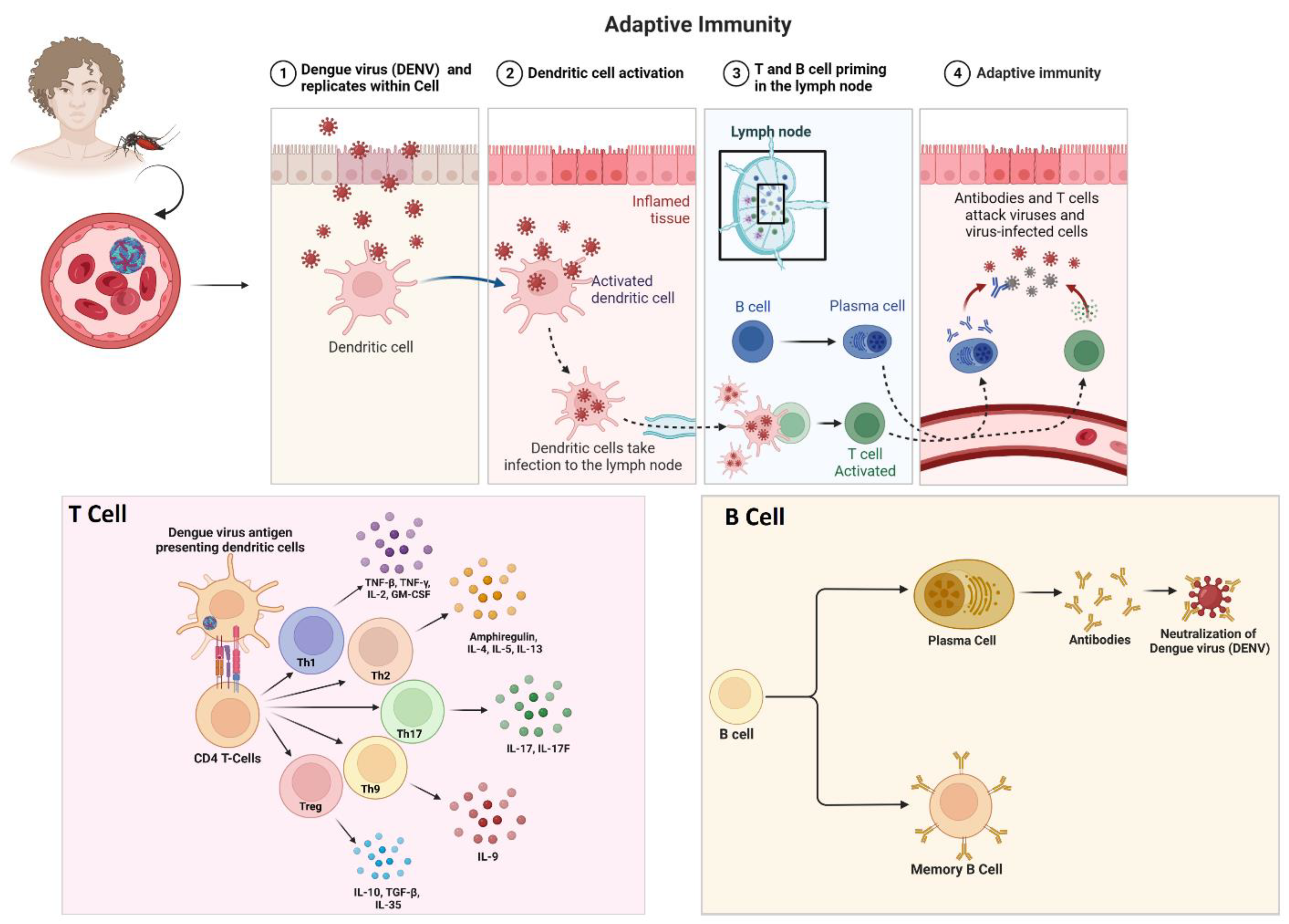
3.3. Dengue Virus and Associated Immune Evasion Strategies
3.3.1. Viral Sensing by the Host’s Immune System
3.3.2. Immune Evasion Strategies of Dengue Virus Strains
3.3.3. Countermeasures to Hijack RLR Signaling
3.3.4. Countermeasures to Hijack the cGAS/STING Pathway and IFN Signaling
3.3.5. Role of microRNA in DENV Pathogenesis
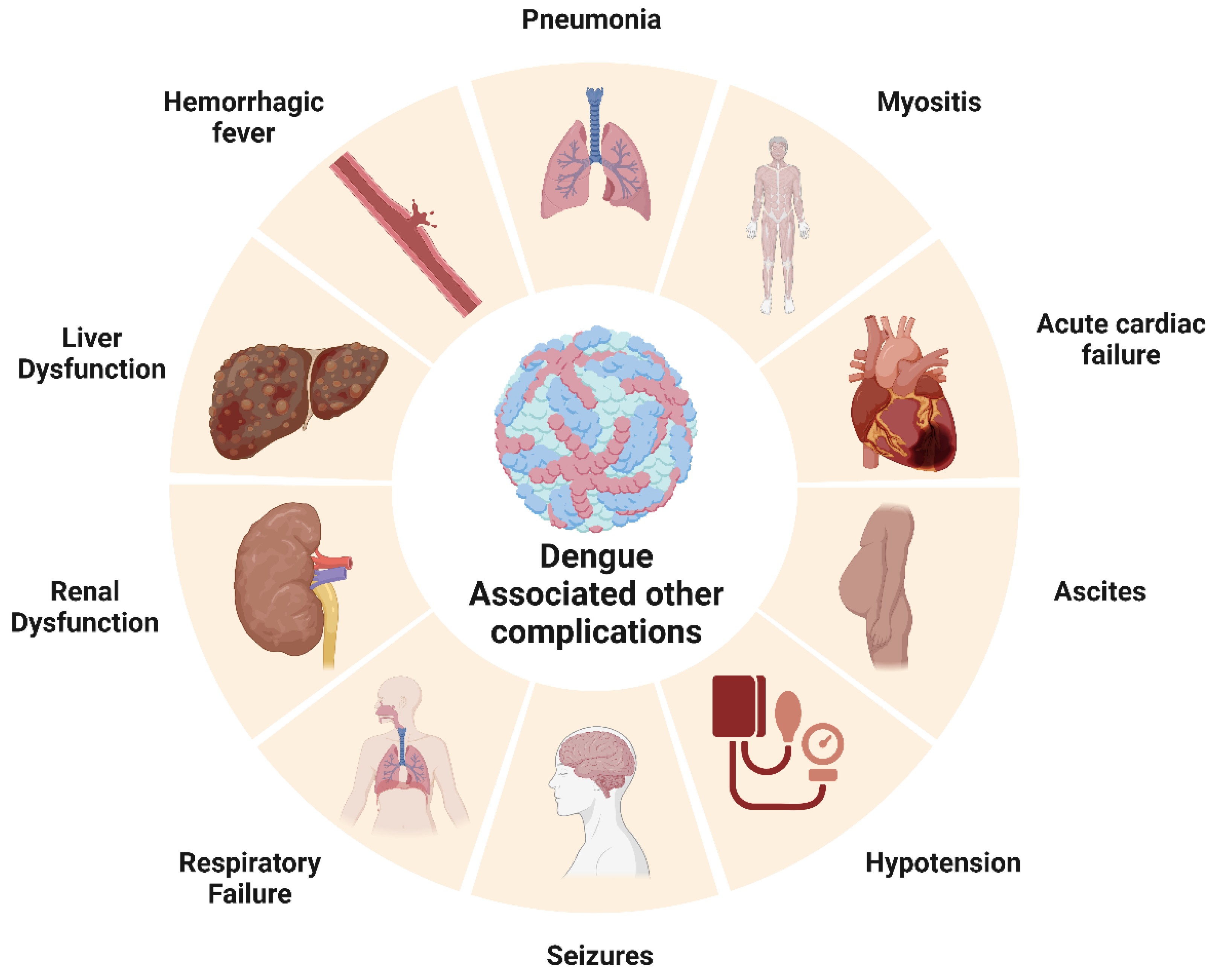
3.3.6. Dengue Fever and Its Association with Other Disease Outcomes
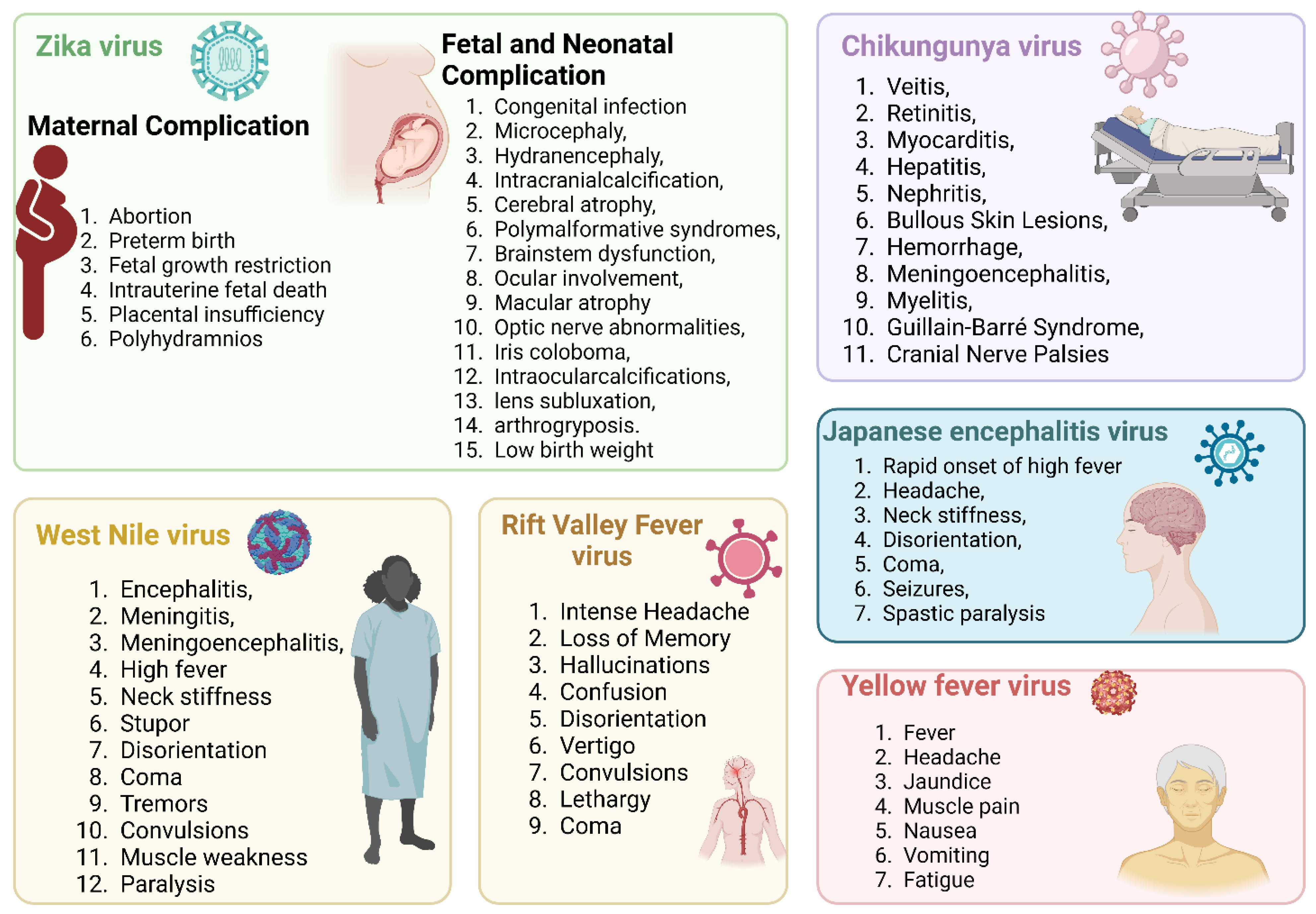
3.4. Zika Virus and Associated Immune Evasion Strategies
3.4.1. Hijacking the RNA Interference (RNAi) Pathway
3.4.2. Zika Virus Escapes NK Cell and DC Detection
3.4.3. Immune Evasion Strategies for Various Zika Virus Strains
3.4.4. Zika Virus Infection and Its Association with Other Disease Outcomes
3.5. Chikungunya Virus and Associated Immune Evasion Strategies
Immune Evasion Strategies of Various Chikungunya Virus Strains
3.6. Yellow Fever Virus and Associated Immune Evasion Strategies
Immune Evasion by YFV Strains
3.7. Rift Valley Fever Virus and Associated Immune Evasion Strategies
3.7.1. The Role of RVFV Non-Structural Proteins in Immune Evasion
3.7.2. Immune Evasion by Different RVFV Strains
3.8. Japanese Encephalitis Virus and Associated Immune Evasion Strategies
Immune Evasion Strategies of JEV Strains
3.9. West Nile Viruses and Associated Immune Evasion Strategies
Immune Evasion Strategies by WNV Strains

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mosquito-Borne Diseases | Year | Location | Type of Study | Method of Assessment | Disease Manifestation | Reference |
|---|---|---|---|---|---|---|
| Chikungunya | 2005–2006 | Réunion | Prospective study | Fulfilled International Encephalitis Consortium criteria for encephalitis | Perinatal encephalopathy | [317] |
| Chikungunya | 2005–2006 | Mayotte | Prospective study | Unknown | Perinatal encephalopathy | [318] |
| Chikungunya | 2005–2006 | Réunion | Prospective observational study | Seizures; EEG consistent with encephalitis | Perinatal encephalopathy | [319] |
| Chikungunya | 2010 | India | Prospective study | Altered sensorium, apnoeic seizures | Perinatal encephalopathy | [320] |
| Chikungunya | 2014–2015 | Colombia | Case series study | EEG | Perinatal encephalopathy | [321] |
| Chikungunya | 2014–2015 | Colombia | Prospective study | EEG | Perinatal encephalopathy | [322] |
| Chikungunya | 2015 | Brazil | Case study | Seizures; abnormal brain MRI | Perinatal encephalopathy | [323] |
| Chikungunya | 2015 | Honduras | Retrospective study | Unknown | Perinatal encephalopathy | [324] |
| Chikungunya | 2016 | India | Case study | Dizygotic twins; both had seizures, required ventilation, thrombocytopenia; abnormal brain MRI | Perinatal encephalopathy | [325] |
| Chikungunya | 2016 | Brazil | Case study | Prostration, lethargy, seizures, required ventilation, thrombocytopenia; abnormal brain MRI and EEG | Perinatal encephalopathy | [326] |
| Chikungunya | 2005–2006 | Réunion | Retrospective study | DIC, transient scattered parenchymal petechiae, cerebellar hematoma | Perinatal brain hemorrhage | [327] |
| Chikungunya | 2005–2006 | Réunion | Retrospective descriptive study | Unknown | Perinatal brain hemorrhage | [328] |
| Chikungunya | 2015 | Brazil | Case study | Intraventricular bleeding (cranial US), lethargy | Perinatal brain hemorrhage | [329] |
| Chikungunya | 2005–2006 | Réunion | Case study | Seizures; hypotonia | Perinatal other | [328] |
| Chikungunya virus | 2008 | France | Case study | ECG and cardiac MRI | Myopericarditis, pericardial effusion | [330] |
| CHIKV infection | 2015 | Honduras | Retrospective study | CSF analysis, EEG, CAT (brain) scan, MRI (brain) | Meningoencephalitis, seizures. CSF analysis shows enhanced leukocyte presence. | [324] |
| CHIKV infection | Not specified | India | Case study | Neurologic examination, sensory examination, electrophysiological studies | Global areflexia and quadriparesis. Guillain–Barre syndrome | [331] |
| CHIKV infection | 2020 | India | Case study | MRI, nerve conduction studies | Sensorimotor axonopathy, myelopathy. | [332] |
| Dengue | 1987 to 1998 | Thailand | Observational study | MRI and autopsy | Seizure, mental confusion, nuchal rigidity, spasticity of limbs, positive clonus, hyponatremia, abnormal liver enzymes and CSF pleocytosis, hemiplegia and positive Kernig | [333] |
| Dengue | 2017 | Brazil | Case study | Brain MRI | Loss of vision, generalized myalgia, severe headache, retro-ocular pain, and cutaneous rash | [206] |
| Dengue | 1997–1999 | Vietnam | Case–control study | MRI and ultrasound | Cerebral edema, seizures, hemiplegia, hemorrhage, hyponatremia, hepatic failure, and microcapillary | [205] |
| Dengue | 2019 | India | Case study | MRI (brain), cerebrospinal fluid (CSF) examination | Bradyphonia, unclear speech, gait instability, cogwheel rigidity, mask-like facies and stooped posture while walking | [204] |
| Dengue | 2017 | Sri Lanka | Case study | Electroencephalogram and cerebrospinal fluid (CSF) examination | Encephalitis | [334] |
| Dengue | 2019 | India | Prospective study | Echocardiogram, ECG | Asymptomatic sinus bradycardia, symptomatic bradyarrhythmias, left ventricular systolic dysfunction (ejection fraction 35–45%), pericardial effusion, atrial fibrillation | [335] |
| Dengue | 1998 | India | Cohort study | Echocardiogram, ECG | Ejection fraction, global hypokinesia, ST and T changes in the ECG | [336] |
| Dengue | 1998 | India | Observational study | Echocardiogram | Sinus bradycardia | [337] |
| Dengue | 2007 | Sri Lanka | Cohort study | Echocardiogram, ECG | T inversion, ST depression and bundle branch blocks, hypotension and tachycardia and bradycardia, suggestive of significant cardiac dysfunction | [338] |
| Dengue | 2012 | Brazil | Case series study | Echocardiogram, ECG | Ileo-femoral deep vein thrombosis, pulmonary thromboembolism, mesenteric vein thrombosis | [339] |
| Dengue | 2014 | México | Case report | Echocardiogram, ECG | Myocarditis characterised by: S3 gallop rhythm, generalized lung rales and shock; ECG showed sinus tachycardia, ST depression in V1-V3, and ST elevation in a VR and aVL | [340] |
| Dengue | 2018 | Sri Lanka | Case series | CT angiography, ECG | Tachycardia in three cases. Myocarditis confirmed by troponin estimation and echocardiogram in one case, and in the other two, this was also confirmed by histopathology | [341] |
| Dengue | 2013 | Singapore | Case reports | Echocardiogram, ECG | Myocarditis | [342] |
| Dengue | 2015 | Vietnam | Case report | Echocardiogram, ECG | Acute cardiac failure | [343] |
| Dengue | 2008 | India | Case report | Echocardiogram, ECG | Myocarditis, Takotsubo cardiomyopathy, ECG showed sinus bradycardia | [344] |
| Dengue | 2016 | Taiwan | Case report | Echocardiogram, cardiac biomarkers | Elevated cardiac enzymes | [345] |
| Dengue | 2015 | Singapore | Case report | Echocardiogram, ECG | Acute myocardial infarction, elevated serum troponin I levels | [346] |
| Dengue | 2011 | France | Case report | Echocardiogram, ECG | Acute pericarditis, Consulting with acute chest pain; ECG revealed negative anterolateral T waves with long QT segment | [347] |
| Dengue | 2011 | Thailand | Prospective study | Holtrer | Sinus pause, first-degree and Mobitz type I second-degree atrioventricular block (Wenckebach) and atrial and ventricular ectopic beats | [196] |
| Dengue | 2016 | India | Case report | CT angiography, ECG | Repeated symptomatic episodes of a high-degree atrioventricular block with ventricular asystole | [348] |
| Dengue | 2015 | Sri Lanka | Case report | Echocardiogram, ECG | Bradycardia and heart block with atrioventricular dissociation | [349] |
| Dengue | 2013 | India | Case series | Echocardiography | Sinus bradycardia, echocardiography showed a decreased ejection fraction | [350] |
| Dengue | 2000 | Thailand | Case reports | CT angiography, ECG | Morbitz type I second-degree atrioventricular block | [351] |
| Dengue | 2004 | Thailand | Case report | Echocardiography and ECG | Myocarditis with bradycardia | [351] |
| Dengue | 2010 | India | Case report | Echocardiography and ECG | Sino-atrial block and atrioventricular dissociation, atrial fibrillation, atrial fibrillation | [352] |
| Dengue | 2009 | Malaysia | Case report | Echocardiography and ECG | Sino-atrial block and atrioventricular dissociation, atrial fibrillation | [353] |
| Dengue | 2003 | Brazil | Case report | Echocardiography and ECG | Sino-atrial block and atrioventricular dissociation, atrial fibrillation | [354] |
| Dengue | 2008 | Saudi Arabia | Cohort study | Not specified | Acute kidney injury | [355] |
| Dengue | 2008 | Taiwan | Retrospective study | eGFR estimation | Acute kidney injury | [356] |
| Dengue | 2009 | Taiwan | Retrospective study | SCr > 2 mg/dL | Acute kidney injury | [357] |
| Dengue | 2010 | Thailand | Retrospective study | SCr > 2 mg/dL | Acute kidney injury | [358] |
| Dengue | 2011 | India | Prospective study | RIFLE | Acute kidney injury | [359] |
| Dengue | 2012 | Pakistan | Case series study | AKIN | Acute kidney injury | [360] |
| Dengue | 2012 | Taiwan | Retrospective case–control study | SCr increase ≥ 0.5 mg/dL | Acute kidney injury | [361] |
| Dengue | 2012 | India | Prospective study | AKIN | Acute kidney injury | [362] |
| Dengue | 2015 | Taiwan | Retrospective study | AKIN | Acute kidney injury | [363] |
| Dengue | 2015 | Malaysia | Retrospective study | AKIN | Acute kidney injury | [364] |
| Dengue | 2016 | India | Cohort study | KDIGO | Acute kidney injury | [208] |
| Dengue | 2017 | Taiwan | Retrospective case–control study | SCr > 2 mg/dL | Acute kidney injury | [365] |
| Dengue | 2017 | India | Observational study | SCr > 1.2 mg/dL | Acute kidney injury | [366] |
| Dengue | 2018 | India | Retrospective study | eGFR < 60 mL/min/1.73 m2 | Acute kidney injury | [367] |
| Dengue | 2018 | Taiwan | Retrospective study | KDIGO | Acute kidney injury | [368] |
| Dengue | 2018 | Malaysia | Follow-up prospective cohort | AKIN | Acute kidney injury | [369] |
| Dengue | 2018 | India | Observational prospective study | AKIN | Acute kidney injury | [370] |
| Dengue | 2019 | Thailand | Retrospective single-center study | KDIGO | Acute kidney injury | [371] |
| Dengue | 2019 | India | Retrospective single-center study | Not specified | Acute kidney injury | [372] |
| Dengue | 2019 | India | Retrospective study | AKIN | Acute kidney injury | [373] |
| Japanese encephalitis | 2014 | China | Case study | EEG, MRI, electromyography, and CSF analysis | Guillain–Barre syndrome | [374] |
| Japanese encephalitis | 2012 | India | Case study | MRI (cervico-thoracic spine) | Acute transverse myelitis | [375] |
| Malaria | 2014 | India | Observational study | Neurological and neuropsychiatric evaluation by MSME (Mini-Mental Status Examination) and BRPS (Brief Psychiatric Rating Scale) | Cerebellar ataxia, psychosis, convolution | [376] |
| Malaria | 2011 | Sierra Leone | Case study | MRI and autoimmune screen | Extensive abnormal high signal and swelling in brainstem and spinal cord | [377] |
| Malaria | 1965–1994, 2010–2015 | Ghana and Uganda | Case–control study | Multiplex Luminex bead assay, histological and cytological investigations | Burkitt Lymphoma | [378] |
| Malaria | 2005–2010 | Malawi | Case–control study | Histological and cytological laboratory investigations | Burkitt Lymphoma | [379] |
| Malaria | 2011–2015 | Uganda | Case–control study | Histological and cytological laboratory investigations | Burkitt Lymphoma | [380] |
| Malaria | 2010–2016 | Uganda, Tanzania, and Kenya | Case–control study | Histological and cytological laboratory investigations | Burkitt Lymphoma | [380] |
| Malaria | 1987–2015 | Sweden | Cohort study | Histological and cytological laboratory investigations | Burkitt Lymphoma | [381] |
| Malaria | 2017 | South Korea | Case Study | Brain MRI | Parkinsonian features | [382] |
| Malaria | 2015 | India | Case study | CT scan of the brain, MRI, and electroencephalography | Cognitive dysfunction, calcification (lesion) in the tempo-parietal region, temporal and hippocampal hyperintensities, microhaemorrhages | [383] |
| Malaria | 1999 | India | Case study | Neurological (nerve conduction) examination | Guillain–Barre syndrome | [384] |
| West Nile Virus | 2022 | USA | Case study | ECG, transthoracic echocardiogram (TTE), X-ray study | Reduced LV systolic | [385] |
| West Nile Virus | 2022 | USA | Case study | TTE and X-ray | Left ventricular hypokinesis | [386] |
| West Nile virus (WNV) | 2016 | USA | Prospective cohort study | Biomicroscopy | Retinopathy and Neurological and Neurocognitive Sequelae | [387] |
| West Nile virus (WNV) | 2002–2004 | USA | Prospective cohort study | ELISA and anti-WNV IgM antibody detection | Encephalitis/meningoencephalitis/encephalomyelitis (WNE) | [388] |
| Yellow fever virus | 2016 | Portugal | Case study | CT scan; CSF investigation | Acute onset expression aphasia, agraphia, dyscalculia | [389] |
| Yellow fever virus | 2020 | Brazil | Experimental study | Histological and cytological laboratory investigations | Hepatic injury | [390] |
| ZIKV infection | 2016 | France | Case study | MRI (Spinal), electromyography, and CSF examination | Acute myelitis and lesions of the cervical and thoracic spinal cord | [391] |
| ZIKV infection | 2017 | Brazil | Case study | Brain MRI, electroencephalography, and autopsy | Meningoencephalitis. | [392] |
| ZIKV infection | 2015 | Colombia | Observational study | Electromyography and nerve conduction studies, CSF examination | Guillain–Barre syndrome (inflammatory demyelinating polyneuropathy) | [393] |
| ZIKV infection | 2017 | Colombia | Case–control study | MRI and electrophysiological examination | Encephalitis, peripheral facial palsy, thoracolumbosacral myelopathy, and transverse myelitis | [394] |
| ZIKV infection | 2017 | Brazil | Cross-sectional study | MRI (brain) and EEG examination | Epilepsy tended to be early and refractory | [395] |
4. Immunological Aspects Facilitating Therapeutic Development
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Ethics and Vector-Borne Diseases: WHO Guidance; World Health Organization: Geneva, Switzerland, 2020. [Google Scholar]
- Tolle, M.A. Mosquito-borne Diseases. Curr. Probl. Pediatr. Adolesc. Health Care 2009, 39, 97–140. [Google Scholar] [CrossRef]
- Oakley, M.S.; Gerald, N.; McCutchan, T.F.; Aravind, L.; Kumar, S. Clinical and molecular aspects of malaria fever. Trends Parasitol. 2011, 27, 442–449. [Google Scholar] [CrossRef]
- Collins, W.E.; Jeffery, G.M. Plasmodium malariae: Parasite and disease. Clin. Microbiol. Rev. 2007, 20, 579–592. [Google Scholar] [CrossRef]
- Hwang, J.; Cullen, K.A.; Kachur, S.P.; Arguin, P.M.; Baird, J.K. Severe morbidity and mortality risk from malaria in the United States, 1985–2011. Open Forum Infect. Dis. 2014, 1, ofu034. [Google Scholar] [CrossRef]
- Wilder-Smith, A.; Lindsay, S.W.; Scott, T.W.; Ooi, E.E.; Gubler, D.J.; Das, P. The Lancet Commission on dengue and other Aedes-transmitted viral diseases. Lancet 2020, 395, 1890–1891. [Google Scholar] [CrossRef]
- Tsetsarkin, K.A.; Chen, R.; Sherman, M.B.; Weaver, S.C. Chikungunya virus: Evolution and genetic determinants of emergence. Curr. Opin. Virol. 2011, 1, 310–317. [Google Scholar] [CrossRef]
- Couderc, T.; Chretien, F.; Schilte, C.; Disson, O.; Brigitte, M.; Guivel-Benhassine, F.; Touret, Y.; Barau, G.; Cayet, N.; Schuffenecker, I.; et al. A mouse model for Chikungunya: Young age and inefficient type-I interferon signaling are risk factors for severe disease. PLoS Pathog. 2008, 4, e29. [Google Scholar] [CrossRef]
- Monath, T.P.; Vasconcelos, P.F.C. Yellow fever. J. Clin. Virol. 2015, 64, 160–173. [Google Scholar] [CrossRef]
- Douam, F.; Ploss, A. Yellow Fever Virus: Knowledge Gaps Impeding the Fight Against an Old Foe. Trends Microbiol. 2018, 26, 913–928. [Google Scholar] [CrossRef]
- Quaresma, J.A.S.; Pagliari, C.; Medeiros, D.B.A.; Duarte, M.I.S.; Vasconcelos, P.F.C. Immunity and immune response, pathology and pathologic changes: Progress and challenges in the immunopathology of yellow fever. Rev. Med. Virol. 2013, 23, 305–318. [Google Scholar] [CrossRef]
- Lequime, S.; Lambrechts, L. Vertical transmission of arboviruses in mosquitoes: A historical perspective. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2014, 28, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Davies, F. Rift Valley fever. Dis. Cattle Trop. Econ. Zoonotic Relev. 1981, 23, 153–165. [Google Scholar]
- Van den Eynde, C.; Sohier, C.; Matthijs, S.; De Regge, N. Japanese Encephalitis Virus Interaction with Mosquitoes: A Review of Vector Competence, Vector Capacity and Mosquito Immunity. Pathogens 2022, 11, 317. [Google Scholar] [CrossRef]
- Girard, Y.A.; Klingler, K.A.; Higgs, S. West Nile virus dissemination and tissue tropisms in orally infected Culex pipiens quinquefasciatus. Vector Borne Zoonotic Dis. 2004, 4, 109–122. [Google Scholar] [CrossRef]
- Samuel, M.A.; Diamond, M.S. Pathogenesis of West Nile Virus infection: A balance between virulence, innate and adaptive immunity, and viral evasion. J. Virol. 2006, 80, 9349–9360. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, J.; Du, S.; Shan, C.; Nie, K.; Zhang, R.; Li, X.F.; Zhang, R.; Wang, T.; Qin, C.F.; et al. Evolutionary enhancement of Zika virus infectivity in Aedes aegypti mosquitoes. Nature 2017, 545, 482–486. [Google Scholar] [CrossRef]
- Boyer, S.; Calvez, E.; Chouin-Carneiro, T.; Diallo, D.; Failloux, A.B. An overview of mosquito vectors of Zika virus. Microbes Infect. 2018, 20, 646–660. [Google Scholar] [CrossRef]
- Hanley, K.A.; Monath, T.P.; Weaver, S.C.; Rossi, S.L.; Richman, R.L.; Vasilakis, N. Fever versus fever: The role of host and vector susceptibility and interspecific competition in shaping the current and future distributions of the sylvatic cycles of dengue virus and yellow fever virus. Infect. Genet. Evol. 2013, 19, 292–311. [Google Scholar] [CrossRef]
- Caron, M.; Paupy, C.; Grard, G.; Becquart, P.; Mombo, I.; Nso, B.B.; Kassa Kassa, F.; Nkoghe, D.; Leroy, E.M. Recent introduction and rapid dissemination of Chikungunya virus and Dengue virus serotype 2 associated with human and mosquito coinfections in Gabon, central Africa. Clin. Infect. Dis. 2012, 55, e45–e53. [Google Scholar] [CrossRef]
- Hayes, E.B.; Komar, N.; Nasci, R.S.; Montgomery, S.P.; O’Leary, D.R.; Campbell, G.L. Epidemiology and transmission dynamics of West Nile virus disease. Emerg. Infect. Dis. 2005, 11, 1167–1173. [Google Scholar] [CrossRef]
- Shaw, W.R.; Catteruccia, F. Vector biology meets disease control: Using basic research to fight vector-borne diseases. Nat. Microbiol. 2019, 4, 20–34. [Google Scholar] [CrossRef] [PubMed]
- Salazar, M.I.; Richardson, J.H.; Sánchez-Vargas, I.; Olson, K.E.; Beaty, B.J. Dengue virus type 2: Replication and tropisms in orally infected Aedes aegypti mosquitoes. BMC Microbiol. 2007, 7, 9. [Google Scholar] [CrossRef]
- Colpitts, T.M.; Cox, J.; Vanlandingham, D.L.; Feitosa, F.M.; Cheng, G.; Kurscheid, S.; Wang, P.; Krishnan, M.N.; Higgs, S.; Fikrig, E. Alterations in the Aedes aegypti Transcriptome during Infection with West Nile, Dengue and Yellow Fever Viruses. PLoS Pathog. 2011, 7, e1002189. [Google Scholar] [CrossRef] [PubMed]
- Xi, Z.; Ramirez, J.L.; Dimopoulos, G. The Aedes aegypti Toll Pathway Controls Dengue Virus Infection. PLoS Pathog. 2008, 4, e1000098. [Google Scholar] [CrossRef]
- Dong, Y.; Das, S.; Cirimotich, C.; Souza-Neto, J.A.; McLean, K.J.; Dimopoulos, G. Engineered anopheles immunity to Plasmodium infection. PLoS Pathog. 2011, 7, e1002458. [Google Scholar] [CrossRef]
- Clayton, A.M.; Dong, Y.; Dimopoulos, G. The Anopheles innate immune system in the defense against malaria infection. J. Innate Immun. 2014, 6, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Ramphul, U.N.; Garver, L.S.; Molina-Cruz, A.; Canepa, G.E.; Barillas-Mury, C. Plasmodium falciparum evades mosquito immunity by disrupting JNK-mediated apoptosis of invaded midgut cells. Proc. Natl. Acad. Sci. USA 2015, 112, 1273–1280. [Google Scholar] [CrossRef]
- Molina-Cruz, A.; Garver, L.S.; Alabaster, A.; Bangiolo, L.; Haile, A.; Winikor, J.; Ortega, C.; van Schaijk, B.C.; Sauerwein, R.W.; Taylor-Salmon, E.; et al. The human malaria parasite Pfs47 gene mediates evasion of the mosquito immune system. Science 2013, 340, 984–987. [Google Scholar] [CrossRef]
- Sim, S.; Jupatanakul, N.; Ramirez, J.L.; Kang, S.; Romero-Vivas, C.M.; Mohammed, H.; Dimopoulos, G. Transcriptomic Profiling of Diverse Aedes aegypti Strains Reveals Increased Basal-level Immune Activation in Dengue Virus-refractory Populations and Identifies Novel Virus-vector Molecular Interactions. PLoS Negl. Trop. Dis. 2013, 7, e2295. [Google Scholar] [CrossRef]
- Behura, S.K.; Gomez-Machorro, C.; Harker, B.W.; deBruyn, B.; Lovin, D.D.; Hemme, R.R.; Mori, A.; Romero-Severson, J.; Severson, D.W. Global Cross-Talk of Genes of the Mosquito Aedes aegypti in Response to Dengue Virus Infection. PLoS Negl. Trop. Dis. 2011, 5, e1385. [Google Scholar] [CrossRef]
- Sánchez-Vargas, I.; Scott, J.C.; Poole-Smith, B.K.; Franz, A.W.E.; Barbosa-Solomieu, V.; Wilusz, J.; Olson, K.E.; Blair, C.D. Dengue Virus Type 2 Infections of Aedes aegypti Are Modulated by the Mosquito’s RNA Interference Pathway. PLoS Pathog. 2009, 5, e1000299. [Google Scholar] [CrossRef] [PubMed]
- Paradkar, P.N.; Trinidad, L.; Voysey, R.; Duchemin, J.-B.; Walker, P.J. Secreted Vago restricts West Nile virus infection in Culex mosquito cells by activating the Jak-STAT pathway. Proc. Natl. Acad. Sci. USA 2012, 109, 18915–18920. [Google Scholar] [CrossRef] [PubMed]
- Briant, L.; Desprès, P.; Choumet, V.; Missé, D. Role of skin immune cells on the host susceptibility to mosquito-borne viruses. Virology 2014, 464–465, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Sinnis, P.; Zavala, F. The skin: Where malaria infection and the host immune response begin. Semin. Immunopathol. 2012, 34, 787–792. [Google Scholar] [CrossRef] [PubMed]
- Welte, T.; Reagan, K.; Fang, H.; Machain-Williams, C.; Zheng, X.; Mendell, N.; Chang, G.J.; Wu, P.; Blair, C.D.; Wang, T. Toll-like receptor 7-induced immune response to cutaneous West Nile virus infection. J. Gen. Virol. 2009, 90, 2660–2668. [Google Scholar] [CrossRef]
- Chen, Y.; Maguire, T.; Hileman, R.E.; Fromm, J.R.; Esko, J.D.; Linhardt, R.J.; Marks, R.M. Dengue virus infectivity depends on envelope protein binding to target cell heparan sulfate. Nat. Med. 1997, 3, 866–871. [Google Scholar] [CrossRef]
- Cleary, E.; Hetzel, M.W.; Clements, A.C.A. A review of malaria epidemiology and control in Papua New Guinea 1900 to 2021: Progress made and future directions. Front. Epidemiol. 2022, 2, 795. [Google Scholar] [CrossRef]
- Gomes, P.S.; Bhardwaj, J.; Rivera-Correa, J.; Freire-De-Lima, C.G.; Morrot, A. Immune Escape Strategies of Malaria Parasites. Front. Microbiol. 2016, 7, 1617. [Google Scholar] [CrossRef]
- Miller, L.H.; Baruch, D.I.; Marsh, K.; Doumbo, O.K. The pathogenic basis of malaria. Nature 2002, 415, 673–679. [Google Scholar] [CrossRef]
- Yam, X.Y.; Preiser, P.R. Host immune evasion strategies of malaria blood stage parasite. Mol. BioSyst. 2017, 13, 2498–2508. [Google Scholar] [CrossRef]
- Phillips, M.A.; Burrows, J.N.; Manyando, C.; van Huijsduijnen, R.H.; Van Voorhis, W.C.; Wells, T.N.C. Malaria. Nat. Rev. Dis. Prim. 2017, 3, 17050. [Google Scholar] [CrossRef]
- Buffet, P.A.; Safeukui, I.; Deplaine, G.; Brousse, V.; Prendki, V.; Thellier, M.; Turner, G.D.; Mercereau-Puijalon, O. The pathogenesis of Plasmodium falciparum malaria in humans: Insights from splenic physiology. Blood 2011, 117, 381–392. [Google Scholar] [CrossRef]
- White, N.J. Determinants of relapse periodicity in Plasmodium vivax malaria. Malar. J. 2011, 10, 297. [Google Scholar] [CrossRef]
- Price, R.N.; Tjitra, E.; Guerra, C.A.; Yeung, S.; White, N.J.; Anstey, N.M. Vivax malaria: Neglected and not benign. Am. J. Trop. Med. Hyg. 2007, 77, 79–87. [Google Scholar] [CrossRef]
- Lopez, C.; Yepes-Perez, Y.; Hincapie-Escobar, N.; Diaz-Arevalo, D.; Patarroyo, M.A. What Is Known about the Immune Response Induced by Plasmodium vivax Malaria Vaccine Candidates? Front. Immunol. 2017, 8, 126. [Google Scholar] [CrossRef]
- Popa, G.L.; Popa, M.I. Recent Advances in Understanding the Inflammatory Response in Malaria: A Review of the Dual Role of Cytokines. J. Immunol. Res. 2021, 2021, 7785180. [Google Scholar] [CrossRef] [PubMed]
- Funaro, A.; Spagnoli, G.C.; Ausiello, C.M.; Alessio, M.; Roggero, S.; Delia, D.; Zaccolo, M.; Malavasi, F. Involvement of the multilineage CD38 molecule in a unique pathway of cell activation and proliferation. J. Immunol. 1990, 145, 2390–2396. [Google Scholar] [CrossRef] [PubMed]
- Burel, J.G.; Apte, S.H.; McCarthy, J.S.; Doolan, D.L. Plasmodium vivax but Not Plasmodium falciparum Blood-Stage Infection in Humans Is Associated with the Expansion of a CD8+ T Cell Population with Cytotoxic Potential. PLoS Negl. Trop. Dis. 2016, 10, e0005031. [Google Scholar] [CrossRef]
- Flannery, E.L.; Kangwanrangsan, N.; Chuenchob, V.; Roobsoong, W.; Fishbaugher, M.; Zhou, K.; Billman, Z.P.; Martinson, T.; Olsen, T.M.; Schafer, C.; et al. Plasmodium vivax latent liver infection is characterized by persistent hypnozoites, hypnozoite-derived schizonts, and time-dependent efficacy of primaquine. Mol. Ther. Methods Clin. Dev. 2022, 26, 427–440. [Google Scholar] [CrossRef] [PubMed]
- Renia, L.; Goh, Y.S. Malaria Parasites: The Great Escape. Front. Immunol. 2016, 7, 463. [Google Scholar] [CrossRef]
- Mandala, W.L.; Harawa, V.; Dzinjalamala, F.; Tembo, D. The role of different components of the immune system against Plasmodium falciparum malaria: Possible contribution towards malaria vaccine development. Mol. Biochem. Parasitol. 2021, 246, 111425. [Google Scholar] [CrossRef]
- Belachew, E.B. Immune Response and Evasion Mechanisms of Plasmodium falciparum Parasites. J. Immunol. Res. 2018, 2018, 6529681. [Google Scholar] [CrossRef]
- Vallejo, A.F.; Read, R.C.; Arevalo-Herrera, M.; Herrera, S.; Elliott, T.; Polak, M.E. Malaria systems immunology: Plasmodium vivax induces tolerance during primary infection through dysregulation of neutrophils and dendritic cells. J. Infect. 2018, 77, 440–447. [Google Scholar] [CrossRef]
- Zheng, H.; Tan, Z.; Xu, W. Immune Evasion Strategies of Pre-Erythrocytic Malaria Parasites. Mediat. Inflamm. 2014, 2014, 362605. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, B.M.; Fidock, D.A.; Kyle, D.E.; Kappe, S.H.I.; Alonso, P.L.; Collins, F.H.; Duffy, P.E. Malaria: Progress, perils, and prospects for eradication. J. Clin. Investig. 2008, 118, 1266–1276. [Google Scholar] [CrossRef] [PubMed]
- Meis, J.F.G.M.; Verhave, J.P.; Jap, P.H.K.; Meuwissen, J.H.E.T. An ultrastructural study on the role of Kupffer cells in the process of infection by Plasmodium berghei sporozoites in rats. Parasitology 2009, 86, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Ikarashi, M.; Nakashima, H.; Kinoshita, M.; Sato, A.; Nakashima, M.; Miyazaki, H.; Nishiyama, K.; Yamamoto, J.; Seki, S. Distinct development and functions of resident and recruited liver Kupffer cells/macrophages. J. Leukoc. Biol. 2013, 94, 1325–1336. [Google Scholar] [CrossRef]
- Steers, N.; Schwenk, R.; Bacon, D.J.; Berenzon, D.; Williams, J.; Krzych, U. The immune status of Kupffer cells profoundly influences their responses to infectious Plasmodium berghei sporozoites. Eur. J. Immunol. 2005, 35, 2335–2346. [Google Scholar] [CrossRef]
- Mota, M.M.; Pradel, G.; Vanderberg, J.P.; Hafalla, J.C.R.; Frevert, U.; Nussenzweig, R.S.; Nussenzweig, V.; Rodríguez, A. Migration of Plasmodium Sporozoites through Cells before Infection. Science 2001, 291, 141–144. [Google Scholar] [CrossRef]
- Carrolo, M.; Giordano, S.; Cabrita-Santos, L.; Corso, S.; Vigário, A.M.; Silva, S.; Leirião, P.; Carapau, D.; Armas-Portela, R.; Comoglio, P.M.; et al. Hepatocyte growth factor and its receptor are required for malaria infection. Nat. Med. 2003, 9, 1363–1369. [Google Scholar] [CrossRef]
- Leirião, P.; Albuquerque, S.S.; Corso, S.; Van Gemert, G.-J.; Sauerwein, R.W.; Rodriguez, A.; Giordano, S.; Mota, M.M. HGF/MET signalling protects Plasmodium-infected host cells from apoptosis. Cell. Microbiol. 2005, 7, 603–609. [Google Scholar] [CrossRef]
- Coppi, A.; Pinzon-Ortiz, C.; Hutter, C.; Sinnis, P. The Plasmodium circumsporozoite protein is proteolytically processed during cell invasion. J. Exp. Med. 2005, 201, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Coppi, A.; Tewari, R.; Bishop, J.R.; Bennett, B.L.; Lawrence, R.; Esko, J.D.; Billker, O.; Sinnis, P. Heparan Sulfate Proteoglycans Provide a Signal to Plasmodium Sporozoites to Stop Migrating and Productively Invade Host Cells. Cell Host Microbe 2007, 2, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.P.; Buscaglia, C.A.; Wang, Q.; Levay, A.; Nussenzweig, D.R.; Walker, J.R.; Winzeler, E.A.; Fujii, H.; Fontoura, B.M.A.; Nussenzweig, V. Plasmodium Circumsporozoite Protein Promotes the Development of the Liver Stages of the Parasite. Cell 2007, 131, 492–504. [Google Scholar] [CrossRef]
- Frevert, U.; Galinski, M.R.; Hügel, F.-U.; Allon, N.; Schreier, H.; Smulevitch, S.; Shakibaei, M.; Clavijo, P. Malaria circumsporozoite protein inhibits protein synthesis in mammalian cells. EMBO J. 1998, 17, 3816–3826. [Google Scholar] [CrossRef]
- Kaushansky, A.; Ye, A.S.; Austin, L.S.; Mikolajczak, S.A.; Vaughan, A.M.; Camargo, N.; Metzger, P.G.; Douglass, A.N.; MacBeath, G.; Kappe, S.H. Suppression of Host p53 Is Critical for Plasmodium Liver-Stage Infection. Cell Rep. 2013, 3, 630–637. [Google Scholar] [CrossRef]
- Prudêncio, M.; Rodriguez, A.; Mota, M.M. The silent path to thousands of merozoites: The Plasmodium liver stage. Nat. Rev. Microbiol. 2006, 4, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Sturm, A.; Amino, R.; van de Sand, C.; Regen, T.; Retzlaff, S.; Rennenberg, A.; Krueger, A.; Pollok, J.-M.; Menard, R.; Heussler, V.T. Manipulation of Host Hepatocytes by the Malaria Parasite for Delivery into Liver Sinusoids. Science 2006, 313, 1287–1290. [Google Scholar] [CrossRef]
- Garg, S.; Agarwal, S.; Kumar, S.; Shams Yazdani, S.; Chitnis, C.E.; Singh, S. Calcium-dependent permeabilization of erythrocytes by a perforin-like protein during egress of malaria parasites. Nat. Commun. 2013, 4, 1736. [Google Scholar] [CrossRef]
- Howard, R.J.; Barnwell, J.W.; Kao, V. Antigenic variation of Plasmodium knowlesi malaria: Identification of the variant antigen on infected erythrocytes. Proc. Natl. Acad. Sci. USA 1983, 80, 4129–4133. [Google Scholar] [CrossRef]
- Su, X.Z.; Heatwole, V.M.; Wertheimer, S.P.; Guinet, F.; Herrfeldt, J.A.; Peterson, D.S.; Ravetch, J.A.; Wellems, T.E. The large diverse gene family var encodes proteins involved in cytoadherence and antigenic variation of Plasmodium falciparum-infected erythrocytes. Cell 1995, 82, 89–100. [Google Scholar] [CrossRef]
- Smith, J.D.; Chitnis, C.E.; Craig, A.G.; Roberts, D.J.; Hudson-Taylor, D.E.; Peterson, D.S.; Pinches, R.; Newbold, C.I.; Miller, L.H. Switches in expression of plasmodium falciparum var genes correlate with changes in antigenic and cytoadherent phenotypes of infected erythrocytes. Cell 1995, 82, 101–110. [Google Scholar] [CrossRef]
- Handunnetti, S.M.; Mendis, K.N.; David, P.H. Antigenic variation of cloned Plasmodium fragile in its natural host Macaca sinica. Sequential appearance of successive variant antigenic types. J. Exp. Med. 1987, 165, 1269–1283. [Google Scholar] [CrossRef]
- Roberts, D.J.; Craig, A.G.; Berendt, A.R.; Pinches, R.; Nash, G.; Marsh, K.; Newbold, C.I. Rapid switching to multiple antigenic and adhesive phenotypes in malaria. Nature 1992, 357, 689–692. [Google Scholar] [CrossRef] [PubMed]
- Baruch, D.I.; Pasloske, B.L.; Singh, H.B.; Bi, X.; Ma, X.C.; Feldman, M.; Taraschi, T.F.; Howard, R.J. Cloning the P. falciparum gene encoding PfEMP1, a malarial variant antigen and adherence receptor on the surface of parasitized human erythrocytes. Cell 1995, 82, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Gardner, M.J.; Hall, N.; Fung, E.; White, O.; Berriman, M.; Hyman, R.W.; Carlton, J.M.; Pain, A.; Nelson, K.E.; Bowman, S.; et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 2002, 419, 498–511. [Google Scholar] [CrossRef]
- Chan, J.-A.; Howell, K.B.; Reiling, L.; Ataide, R.; Mackintosh, C.L.; Fowkes, F.J.I.; Petter, M.; Chesson, J.M.; Langer, C.; Warimwe, G.M.; et al. Targets of antibodies against Plasmodium falciparum–infected erythrocytes in malaria immunity. J. Clin. Investig. 2012, 122, 3227–3238. [Google Scholar] [CrossRef]
- Fernandez, V.; Hommel, M.; Chen, Q.; Hagblom, P.; Wahlgren, M. Small, Clonally Variant Antigens Expressed on the Surface of the Plasmodium falciparum–Infected Erythrocyte Are Encoded by the rif Gene Family and Are the Target of Human Immune Responses. J. Exp. Med. 1999, 190, 1393–1404. [Google Scholar] [CrossRef]
- Kyes, S.A.; Rowe, J.A.; Kriek, N.; Newbold, C.I. Rifins: A second family of clonally variant proteins expressed on the surface of red cells infected with Plasmodium falciparum. Proc. Natl. Acad. Sci. USA 1999, 96, 9333–9338. [Google Scholar] [CrossRef]
- Petter, M.; Haeggström, M.; Khattab, A.; Fernandez, V.; Klinkert, M.-Q.; Wahlgren, M. Variant proteins of the Plasmodium falciparum RIFIN family show distinct subcellular localization and developmental expression patterns. Mol. Biochem. Parasitol. 2007, 156, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, A.; Petter, M.; Tilly, A.-K.; Biller, L.; Uliczka, K.A.; Duffy, M.F.; Tannich, E.; Bruchhaus, I. Temporal Expression and Localization Patterns of Variant Surface Antigens in Clinical Plasmodium falciparum Isolates during Erythrocyte Schizogony. PLoS ONE 2012, 7, e49540. [Google Scholar] [CrossRef]
- Hiller, N.L.; Bhattacharjee, S.; van Ooij, C.; Liolios, K.; Harrison, T.; Lopez-Estraño, C.; Haldar, K. A Host-Targeting Signal in Virulence Proteins Reveals a Secretome in Malarial Infection. Science 2004, 306, 1934–1937. [Google Scholar] [CrossRef]
- Howard, R.J.; Barnwell, J.W.; Rock, E.P.; Neequaye, J.; Ofori-Adjei, D.; Lee Maloy, W.; Lyon, J.A.; Saul, A. Two approximately 300 kilodalton Plasmodium falciparum proteins at the surface membrane of infected erythrocytes. Mol. Biochem. Parasitol. 1988, 27, 207–223. [Google Scholar] [CrossRef] [PubMed]
- Biggs, B.A.; Goozé, L.; Wycherley, K.; Wollish, W.; Southwell, B.; Leech, J.H.; Brown, G.V. Antigenic variation in Plasmodium falciparum. Proc. Natl. Acad. Sci. USA 1991, 88, 9171–9174. [Google Scholar] [CrossRef]
- Bull, P.C.; Abdi, A.I. The role of PfEMP1 as targets of naturally acquired immunity to childhood malaria: Prospects for a vaccine. Parasitology 2016, 143, 171–186. [Google Scholar] [CrossRef] [PubMed]
- Deitsch, K.W.; Hviid, L. Variant surface antigens, virulence genes and the pathogenesis of malaria. Trends Parasitol. 2004, 20, 562–566. [Google Scholar] [CrossRef] [PubMed]
- Waller, K.L.; Cooke, B.M.; Coppel, R.L.; Nunomura, W.; Mohandas, N. Mapping the Binding Domains Involved in the Interaction between the Plasmodium falciparum Knob-associated Histidine-rich Protein (KAHRP) and the Cytoadherence Ligand P. falciparum Erythrocyte Membrane Protein 1 (PfEMP1). J. Biol. Chem. 1999, 274, 23808–23813. [Google Scholar] [CrossRef] [PubMed]
- Turner, L.; Lavstsen, T.; Berger, S.S.; Wang, C.W.; Petersen, J.E.V.; Avril, M.; Brazier, A.J.; Freeth, J.; Jespersen, J.S.; Nielsen, M.A.; et al. Severe malaria is associated with parasite binding to endothelial protein C receptor. Nature 2013, 498, 502–505. [Google Scholar] [CrossRef]
- Howell, D.P.-G.; Levin, E.A.; Springer, A.L.; Kraemer, S.M.; Phippard, D.J.; Schief, W.R.; Smith, J.D. Mapping a common interaction site used by Plasmodium falciparum Duffy binding-like domains to bind diverse host receptors. Mol. Microbiol. 2008, 67, 78–87. [Google Scholar] [CrossRef]
- Fried, M.; Duffy, P.E. Adherence of Plasmodium falciparum to Chondroitin Sulfate A in the Human Placenta. Science 1996, 272, 1502–1504. [Google Scholar] [CrossRef]
- Baruch, D.I.; Gormely, J.A.; Ma, C.; Howard, R.J.; Pasloske, B.L. Plasmodium falciparum erythrocyte membrane protein 1 is a parasitized erythrocyte receptor for adherence to CD36, thrombospondin, and intercellular adhesion molecule 1. Proc. Natl. Acad. Sci. USA 1996, 93, 3497–3502. [Google Scholar] [CrossRef]
- Vogt, A.M.; Barragan, A.; Chen, Q.; Kironde, F.; Spillmann, D.; Wahlgren, M. Heparan sulfate on endothelial cells mediates the binding ofPlasmodium falciparum–infected erythrocytes via the DBL1α domain of PfEMP1. Blood 2003, 101, 2405–2411. [Google Scholar] [CrossRef]
- Stevenson, L.; Laursen, E.; Cowan, G.J.; Bandoh, B.; Barfod, L.; Cavanagh, D.R.; Andersen, G.R.; Hviid, L. α2-Macroglobulin Can Crosslink Multiple Plasmodium falciparum Erythrocyte Membrane Protein 1 (PfEMP1) Molecules and May Facilitate Adhesion of Parasitized Erythrocytes. PLoS Pathog. 2015, 11, e1005022. [Google Scholar] [CrossRef]
- Ghumra, A.; Semblat, J.-P.; McIntosh, R.S.; Raza, A.; Rasmussen, I.B.; Braathen, R.; Johansen, F.-E.; Sandlie, I.; Mongini, P.K.; Rowe, J.A.; et al. Identification of Residues in the Cμ4 Domain of Polymeric IgM Essential for Interaction with Plasmodium falciparum Erythrocyte Membrane Protein 1 (PfEMP1)1. J. Immunol. 2008, 181, 1988–2000. [Google Scholar] [CrossRef] [PubMed]
- Adams, Y.; Kuhnrae, P.; Higgins, M.K.; Ghumra, A.; Rowe, J.A. Rosetting Plasmodium falciparum-Infected Erythrocytes Bind to Human Brain Microvascular Endothelial Cells In Vitro, Demonstrating a Dual Adhesion Phenotype Mediated by Distinct P. falciparum Erythrocyte Membrane Protein 1 Domains. Infect. Immun. 2014, 82, 949–959. [Google Scholar] [CrossRef] [PubMed]
- Deans, A.-M.; Rowe, J.A. Plasmodium falciparum: Rosettes do not protect merozoites from invasion-inhibitory antibodies. Exp. Parasitol. 2006, 112, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Wahlgren, M.; Carlson, J.; Udomsangpetch, R.; Perlmann, P. Why do Plasmodium falciparumm-infected erythrocytes form spontaneous erythrocyte rosettes? Parasitol. Today 1989, 5, 183–185. [Google Scholar] [CrossRef] [PubMed]
- Yam, X.Y.; Niang, M.; Madnani, K.G.; Preiser, P.R. Three Is a Crowd–New Insights into Rosetting in Plasmodium falciparum. Trends Parasitol. 2017, 33, 309–320. [Google Scholar] [CrossRef]
- Rowe, J.A.; Moulds, J.M.; Newbold, C.I.; Miller, L.H.P. falciparum rosetting mediated by a parasite-variant erythrocyte membrane protein and complement-receptor 1. Nature 1997, 388, 292–295. [Google Scholar] [CrossRef]
- Chen, Q.; Barragan, A.; Fernandez, V.; Sundström, A.; Schlichtherle, M.; Sahlén, A.; Carlson, J.; Datta, S.; Wahlgren, M. Identification of Plasmodium falciparum Erythrocyte Membrane Protein 1 (PfEMP1) as the Rosetting Ligand of the Malaria Parasite, P. falciparum. J. Exp. Med. 1998, 187, 15–23. [Google Scholar] [CrossRef]
- Albrecht, L.; Moll, K.; Blomqvist, K.; Normark, J.; Chen, Q.; Wahlgren, M. var gene transcription and PfEMP1 expression in the rosetting and cytoadhesive Plasmodium falciparum clone FCR3S1.2. Malar. J. 2011, 10, 17. [Google Scholar] [CrossRef] [PubMed]
- Vigan-Womas, I.; Guillotte, M.; Juillerat, A.; Vallieres, C.; Lewit-Bentley, A.; Tall, A.; Baril, L.; Bentley, G.A.; Mercereau-Puijalon, O. Allelic Diversity of the Plasmodium falciparum Erythrocyte Membrane Protein 1 Entails Variant-Specific Red Cell Surface Epitopes. PLoS ONE 2011, 6, e16544. [Google Scholar] [CrossRef] [PubMed]
- Wahlgren, M.; Goel, S.; Akhouri, R.R. Variant surface antigens of Plasmodium falciparum and their roles in severe malaria. Nat. Rev. Microbiol. 2017, 15, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Akhouri, R.R.; Goel, S.; Furusho, H.; Skoglund, U.; Wahlgren, M. Architecture of Human IgM in Complex with P. falciparum Erythrocyte Membrane Protein 1. Cell Rep. 2016, 14, 723–736. [Google Scholar] [CrossRef] [PubMed]
- Goel, S.; Palmkvist, M.; Moll, K.; Joannin, N.; Lara, P.; R Akhouri, R.; Moradi, N.; Öjemalm, K.; Westman, M.; Angeletti, D.; et al. RIFINs are adhesins implicated in severe Plasmodium falciparum malaria. Nat. Med. 2015, 21, 314–317. [Google Scholar] [CrossRef]
- Moll, K.; Palmkvist, M.; Ch’ng, J.; Kiwuwa, M.S.; Wahlgren, M. Evasion of Immunity to Plasmodium falciparum: Rosettes of Blood Group A Impair Recognition of PfEMP1. PLoS ONE 2016, 10, e0145120. [Google Scholar] [CrossRef]
- Niang, M.; Bei, A.K.; Madnani, K.G.; Pelly, S.; Dankwa, S.; Kanjee, U.; Gunalan, K.; Amaladoss, A.; Yeo, K.P.; Bob, N.S.; et al. STEVOR Is a Plasmodium falciparum Erythrocyte Binding Protein that Mediates Merozoite Invasion and Rosetting. Cell Host Microbe 2014, 16, 81–93. [Google Scholar] [CrossRef]
- Singh, H.; Madnani, K.; Lim, Y.B.; Cao, J.; Preiser, P.R.; Lim, C.T. Expression dynamics and physiologically relevant functional study of STEVOR in asexual stages of Plasmodium falciparum infection. Cell. Microbiol. 2017, 19, e12715. [Google Scholar] [CrossRef]
- Sylla, B.S.; Wild, C.P. A million africans a year dying from cancer by 2030: What can cancer research and control offer to the continent? Int. J. Cancer 2012, 130, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Artavanis-Tsakonas, K.; Tongren, J.E.; Riley, E.M. The war between the malaria parasite and the immune system: Immunity, immunoregulation and immunopathology. Clin. Exp. Immunol. 2003, 133, 145–152. [Google Scholar] [CrossRef]
- Yao, S.; Hong, C.-C.; Ruiz-Narváez, E.A.; Evans, S.S.; Zhu, Q.; Schaefer, B.A.; Yan, L.; Coignet, M.V.; Lunetta, K.L.; Sucheston-Campbell, L.E.; et al. Genetic ancestry and population differences in levels of inflammatory cytokines in women: Role for evolutionary selection and environmental factors. PLoS Genet. 2018, 14, e1007368. [Google Scholar] [CrossRef]
- Wang, J.; Ou, Z.L.; Hou, Y.F.; Luo, J.M.; Shen, Z.Z.; Ding, J.; Shao, Z.M. Enhanced expression of Duffy antigen receptor for chemokines by breast cancer cells attenuates growth and metastasis potential. Oncogene 2006, 25, 7201–7211. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Zhan, R.; Chaudhuri, A.; Watabe, M.; Pai, S.K.; Hirota, S.; Hosobe, S.; Tsukada, T.; Miura, K.; Takano, Y.; et al. Interaction of KAI1 on tumor cells with DARC on vascular endothelium leads to metastasis suppression. Nat. Med. 2006, 12, 933–938. [Google Scholar] [CrossRef]
- Bentley, G.A.; Gamain, B. How does Plasmodium falciparum stick to CSA? Let’s see in the crystal. Nat. Struct. Mol. Biol. 2008, 15, 895–897. [Google Scholar] [CrossRef] [PubMed]
- Cooney, C.A.; Jousheghany, F.; Yao-Borengasser, A.; Phanavanh, B.; Gomes, T.; Kieber-Emmons, A.M.; Siegel, E.R.; Suva, L.J.; Ferrone, S.; Kieber-Emmons, T.; et al. Chondroitin sulfates play a major role in breast cancer metastasis: A role for CSPG4 and CHST11gene expression in forming surface P-selectin ligands in aggressive breast cancer cells. Breast Cancer Res. 2011, 13, R58. [Google Scholar] [CrossRef] [PubMed]
- Thorley-Lawson, D.; Deitsch, K.W.; Duca, K.A.; Torgbor, C. The Link between Plasmodium falciparum Malaria and Endemic Burkitt’s Lymphoma-New Insight into a 50-Year-Old Enigma. PLoS Pathog. 2016, 12, e1005331. [Google Scholar] [CrossRef] [PubMed]
- Robbiani, D.F.; Deroubaix, S.; Feldhahn, N.; Oliveira, T.Y.; Callen, E.; Wang, Q.; Jankovic, M.; Silva, I.T.; Rommel, P.C.; Bosque, D.; et al. Plasmodium Infection Promotes Genomic Instability and AID-Dependent B Cell Lymphoma. Cell 2015, 162, 727–737. [Google Scholar] [CrossRef]
- Malagon, F.; Gonzalez-Angulo, J.; Carrasco, E.; Robert, L. Etiopathogenesis of Burkitt’s lymphoma: A lesson from a BL-like in CD1 mouse immune to Plasmodium yoelii yoelii. Infect. Agents Cancer 2011, 6, 10. [Google Scholar] [CrossRef]
- Alencar Filho, A.C.; Lacerda, M.V.; Okoshi, K.; Okoshi, M.P. Malaria and vascular endothelium. Arq. Bras. Cardiol. 2014, 103, 165–169. [Google Scholar] [CrossRef]
- Holm, A.E.; Gomes, L.C.; Marinho, C.R.F.; Silvestre, O.M.; Vestergaard, L.S.; Biering-Sorensen, T.; Brainin, P. Prevalence of Cardiovascular Complications in Malaria: A Systematic Review and Meta-Analysis. Am. J. Trop. Med. Hyg. 2021, 104, 1643–1650. [Google Scholar] [CrossRef]
- Brainin, P.; Mohr, G.H.; Modin, D.; Claggett, B.; Silvestre, O.M.; Shah, A.; Vestergaard, L.S.; Jensen, J.U.S.; Hviid, L.; Torp-Pedersen, C.; et al. Heart failure associated with imported malaria: A nationwide Danish cohort study. ESC Heart Fail. 2021, 8, 3521–3529. [Google Scholar] [CrossRef]
- Pain, A.; Ferguson, D.J.P.; Kai, O.; Urban, B.C.; Lowe, B.; Marsh, K.; Roberts, D.J. Platelet-mediated clumping of Plasmodium falciparum-infected erythrocytes is a common adhesive phenotype and is associated with severe malaria. Proc. Natl. Acad. Sci. USA 2001, 98, 1805–1810. [Google Scholar] [CrossRef] [PubMed]
- Wennicke, K.; Debierre-Grockiego, F.; Wichmann, D.; Brattig, N.W.; Pankuweit, S.; Maisch, B.; Schwarz, R.T.; Ruppert, V. Glycosylphosphatidylinositol-induced cardiac myocyte death might contribute to the fatal outcome of Plasmodium falciparum malaria. Apoptosis 2008, 13, 857–866. [Google Scholar] [CrossRef]
- Huber, S.A.; Sartini, D. Roles of Tumor Necrosis Factor Alpha (TNF-α) and the p55 TNF Receptor in CD1d Induction and Coxsackievirus B3-Induced Myocarditis. J. Virol. 2005, 79, 2659–2665. [Google Scholar] [CrossRef]
- Tatsumi, T.; Akashi, K.; Keira, N.; Matoba, S.; Mano, A.; Shiraishi, J.; Yamanaka, S.; Kobara, M.; Hibino, N.; Hosokawa, S.; et al. Cytokine-induced nitric oxide inhibits mitochondrial energy production and induces myocardial dysfunction in endotoxin-treated rat hearts. J. Mol. Cell. Cardiol. 2004, 37, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Colomba, C.; Trizzino, M.; Gioè, C.; Coelho, F.; Lopo, I.; Pinheiro, P.; Sousa, J.; Cascio, A. Malaria and the heart: Two rare case reports of Plasmodium falciparumassociated pericarditis. J. Vector Borne Dis. 2017, 54, 372–374. [Google Scholar] [CrossRef] [PubMed]
- Mohsen, A.H.; Green, S.T.; West, J.N.; McKendrick, M.W. Myocarditis Associated with Plasmodium falciparum Malaria: A Case Report and a Review of the Literature. J. Travel Med. 2006, 8, 219–220. [Google Scholar] [CrossRef]
- Cantalupo, A.; Gargiulo, A.; Dautaj, E.; Liu, C.; Zhang, Y.; Hla, T.; Lorenzo, A.D. S1PR1 (Sphingosine-1-Phosphate Receptor 1) Signaling Regulates Blood Flow and Pressure. Hypertension 2017, 70, 426–434. [Google Scholar] [CrossRef]
- Dhangadamajhi, G.; Singh, S. Malaria link of hypertension: A hidden syndicate of angiotensin II, bradykinin and sphingosine 1-phosphate. Hum. Cell 2021, 34, 734–744. [Google Scholar] [CrossRef] [PubMed]
- Kingston, H.W.F.; Ghose, A.; Rungpradubvong, V.; Satitthummanid, S.; Herdman, M.T.; Plewes, K.; Leopold, S.J.; Ishioka, H.; Mohanty, S.; Maude, R.J.; et al. Reduced Cardiac Index Reserve and Hypovolemia in Severe Falciparum Malaria. J. Infect. Dis. 2019, 221, 1518–1527. [Google Scholar] [CrossRef]
- Akide Ndunge, O.B.; Kilian, N.; Salman, M.M. Cerebral Malaria and Neuronal Implications of Plasmodium Falciparum Infection: From Mechanisms to Advanced Models. Adv. Sci. 2022, 9, e2202944. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.-U.; Lao, Y.; Spicer, V.; Coombs, K.M. Zika Virus Infection of Sertoli Cells Alters Protein Expression Involved in Activated Immune and Antiviral Response Pathways, Carbohydrate Metabolism and Cardiovascular Disease. Viruses 2022, 14, 377. [Google Scholar] [CrossRef] [PubMed]
- Gorbatyuk, O.S.; Li, S.; Nguyen, F.N.; Manfredsson, F.P.; Kondrikova, G.; Sullivan, L.F.; Meyers, C.; Chen, W.; Mandel, R.J.; Muzyczka, N. α-Synuclein Expression in Rat Substantia Nigra Suppresses Phospholipase D2 Toxicity and Nigral Neurodegeneration. Mol. Ther. 2010, 18, 1758–1768. [Google Scholar] [CrossRef]
- Gandarilla-Martínez, N.; Carbayo-Viejo, Á.; Sánchez-Gómez, A.; Valldeoriola-Serra, F. Parkinsonism in a patient with Plasmodium falciparum malaria. Park. Relat. Disord. 2020, 80, 181–183. [Google Scholar] [CrossRef]
- Wilhelmus, M.M.M.; van der Pol, S.M.A.; Jansen, Q.; Witte, M.E.; van der Valk, P.; Rozemuller, A.J.M.; Drukarch, B.; de Vries, H.E.; Van Horssen, J. Association of Parkinson disease-related protein PINK1 with Alzheimer disease and multiple sclerosis brain lesions. Free Radic. Biol. Med. 2011, 50, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Rejdak, K.; Kuhle, J.; Rüegg, S.; Lindberg, R.L.P.; Petzold, A.; Sulejczak, D.; Papuc, E.; Rejdak, R.; Stelmasiak, Z.; Grieb, P. Neurofilament heavy chain and heat shock protein 70 as markers of seizure-related brain injury. Epilepsia 2012, 53, 922–927. [Google Scholar] [CrossRef]
- Anstey, N.M.; Douglas, N.M.; Poespoprodjo, J.R.; Price, R.N. Chapter Three—Plasmodium vivax: Clinical Spectrum, Risk Factors and Pathogenesis. In Advances in Parasitology; Hay, S.I., Price, R., Baird, J.K., Eds.; Academic Press: New York, NY, USA, 2012; Volume 80, pp. 151–201. [Google Scholar]
- de Silva, H.J.; Hoang, P.; Dalton, H.; de Silva, N.R.; Jewell, D.P.; Peiris, J.B. Immune activation during cerebellar dysfunction following Plasmodium falciparum malaria. Trans. R. Soc. Trop. Med. Hyg. 1992, 86, 129–131. [Google Scholar] [CrossRef]
- Bergmark, B.; Bergmark, R.; Beaudrap, P.D.; Boum, Y.; Mwanga-Amumpaire, J.; Carroll, R.; Zapol, W. Inhaled Nitric Oxide and Cerebral Malaria: Basis of a Strategy for Buying Time for Pharmacotherapy. Pediatr. Infect. Dis. J. 2012, 31, e250–e254. [Google Scholar] [CrossRef]
- Turner, G.D.; Morrison, H.; Jones, M.; Davis, T.M.; Looareesuwan, S.; Buley, I.D.; Gatter, K.C.; Newbold, C.I.; Pukritayakamee, S.; Nagachinta, B.; et al. An immunohistochemical study of the pathology of fatal malaria. Evidence for widespread endothelial activation and a potential role for intercellular adhesion molecule-1 in cerebral sequestration. Am. J. Pathol. 1994, 145, 1057–1069. [Google Scholar]
- Castaldo, N.; Tascini, C.; Della Siega, P.; Peghin, M.; Pecori, D. Clinical presentation and immunological features of Post-Malaria Neurologic Syndrome: A case report and review of literature. Malar. J. 2020, 19, 419. [Google Scholar] [CrossRef]
- Carter, J.A.; Neville, B.G.R.; White, S.; Ross, A.J.; Otieno, G.; Mturi, N.; Musumba, C.; Newton, C.R.J.C. Increased Prevalence of Epilepsy Associated with Severe Falciparum Malaria in Children. Epilepsia 2004, 45, 978–981. [Google Scholar] [CrossRef]
- Birbeck, G.L.; Molyneux, M.E.; Kaplan, P.W.; Seydel, K.B.; Chimalizeni, Y.F.; Kawaza, K.; Taylor, T.E. Blantyre Malaria Project Epilepsy Study (BMPES) of neurological outcomes in retinopathy-positive paediatric cerebral malaria survivors: A prospective cohort study. Lancet Neurol. 2010, 9, 1173–1181. [Google Scholar] [CrossRef] [PubMed]
- Katsoulis, O.; Georgiadou, A.; Cunnington, A.J. Immunopathology of Acute Kidney Injury in Severe Malaria. Front. Immunol. 2021, 12, 651739. [Google Scholar] [CrossRef]
- Eiam-Ong, S.; Sitprija, V. Falciparum malaria and the kidney: A model of inflammation. Am. J. Kidney Dis. 1998, 32, 361–375. [Google Scholar] [CrossRef]
- Plewes, K.; Turner, G.D.H.; Dondorp, A.M. Pathophysiology, clinical presentation, and treatment of coma and acute kidney injury complicating falciparum malaria. Curr. Opin. Infect. Dis. 2018, 31, 69–77. [Google Scholar] [CrossRef]
- Kanodia, K.V.; Shah, P.R.; Vanikar, A.V.; Kasat, P.; Gumber, M.; Trivedi, H.L. Malaria induced acute renal failure: A single center experience. Saudi J. Kidney Dis. Transplant. 2010, 21, 1088–1091. [Google Scholar]
- Lyke, K.E.; Burges, R.; Cissoko, Y.; Sangare, L.; Dao, M.; Diarra, I.; Kone, A.; Harley, R.; Plowe, C.V.; Doumbo, O.K.; et al. Serum levels of the proinflammatory cytokines interleukin-1 beta (IL-1beta), IL-6, IL-8, IL-10, tumor necrosis factor alpha, and IL-12(p70) in Malian children with severe Plasmodium falciparum malaria and matched uncomplicated malaria or healthy controls. Infect. Immun. 2004, 72, 5630–5637. [Google Scholar] [CrossRef]
- Sinniah, R.; Rui-Mei, L.; Kara, A. Up-regulation of cytokines in glomerulonephritis associated with murine malaria infection. Int. J. Exp. Pathol. 1999, 80, 87–95. [Google Scholar] [CrossRef]
- Barber, B.E.; Grigg, M.J.; Piera, K.A.; William, T.; Cooper, D.J.; Plewes, K.; Dondorp, A.M.; Yeo, T.W.; Anstey, N.M. Intravascular haemolysis in severe Plasmodium knowlesi malaria: Association with endothelial activation, microvascular dysfunction, and acute kidney injury. Emerg. Microbes Infect. 2018, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, P.H.; Morris-Jones, S.D.; Hviid, L.; Theander, T.G.; Høier-Madsen, M.; Bayoumi, R.A.; Greenwood, B.M. Anti-phospholipid antibodies in patients with Plasmodium falciparum malaria. Immunology 1993, 79, 653–657. [Google Scholar] [PubMed]
- de Alwis, R.; Williams, K.L.; Schmid, M.A.; Lai, C.Y.; Patel, B.; Smith, S.A.; Crowe, J.E.; Wang, W.K.; Harris, E.; de Silva, A.M. Dengue viruses are enhanced by distinct populations of serotype cross-reactive antibodies in human immune sera. PLoS Pathog. 2014, 10, e1004386. [Google Scholar] [CrossRef]
- Loo, Y.-M.; Gale, M., Jr. Immune Signaling by RIG-I-like Receptors. Immunity 2011, 34, 680–692. [Google Scholar] [CrossRef]
- Nasirudeen, A.M.A.; Wong, H.H.; Thien, P.; Xu, S.; Lam, K.-P.; Liu, D.X. RIG-I, MDA5 and TLR3 Synergistically Play an Important Role in Restriction of Dengue Virus Infection. PLoS Negl. Trop. Dis. 2011, 5, e926. [Google Scholar] [CrossRef]
- Said, E.A.; Tremblay, N.; Al-Balushi, M.S.; Al-Jabri, A.A.; Lamarre, D. Viruses Seen by Our Cells: The Role of Viral RNA Sensors. J. Immunol. Res. 2018, 2018, 9480497. [Google Scholar] [CrossRef]
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.-T.; Grishin, N.V.; et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 2015, 347, aaa2630. [Google Scholar] [CrossRef]
- Liang, Z.; Wu, S.; Li, Y.; He, L.; Wu, M.; Jiang, L.; Feng, L.; Zhang, P.; Huang, X. Activation of Toll-Like Receptor 3 Impairs the Dengue Virus Serotype 2 Replication through Induction of IFN-β in Cultured Hepatoma Cells. PLoS ONE 2011, 6, e23346. [Google Scholar] [CrossRef]
- Sun, B.; Sundström, K.B.; Chew, J.J.; Bist, P.; Gan, E.S.; Tan, H.C.; Goh, K.C.; Chawla, T.; Tang, C.K.; Ooi, E.E. Dengue virus activates cGAS through the release of mitochondrial DNA. Sci. Rep. 2017, 7, 3594. [Google Scholar] [CrossRef] [PubMed]
- Holm, C.K.; Rahbek, S.H.; Gad, H.H.; Bak, R.O.; Jakobsen, M.R.; Jiang, Z.; Hansen, A.L.; Jensen, S.K.; Sun, C.; Thomsen, M.K.; et al. Influenza A virus targets a cGAS-independent STING pathway that controls enveloped RNA viruses. Nat. Commun. 2016, 7, 10680. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; MacDuff, D.A.; Imanaka, N.; Gainey, M.D.; Shrestha, B.; Eitson, J.L.; Mar, K.B.; Richardson, R.B.; Ratushny, A.V.; Litvak, V.; et al. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 2014, 505, 691–695. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, S.; Luthra, P.; Sanchez-Aparicio, M.T.; Maestre, A.M.; Patel, J.; Lamothe, F.; Fredericks, A.C.; Tripathi, S.; Zhu, T.; Pintado-Silva, J.; et al. Dengue virus NS2B protein targets cGAS for degradation and prevents mitochondrial DNA sensing during infection. Nat. Microbiol. 2017, 2, 17037. [Google Scholar] [CrossRef]
- Aarreberg, L.D.; Esser-Nobis, K.; Driscoll, C.; Shuvarikov, A.; Roby, J.A.; Gale, M., Jr. Interleukin-1β Induces mtDNA Release to Activate Innate Immune Signaling via cGAS-STING. Mol. Cell 2019, 74, 801–815.e6. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.-H.; Wang, M.-Y.; Huang, C.-Y.; Wu, C.-H.; Hung, L.-F.; Yang, C.-Y.; Ke, P.-Y.; Luo, S.-F.; Liu, S.-J.; Ho, L.-J. Infection with the dengue RNA virus activates TLR9 signaling in human dendritic cells. EMBO Rep. 2018, 19, e46182. [Google Scholar] [CrossRef] [PubMed]
- Murugesan, A.; Manoharan, M. Chapter 16—Dengue Virus. In Emerging and Reemerging Viral Pathogens; Ennaji, M.M., Ed.; Academic Press: New York, NY, USA, 2020; pp. 281–359. [Google Scholar]
- Reich, N.G.; Shrestha, S.; King, A.A.; Rohani, P.; Lessler, J.; Kalayanarooj, S.; Yoon, I.K.; Gibbons, R.V.; Burke, D.S.; Cummings, D.A. Interactions between serotypes of dengue highlight epidemiological impact of cross-immunity. J. R. Soc. Interface 2013, 10, 20130414. [Google Scholar] [CrossRef] [PubMed]
- Weiskopf, D.; Angelo, M.A.; Sidney, J.; Peters, B.; Shresta, S.; Sette, A. Immunodominance changes as a function of the infecting dengue virus serotype and primary versus secondary infection. J. Virol. 2014, 88, 11383–11394. [Google Scholar] [CrossRef] [PubMed]
- Gubler, D.J. Dengue and dengue hemorrhagic fever. Clin. Microbiol. Rev. 1998, 11, 480–496. [Google Scholar] [CrossRef]
- Srikiatkhachorn, A. Plasma leakage in dengue haemorrhagic fever. Thromb. Haemost. 2009, 102, 1042–1049. [Google Scholar] [CrossRef]
- Martina, B.E.; Koraka, P.; Osterhaus, A.D. Dengue virus pathogenesis: An integrated view. Clin. Microbiol. Rev. 2009, 22, 564–581. [Google Scholar] [CrossRef]
- Chang, D.C.; Hoang, L.T.; Mohamed Naim, A.N.; Dong, H.; Schreiber, M.J.; Hibberd, M.L.; Tan, M.J.A.; Shi, P.-Y. Evasion of early innate immune response by 2′-O-methylation of dengue genomic RNA. Virology 2016, 499, 259–266. [Google Scholar] [CrossRef]
- Dong, H.; Chang, D.C.; Hua, M.H.C.; Lim, S.P.; Chionh, Y.H.; Hia, F.; Lee, Y.H.; Kukkaro, P.; Lok, S.-M.; Dedon, P.C.; et al. 2′-O Methylation of Internal Adenosine by Flavivirus NS5 Methyltransferase. PLoS Pathog. 2012, 8, e1002642. [Google Scholar] [CrossRef]
- Manokaran, G.; Finol, E.; Wang, C.; Gunaratne, J.; Bahl, J.; Ong, E.Z.; Tan, H.C.; Sessions, O.M.; Ward, A.M.; Gubler, D.J.; et al. Dengue subgenomic RNA binds TRIM25 to inhibit interferon expression for epidemiological fitness. Science 2015, 350, 217–221. [Google Scholar] [CrossRef]
- Gack, M.U.; Albrecht, R.A.; Urano, T.; Inn, K.-S.; Huang, I.C.; Carnero, E.; Farzan, M.; Inoue, S.; Jung, J.U.; García-Sastre, A. Influenza A Virus NS1 Targets the Ubiquitin Ligase TRIM25 to Evade Recognition by the Host Viral RNA Sensor RIG-I. Cell Host Microbe 2009, 5, 439–449. [Google Scholar] [CrossRef]
- Turtle, L.; Bali, T.; Buxton, G.; Chib, S.; Chan, S.; Soni, M.; Hussain, M.; Isenman, H.; Fadnis, P.; Venkataswamy, M.M.; et al. Human T cell responses to Japanese encephalitis virus in health and disease. J. Exp. Med. 2016, 213, 1331–1352. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.M.; Loo, Y.-M.; Horner, S.M.; Zornetzer, G.A.; Katze, M.G.; Gale, M., Jr. The Mitochondrial Targeting Chaperone 14-3-3ε; Regulates a RIG-I Translocon that Mediates Membrane Association and Innate Antiviral Immunity. Cell Host Microbe 2012, 11, 528–537. [Google Scholar] [CrossRef]
- Jacobs, J.L.; Coyne, C.B. Mechanisms of MAVS Regulation at the Mitochondrial Membrane. J. Mol. Biol. 2013, 425, 5009–5019. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-J.; Syed, G.H.; Khan, M.; Chiu, W.-W.; Sohail, M.A.; Gish, R.G.; Siddiqui, A. Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc. Natl. Acad. Sci. USA 2014, 111, 6413–6418. [Google Scholar] [CrossRef]
- Dalrymple, N.A.; Cimica, V.; Mackow, E.R. Dengue Virus NS Proteins Inhibit RIG-I/MAVS Signaling by Blocking TBK1/IRF3 Phosphorylation: Dengue Virus Serotype 1 NS4A Is a Unique Interferon-Regulating Virulence Determinant. mBio 2015, 6, e00553-15. [Google Scholar] [CrossRef]
- Yu, C.Y.; Chang, T.H.; Liang, J.J.; Chiang, R.L.; Lee, Y.L.; Liao, C.L.; Lin, Y.L. P122 Dengue virus targets the adaptor protein MITA to subvert host innate immunity. Cytokine 2012, 59, 558. [Google Scholar] [CrossRef]
- Muñoz-Jordán, J.L.; Laurent-Rolle, M.; Ashour, J.; Martínez-Sobrido, L.; Ashok, M.; Lipkin, W.I.; García-Sastre, A. Inhibition of Alpha/Beta Interferon Signaling by the NS4B Protein of Flaviviruses. J. Virol. 2005, 79, 8004–8013. [Google Scholar] [CrossRef]
- Morrison, J.; Laurent-Rolle, M.; Maestre, A.M.; Rajsbaum, R.; Pisanelli, G.; Simon, V.; Mulder, L.C.F.; Fernandez-Sesma, A.; García-Sastre, A. Dengue Virus Co-opts UBR4 to Degrade STAT2 and Antagonize Type I Interferon Signaling. PLoS Pathog. 2013, 9, e1003265. [Google Scholar] [CrossRef] [PubMed]
- Tambyah, P.A.; Ching, C.S.; Sepramaniam, S.; Ali, J.M.; Armugam, A.; Jeyaseelan, K. microRNA expression in blood of dengue patients. Ann. Clin. Biochem. 2016, 53, 466–476. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, X.; Jiang, X.; Gu, D.; Zhang, Y.; Kong, S.K.; Jiang, C.; Xie, W. Dysregulated Serum MiRNA Profile and Promising Biomarkers in Dengue-infected Patients. Int. J. Med. Sci. 2016, 13, 195–205. [Google Scholar] [CrossRef]
- Kanokudom, S.; Vilaivan, T.; Wikan, N.; Thepparit, C.; Smith, D.R.; Assavalapsakul, W. miR-21 promotes dengue virus serotype 2 replication in HepG2 cells. Antivir. Res. 2017, 142, 169–177. [Google Scholar] [CrossRef]
- Miranda, K.C.; Huynh, T.; Tay, Y.; Ang, Y.-S.; Tam, W.-L.; Thomson, A.M.; Lim, B.; Rigoutsos, I. A Pattern-Based Method for the Identification of MicroRNA Binding Sites and Their Corresponding Heteroduplexes. Cell 2006, 126, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; He, L.; Li, Y.; Wang, T.; Feng, L.; Jiang, L.; Zhang, P.; Huang, X. miR-146a facilitates replication of dengue virus by dampening interferon induction by targeting TRAF6. J. Infect. 2013, 67, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Ho, B.-C.; Yu, I.S.; Lu, L.-F.; Rudensky, A.; Chen, H.-Y.; Tsai, C.-W.; Chang, Y.-L.; Wu, C.-T.; Chang, L.-Y.; Shih, S.-R.; et al. Inhibition of miR-146a prevents enterovirus-induced death by restoring the production of type I interferon. Nat. Commun. 2014, 5, 3344. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Chen, L.; Zeng, Y.; Si, L.; Guo, X.; Zhou, J.; Fang, D.; Zeng, G.; Jiang, L. Suppressed expression of miR-378 targeting gzmb in NK cells is required to control dengue virus infection. Cell. Mol. Immunol. 2016, 13, 700–708. [Google Scholar] [CrossRef]
- Chien, Y.-W.; Wang, C.-C.; Wang, Y.-P.; Lee, C.-Y.; Perng, G.C. Risk of Leukemia after Dengue Virus Infection: A Population-Based Cohort Study. Cancer Epidemiol. Biomark. Prev. 2020, 29, 558–564. [Google Scholar] [CrossRef]
- Talwar, V.; Goel, V.; Raina, S.; Talwar, J.; Patnaik, N.; Doval, D.C. Dengue fever in cancer patients: Retrospective analysis. Curr. Med. Res. Pract. 2016, 6, 157–159. [Google Scholar] [CrossRef]
- Gan, E.S.; Tan, H.C.; Le, D.H.T.; Huynh, T.T.; Wills, B.; Seidah, N.G.; Ooi, E.E.; Yacoub, S. Dengue virus induces PCSK9 expression to alter antiviral responses and disease outcomes. J. Clin. Investig. 2020, 130, 5223–5234. [Google Scholar] [CrossRef]
- Almontashiri, N.A.M.; Vilmundarson, R.O.; Ghasemzadeh, N.; Dandona, S.; Roberts, R.; Quyyumi, A.A.; Chen, H.-H.; Stewart, A.F.R. Plasma PCSK9 Levels Are Elevated with Acute Myocardial Infarction in Two Independent Retrospective Angiographic Studies. PLoS ONE 2014, 9, e106294. [Google Scholar] [CrossRef]
- Miranda, C.H.; Borges, M.d.C.; Matsuno, A.K.; Vilar, F.C.; Gali, L.G.; Volpe, G.J.; Schmidt, A.; Pazin-Filho, A.; da Silva, F.M.F.; de Castro-Jorge, L.A.; et al. Evaluation of Cardiac Involvement During Dengue Viral Infection. Clin. Infect. Dis. 2013, 57, 812–819. [Google Scholar] [CrossRef] [PubMed]
- Salgado, D.M.; Eltit, J.M.; Mansfield, K.; Panqueba, C.; Castro, D.; Vega, M.R.; Xhaja, K.; Schmidt, D.; Martin, K.J.; Allen, P.D.; et al. Heart and skeletal muscle are targets of dengue virus infection. Pediatr. Infect. Dis. J. 2010, 29, 238–242. [Google Scholar] [CrossRef]
- La-Orkhun, V.; Supachokchaiwattana, P.; Lertsapcharoen, P.; Khongphatthanayothin, A. Spectrum of cardiac rhythm abnormalities and heart rate variability during the convalescent stage of dengue virus infection: A Holter study. Ann. Trop. Paediatr. 2011, 31, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Thacker, M.M.; Makwana, P.V. Ventricular Tachycardia in Primary Dengue Fever. J. Assoc. Physicians India 2018, 66, 100. [Google Scholar] [PubMed]
- Chaturvedi, U.C.; Mathur, A.; Mehrotra, R.M. Experimentally produced cardiac injury following dengue virus infection. Indian J. Pathol. Bacteriol. 1974, 17, 218–220. [Google Scholar]
- Araújo, F.M.C.; Araújo, M.S.; Nogueira, R.M.R.; Brilhante, R.S.N.; Oliveira, D.N.; Rocha, M.F.G.; Cordeiro, R.A.; Araújo, R.M.C.; Sidrim, J.J.C. Central nervous system involvement in dengue. Neurology 2012, 78, 736–742. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, U.C.; Dhawan, R.; Khanna, M.; Mathur, A. Breakdown of the blood-brain barrier during dengue virus infection of mice. J. Gen. Virol. 1991, 72, 859–866. [Google Scholar] [CrossRef]
- Bordignon, J.; Strottmann, D.M.; Mosimann, A.L.; Probst, C.M.; Stella, V.; Noronha, L.; Zanata, S.M.; Dos Santos, C.N. Dengue neurovirulence in mice: Identification of molecular signatures in the E and NS3 helicase domains. J. Med. Virol. 2007, 79, 1506–1517. [Google Scholar] [CrossRef]
- Modhiran, N.; Watterson, D.; Muller, D.A.; Panetta, A.K.; Sester, D.P.; Liu, L.; Hume, D.A.; Stacey, K.J.; Young, P.R. Dengue virus NS1 protein activates cells via Toll-like receptor 4 and disrupts endothelial cell monolayer integrity. Sci. Transl. Med. 2015, 7, 304ra142. [Google Scholar] [CrossRef]
- Mathew, S.; Pandian, J.D. Stroke in Patients with Dengue. J. Stroke Cerebrovasc. Dis. 2010, 19, 253–256. [Google Scholar] [CrossRef]
- Panda, P.K.; Sharawat, I.K.; Bolia, R.; Shrivastava, Y. Case Report: Dengue Virus-Triggered Parkinsonism in an Adolescent. Am. J. Trop. Med. Hyg. 2020, 103, 851–854. [Google Scholar] [CrossRef]
- Cam, B.V.; Fonsmark, L.; Hue, N.B.; Phuong, N.T.; Poulsen, A.; Heegaard, E.D. Prospective case-control study of encephalopathy in children with dengue hemorrhagic fever. Am. J. Trop. Med. Hyg. 2001, 65, 848–851. [Google Scholar] [CrossRef]
- Lana-Peixoto, M.A.; Pedrosa, D.; Talim, N.; Amaral, J.; Horta, A.; Kleinpaul, R. Neuromyelitis optica spectrum disorder associated with dengue virus infection. J. Neuroimmunol. 2018, 318, 53–55. [Google Scholar] [CrossRef] [PubMed]
- Kamel, M.G.; Nam, N.T.; Han, N.H.B.; El-Shabouny, A.-E.; Makram, A.-E.M.; Abd-Elhay, F.A.-E.; Dang, T.N.; Hieu, N.L.T.; Huong, V.T.Q.; Tung, T.H.; et al. Post-dengue acute disseminated encephalomyelitis: A case report and meta-analysis. PLoS Negl. Trop. Dis. 2017, 11, e0005715. [Google Scholar] [CrossRef]
- Nair, J.J.; Bhat, A.; Prabhu, M.V. A Clinical Study of Acute Kidney Injury in Tropical Acute Febrile Illness. J. Clin. Diagn. Res. 2016, 10, OC01–OC05. [Google Scholar] [CrossRef] [PubMed]
- Pagliari, C.; Simões Quaresma, J.A.; Kanashiro-Galo, L.; de Carvalho, L.V.; Vitoria, W.O.; da Silva, W.L.F.; Penny, R.; Vasconcelos, B.C.B.; da Costa Vasconcelos, P.F.; Duarte, M.I.S. Human kidney damage in fatal dengue hemorrhagic fever results of glomeruli injury mainly induced by IL17. J. Clin. Virol. 2016, 75, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Horvath, R.L.; McBride, W.J.H.; Hanna, J.N. Clinical Features of Hospitalized Patients During Dengue-3 Epidemic in Far North Queensland, 1997–1999. Dengue Bull. 2002, 23, 24–29. [Google Scholar]
- Jayarajah, U.; Madarasinghe, M.; Hapugoda, D.; Dissanayake, U.; Perera, L.; Kannangara, V.; Udayangani, C.; Peiris, R.; Yasawardene, P.; De Zoysa, I.; et al. Clinical and Biochemical Characteristics of Dengue Infections in Children From Sri Lanka. Glob. Pediatr. Health 2020, 7, 2333794X20974207. [Google Scholar] [CrossRef] [PubMed]
- Talib, S.; Bhattu, S.; Bhattu, R.; Deshpande, S.; Dahiphale, D. Dengue fever triggering systemic lupus erythematosus and lupus nephritis: A case report. Int. Med. Case Rep. J. 2013, 6, 71. [Google Scholar] [CrossRef] [PubMed]
- Lizarraga, K.J.; Florindez, J.A.; Daftarian, P.; Andrews, D.M.; Ortega, L.M.; Mendoza, J.M.; Contreras, G.N.; Nayer, A. Anti-GBM disease and ANCA during dengue infection. Clin. Nephrol. 2015, 83, 104–110. [Google Scholar] [CrossRef]
- Gutiérrez-Bugallo, G.; Piedra, L.A.; Rodriguez, M.; Bisset, J.A.; Lourenço-de-Oliveira, R.; Weaver, S.C.; Vasilakis, N.; Vega-Rúa, A. Vector-borne transmission and evolution of Zika virus. Nat. Ecol. Evol. 2019, 3, 561–569. [Google Scholar] [CrossRef]
- Calvet, G.; Aguiar, R.S.; Melo, A.S.O.; Sampaio, S.A.; de Filippis, I.; Fabri, A.; Araujo, E.S.M.; de Sequeira, P.C.; de Mendonça, M.C.L.; de Oliveira, L.; et al. Detection and sequencing of Zika virus from amniotic fluid of fetuses with microcephaly in Brazil: A case study. Lancet Infect. Dis. 2016, 16, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Sirohi, D.; Kuhn, R.J. Zika Virus Structure, Maturation, and Receptors. J. Infect. Dis. 2017, 216, S935–S944. [Google Scholar] [CrossRef]
- Donald, C.L.; Brennan, B.; Cumberworth, S.L.; Rezelj, V.V.; Clark, J.J.; Cordeiro, M.T.; Freitas de Oliveira França, R.; Pena, L.J.; Wilkie, G.S.; Da Silva Filipe, A.; et al. Full Genome Sequence and sfRNA Interferon Antagonist Activity of Zika Virus from Recife, Brazil. PLoS Negl. Trop. Dis. 2016, 10, e0005048. [Google Scholar] [CrossRef]
- Dang, J.W.; Tiwari, S.K.; Qin, Y.; Rana, T.M. Genome-wide Integrative Analysis of Zika-Virus-Infected Neuronal Stem Cells Reveals Roles for MicroRNAs in Cell Cycle and Stemness. Cell Rep. 2019, 27, 3618–3628.e5. [Google Scholar] [CrossRef] [PubMed]
- Baril, M.; Racine, M.-E.; Penin, F.; Lamarre, D. MAVS Dimer Is a Crucial Signaling Component of Innate Immunity and the Target of Hepatitis C Virus NS3/4A Protease. J. Virol. 2009, 83, 1299–1311. [Google Scholar] [CrossRef]
- Glasner, A.; Oiknine-Djian, E.; Weisblum, Y.; Diab, M.; Panet, A.; Wolf, D.G.; Mandelboim, O. Zika Virus Escapes NK Cell Detection by Upregulating Major Histocompatibility Complex Class I Molecules. J. Virol. 2017, 91, e00785-17. [Google Scholar] [CrossRef]
- Branche, E.; Wang, Y.-T.; Viramontes, K.M.; Valls Cuevas, J.M.; Xie, J.; Ana-Sosa-Batiz, F.; Shafee, N.; Duttke, S.H.; McMillan, R.E.; Clark, A.E.; et al. SREBP2-dependent lipid gene transcription enhances the infection of human dendritic cells by Zika virus. Nat. Commun. 2022, 13, 5341. [Google Scholar] [CrossRef]
- Heinz, F.X.; Stiasny, K. The Antigenic Structure of Zika Virus and Its Relation to Other Flaviviruses: Implications for Infection and Immunoprophylaxis. Microbiol. Mol. Biol. Rev. 2017, 81, e00055-16. [Google Scholar] [CrossRef] [PubMed]
- Estevez-Herrera, J.; Perez-Yanes, S.; Cabrera-Rodriguez, R.; Marquez-Arce, D.; Trujillo-Gonzalez, R.; Machado, J.D.; Madrid, R.; Valenzuela-Fernandez, A. Zika Virus Pathogenesis: A Battle for Immune Evasion. Vaccines 2021, 9, 294. [Google Scholar] [CrossRef]
- Slonchak, A.; Hugo, L.E.; Freney, M.E.; Hall-Mendelin, S.; Amarilla, A.A.; Torres, F.J.; Setoh, Y.X.; Peng, N.Y.G.; Sng, J.D.J.; Hall, R.A.; et al. Zika virus noncoding RNA suppresses apoptosis and is required for virus transmission by mosquitoes. Nat. Commun. 2020, 11, 2205. [Google Scholar] [CrossRef]
- Gratton, R.; Agrelli, A.; Tricarico, P.M.; Brandao, L.; Crovella, S. Autophagy in Zika Virus Infection: A Possible Therapeutic Target to Counteract Viral Replication. Int. J. Mol. Sci. 2019, 20, 1048. [Google Scholar] [CrossRef] [PubMed]
- Brasil, P.; Pereira, J.P.; Moreira, M.E.; Ribeiro Nogueira, R.M.; Damasceno, L.; Wakimoto, M.; Rabello, R.S.; Valderramos, S.G.; Halai, U.-A.; Salles, T.S.; et al. Zika Virus Infection in Pregnant Women in Rio de Janeiro. N. Engl. J. Med. 2016, 375, 2321–2334. [Google Scholar] [CrossRef]
- Retallack, H.; Di Lullo, E.; Arias, C.; Knopp, K.A.; Laurie, M.T.; Sandoval-Espinosa, C.; Mancia Leon, W.R.; Krencik, R.; Ullian, E.M.; Spatazza, J.; et al. Zika virus cell tropism in the developing human brain and inhibition by azithromycin. Proc. Natl. Acad. Sci. USA 2016, 113, 14408–14413. [Google Scholar] [CrossRef]
- Woods, C.G.; Parker, A. Investigating microcephaly. Arch. Dis. Child. 2013, 98, 707–713. [Google Scholar] [CrossRef]
- Vannucci, R.C.; Barron, T.F.; Vannucci, S.J. Craniometric measures of microcephaly using MRI. Early Hum. Dev. 2012, 88, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Hammack, C.; Ogden, S.C.; Wen, Z.; Qian, X.; Li, Y.; Yao, B.; Shin, J.; Zhang, F.; Lee, E.M.; et al. Zika Virus Infects Human Cortical Neural Progenitors and Attenuates Their Growth. Cell Stem Cell 2016, 18, 587–590. [Google Scholar] [CrossRef]
- Forrest, C.M.; Khalil, O.S.; Pisar, M.; Smith, R.A.; Darlington, L.G.; Stone, T.W. Prenatal activation of Toll-like receptors-3 by administration of the viral mimetic poly(I:C) changes synaptic proteins, N-methyl-D-aspartate receptors and neurogenesis markers in offspring. Mol. Brain 2012, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Santi, L.; Riesgo, R.S.; Quincozes-Santos, A.; Schuler-Faccini, L.; Tureta, E.F.; Rosa, R.L.; Berger, M.; Oliveira, A.C.C.; Beltrão-Braga, P.C.B.; Souza, D.O.; et al. Zika Virus Infection Associated with Autism Spectrum Disorder: A Case Report. Neuroimmunomodulation 2021, 28, 229–232. [Google Scholar] [CrossRef]
- Blackmon, K.; Waechter, R.; Landon, B.; Noël, T.; Macpherson, C.; Donald, T.; Cudjoe, N.; Evans, R.; Burgen, K.S.; Jayatilake, P.; et al. Epilepsy surveillance in normocephalic children with and without prenatal Zika virus exposure. PLoS Negl. Trop. Dis. 2020, 14, e0008874. [Google Scholar] [CrossRef]
- Kung, P.-L.; Chou, T.-W.; Lindman, M.; Chang, N.P.; Estevez, I.; Buckley, B.D.; Atkins, C.; Daniels, B.P. Zika virus-induced TNF-α signaling dysregulates expression of neurologic genes associated with psychiatric disorders. J. Neuroinflamm. 2022, 19, 100. [Google Scholar] [CrossRef]
- Gerakis, Y.; Hetz, C. Emerging roles of ER stress in the etiology and pathogenesis of Alzheimer’s disease. FEBS J. 2018, 285, 995–1011. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-E.; Choi, H.; Shin, N.; Kong, D.; Kim, N.G.; Kim, H.-Y.; Kim, M.-J.; Choi, S.W.; Kim, Y.B.; Kang, K.-S. Zika virus infection accelerates Alzheimer’s disease phenotypes in brain organoids. Cell Death Discov. 2022, 8, 153. [Google Scholar] [CrossRef] [PubMed]
- Janssens, S.; Schotsaert, M.; Karnik, R.; Balasubramaniam, V.; Dejosez, M.; Meissner, A.; García-Sastre, A.; Zwaka, T.P. Zika Virus Alters DNA Methylation of Neural Genes in an Organoid Model of the Developing Human Brain. mSystems 2018, 3, e00219-17. [Google Scholar] [CrossRef]
- Cao-Lormeau, V.-M.; Blake, A.; Mons, S.; Lastère, S.; Roche, C.; Vanhomwegen, J.; Dub, T.; Baudouin, L.; Teissier, A.; Larre, P.; et al. Guillain-Barré Syndrome outbreak associated with Zika virus infection in French Polynesia: A case-control study. Lancet 2016, 387, 1531–1539. [Google Scholar] [CrossRef]
- Lucchese, G.; Kanduc, D. Zika virus and autoimmunity: From microcephaly to Guillain-Barré syndrome, and beyond. Autoimmun. Rev. 2016, 15, 801–808. [Google Scholar] [CrossRef]
- Chen, J.; Yang, Y.-F.; Chen, J.; Zhou, X.; Dong, Z.; Chen, T.; Yang, Y.; Zou, P.; Jiang, B.; Hu, Y.; et al. Zika virus infects renal proximal tubular epithelial cells with prolonged persistency and cytopathic effects. Emerg. Microbes Infect. 2017, 6, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Tang, L.; Tang, H.; Pu, J.; Gong, S.; Fang, D.; Zhang, H.; Li, Y.-P.; Zhu, X.; Wang, W.; et al. Zika Virus Infection Induces Acute Kidney Injury Through Activating NLRP3 Inflammasome Via Suppressing Bcl-2. Front. Immunol. 2019, 10, 1925. [Google Scholar] [CrossRef]
- Costa Monteiro, L.M.; Cruz, G.N.d.O.; Fontes, J.M.; Saad Salles, T.R.D.; Boechat, M.C.B.; Monteiro, A.C.; Moreira, M.E.L. Neurogenic bladder findings in patients with Congenital Zika Syndrome: A novel condition. PLoS ONE 2018, 13, e0193514. [Google Scholar] [CrossRef]
- Davenport, B.J.; Bullock, C.; McCarthy, M.K.; Hawman, D.W.; Murphy, K.M.; Kedl, R.M.; Diamond, M.S.; Morrison, T.E. Chikungunya Virus Evades Antiviral CD8+ T Cell Responses To Establish Persistent Infection in Joint-Associated Tissues. J. Virol. 2020, 94, e02036-19. [Google Scholar] [CrossRef]
- Simmons, J.D.; White, L.J.; Morrison, T.E.; Montgomery, S.A.; Whitmore, A.C.; Johnston, R.E.; Heise, M.T. Venezuelan Equine Encephalitis Virus Disrupts STAT1 Signaling by Distinct Mechanisms Independent of Host Shutoff. J. Virol. 2009, 83, 10571–10581. [Google Scholar] [CrossRef] [PubMed]
- Göertz, G.P.; McNally, K.L.; Robertson, S.J.; Best, S.M.; Pijlman, G.P.; Fros, J.J. The Methyltransferase-Like Domain of Chikungunya Virus nsP2 Inhibits the Interferon Response by Promoting the Nuclear Export of STAT1. J. Virol. 2018, 92, e01008-18. [Google Scholar] [CrossRef]
- Burdeinick-Kerr, R.; Govindarajan, D.; Griffin, D.E. Noncytolytic Clearance of Sindbis Virus Infection from Neurons by Gamma Interferon Is Dependent on Jak/Stat Signaling. J. Virol. 2009, 83, 3429–3435. [Google Scholar] [CrossRef]
- Bae, S.; Lee, J.Y.; Myoung, J. Chikungunya Virus-Encoded nsP2, E2 and E1 Strongly Antagonize the Interferon-β Signaling Pathway. J. Microbiol. Biotechnol. 2019, 29, 1852–1859. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.H.; Maric, M.; Madison, M.N.; Maury, W.; Roller, R.J.; Okeoma, C.M. BST-2/tetherin-mediated restriction of chikungunya (CHIKV) VLP budding is counteracted by CHIKV non-structural protein 1 (nsP1). Virology 2013, 438, 37–49. [Google Scholar] [CrossRef]
- Li, M.M.H.; Aguilar, E.G.; Michailidis, E.; Pabon, J.; Park, P.; Wu, X.; Jong, Y.P.d.; Schneider, W.M.; Molina, H.; Rice, C.M.; et al. Characterization of Novel Splice Variants of Zinc Finger Antiviral Protein (ZAP). J. Virol. 2019, 93, e00715-19. [Google Scholar] [CrossRef]
- Webb, L.G.; Veloz, J.; Pintado-Silva, J.; Zhu, T.; Rangel, M.V.; Mutetwa, T.; Zhang, L.; Bernal-Rubio, D.; Figueroa, D.; Carrau, L.; et al. Chikungunya virus antagonizes cGAS-STING mediated type-I interferon responses by degrading cGAS. PLoS Pathog. 2020, 16, e1008999. [Google Scholar] [CrossRef]
- Labadie, K.; Larcher, T.; Joubert, C.; Mannioui, A.; Delache, B.; Brochard, P.; Guigand, L.; Dubreil, L.; Lebon, P.; Verrier, B.; et al. Chikungunya disease in nonhuman primates involves long-term viral persistence in macrophages. J. Clin. Investig. 2010, 120, 894–906. [Google Scholar] [CrossRef] [PubMed]
- Mathur, K.; Anand, A.; Dubey, S.K.; Sanan-Mishra, N.; Bhatnagar, R.K.; Sunil, S. Analysis of chikungunya virus proteins reveals that non-structural proteins nsP2 and nsP3 exhibit RNA interference (RNAi) suppressor activity. Sci. Rep. 2016, 6, 38065. [Google Scholar] [CrossRef]
- Qian, Q.; Zhou, H.; Shu, T.; Mu, J.; Fang, Y.; Xu, J.; Li, T.; Kong, J.; Qiu, Y.; Zhou, X. The Capsid Protein of Semliki Forest Virus Antagonizes RNA Interference in Mammalian Cells. J. Virol. 2020, 94, e01233-19. [Google Scholar] [CrossRef]
- Rangel, M.V.; McAllister, N.; Dancel-Manning, K.; Noval, M.G.; Silva, L.A.; Stapleford, K.A. Emerging Chikungunya Virus Variants at the E1-E1 Interglycoprotein Spike Interface Impact Virus Attachment and Inflammation. J. Virol. 2022, 96, e0158621. [Google Scholar] [CrossRef]
- Tanabe, I.S.B.; Tanabe, E.L.L.; Santos, E.C.; Martins, W.V.; Araujo, I.; Cavalcante, M.C.A.; Lima, A.R.V.; Camara, N.O.S.; Anderson, L.; Yunusov, D.; et al. Cellular and Molecular Immune Response to Chikungunya Virus Infection. Front. Cell. Infect. Microbiol. 2018, 8, 345. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.A.; Dermody, T.S. Chikungunya virus: Epidemiology, replication, disease mechanisms, and prospective intervention strategies. J. Clin. Investig. 2017, 127, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Morrison, C.R.; Plante, K.S.; Heise, M.T. Chikungunya Virus: Current Perspectives on a Reemerging Virus. Microbiol. Spectr. 2016, 4, 143–161. [Google Scholar] [CrossRef]
- Kleinert, R.D.V.; Montoya-Diaz, E.; Khera, T.; Welsch, K.; Tegtmeyer, B.; Hoehl, S.; Ciesek, S.; Brown, R.J.P. Yellow Fever: Integrating Current Knowledge with Technological Innovations to Identify Strategies for Controlling a Re-Emerging Virus. Viruses 2019, 11, 960. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int. Immunol. 2009, 21, 317–337. [Google Scholar] [CrossRef]
- Suthar, M.S.; Aguirre, S.; Fernandez-Sesma, A. Innate Immune Sensing of Flaviviruses. PLoS Pathog. 2013, 9, e1003541. [Google Scholar] [CrossRef]
- Guo, H.-Y.; Zhang, X.-C.; Jia, R.-Y. Toll-Like Receptors and RIG-I-Like Receptors Play Important Roles in Resisting Flavivirus. J. Immunol. Res. 2018, 2018, 6106582. [Google Scholar] [CrossRef]
- Schuberth-Wagner, C.; Ludwig, J.; Bruder, A.K.; Herzner, A.-M.; Zillinger, T.; Goldeck, M.; Schmidt, T.; Schmid-Burgk, J.L.; Kerber, R.; Wolter, S.; et al. A Conserved Histidine in the RNA Sensor RIG-I Controls Immune Tolerance to N12′O-Methylated Self RNA. Immunity 2015, 43, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Laurent-Rolle, M.; Morrison, J.; Rajsbaum, R.; Macleod, J.M.L.; Pisanelli, G.; Pham, A.; Ayllon, J.; Miorin, L.; Martínez-Romero, C.; Tenoever, B.R.; et al. The Interferon Signaling Antagonist Function of Yellow Fever Virus NS5 Protein Is Activated by Type I Interferon. Cell Host Microbe 2014, 16, 314–327. [Google Scholar] [CrossRef]
- ter Meulen, J.; Sakho, M.; Koulemou, K.; Magassouba, N.F.; Bah, A.; Preiser, W.; Daffis, S.; Klewitz, C.; Bae, H.-G.; Niedrig, M.; et al. Activation of the Cytokine Network and Unfavorable Outcome in Patients with Yellow Fever. J. Infect. Dis. 2004, 190, 1821–1827. [Google Scholar] [CrossRef]
- Quaresma, J.A.S.; Barros, V.L.R.S.; Pagliari, C.; Fernandes, E.R.; Guedes, F.; Takakura, C.F.H.; Andrade, H.F.; Vasconcelos, P.F.C.; Duarte, M.I.S. Revisiting the liver in human yellow fever: Virus-induced apoptosis in hepatocytes associated with TGF-β, TNF-α and NK cells activity. Virology 2006, 345, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Quaresma, J.A.S.; Barros, V.L.R.S.; Fernandes, E.R.; Pagliari, C.; Guedes, F.; Vasconcelos, P.F.d.C.; Andrade Junior, H.F.d.; Duarte, M.I.S. Immunohistochemical examination of the role of Fas ligand and lymphocytes in the pathogenesis of human liver yellow fever. Virus Res. 2006, 116, 91–97. [Google Scholar] [CrossRef]
- Li, M.O.; Wan, Y.Y.; Sanjabi, S.; Robertson, A.-K.L.; Flavell, R.A. Transforming Growth Factor-Β Regulation of Immune Responses. Annu. Rev. Immunol. 2006, 24, 99–146. [Google Scholar] [CrossRef]
- Stock, N.K.; Laraway, H.; Faye, O.; Diallo, M.; Niedrig, M.; Sall, A.A. Biological and phylogenetic characteristics of yellow fever virus lineages from West Africa. J. Virol. 2013, 87, 2895–2907. [Google Scholar] [CrossRef] [PubMed]
- Collins, N.D.; Barrett, A.D. Live Attenuated Yellow Fever 17D Vaccine: A Legacy Vaccine Still Controlling Outbreaks In Modern Day. Curr. Infect. Dis. Rep. 2017, 19, 14. [Google Scholar] [CrossRef] [PubMed]
- Azamor, T.; da Silva, A.M.V.; Melgaco, J.G.; Dos Santos, A.P.; Xavier-Carvalho, C.; Alvarado-Arnez, L.E.; Batista-Silva, L.R.; de Souza Matos, D.C.; Bayma, C.; Missailidis, S.; et al. Activation of an Effective Immune Response after Yellow Fever Vaccination Is Associated with the Genetic Background and Early Response of IFN-gamma and CLEC5A. Viruses 2021, 13, 96. [Google Scholar] [CrossRef]
- Siam, A.L.; Meegan, J.M.; Gharbawi, K.F. Rift Valley fever ocular manifestations: Observations during the 1977 epidemic in Egypt. Br. J. Ophthalmol. 1980, 64, 366–374. [Google Scholar] [CrossRef]
- Wang, X.; Yuan, Y.; Liu, Y.; Zhang, L. Arm race between Rift Valley fever virus and host. Front. Immunol. 2022, 13, 230. [Google Scholar] [CrossRef]
- Brisse, M.; Ly, H. Comparative Structure and Function Analysis of the RIG-I-Like Receptors: RIG-I and MDA5. Front. Immunol. 2019, 10, 1586. [Google Scholar] [CrossRef]
- Havranek, K.E.; White, L.A.; Lanchy, J.-M.; Lodmell, J.S. Transcriptome profiling in Rift Valley fever virus infected cells reveals modified transcriptional and alternative splicing programs. PLoS ONE 2019, 14, e0217497. [Google Scholar] [CrossRef]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Jesus, P.D.D.; Su, V.; Han, S.; Gong, D.; Wu, N.C.; Tian, Y.; Li, X.; Wu, T.-T.; Chanda, S.K.; et al. RIOK3 Is an Adaptor Protein Required for IRF3-Mediated Antiviral Type I Interferon Production. J. Virol. 2014, 88, 7987–7997. [Google Scholar] [CrossRef]
- Bisom, T.C.; White, L.A.; Lanchy, J.-M.; Lodmell, J.S. RIOK3 and Its Alternatively Spliced Isoform Have Disparate Roles in the Innate Immune Response to Rift Valley Fever Virus (MP12) Infection. Viruses 2022, 14, 2064. [Google Scholar] [CrossRef]
- White, L.A.; Bisom, T.C.; Grimes, H.L.; Hayashi, M.; Lanchy, J.-M.; Lodmell, J.S. Tra2beta-Dependent Regulation of RIO Kinase 3 Splicing During Rift Valley Fever Virus Infection Underscores the Links between Alternative Splicing and Innate Antiviral Immunity. Front. Cell. Infect. Microbiol. 2022, 11, 24. [Google Scholar] [CrossRef] [PubMed]
- Ly, H.J.; Ikegami, T. Rift Valley fever virus NSs protein functions and the similarity to other bunyavirus NSs proteins. Virol. J. 2016, 13, 118. [Google Scholar] [CrossRef]
- Le May, N.; Mansuroglu, Z.; Léger, P.; Josse, T.; Blot, G.; Billecocq, A.; Flick, R.; Jacob, Y.; Bonnefoy, E.; Bouloy, M. A SAP30 Complex Inhibits IFN-β Expression in Rift Valley Fever Virus Infected Cells. PLoS Pathog. 2008, 4, e13. [Google Scholar] [CrossRef]
- Le May, N.; Dubaele, S.; De Santis, L.P.; Billecocq, A.; Bouloy, M.; Egly, J.-M. TFIIH Transcription Factor, a Target for the Rift Valley Hemorrhagic Fever Virus. Cell 2004, 116, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Kalveram, B.; Lihoradova, O.; Ikegami, T. NSs Protein of Rift Valley Fever Virus Promotes Posttranslational Downregulation of the TFIIH Subunit p62. J. Virol. 2011, 85, 234–6243. [Google Scholar] [CrossRef]
- Kainulainen, M.; Habjan, M.; Hubel, P.; Busch, L.; Lau, S.; Colinge, J.; Superti-Furga, G.; Pichlmair, A.; Weber, F. Virulence Factor NSs of Rift Valley Fever Virus Recruits the F-Box Protein FBXO3 To Degrade Subunit p62 of General Transcription Factor TFIIH. J. Virol. 2014, 88, 3464–3473. [Google Scholar] [CrossRef]
- Mansuroglu, Z.; Josse, T.; Gilleron, J.; Billecocq, A.; Leger, P.; Bouloy, M.; Bonnefoy, E. Nonstructural NSs Protein of Rift Valley Fever Virus Interacts with Pericentromeric DNA Sequences of the Host Cell, Inducing Chromosome Cohesion and Segregation Defects. J. Virol. 2010, 84, 928–939. [Google Scholar] [CrossRef] [PubMed]
- Baer, A.; Austin, D.; Narayanan, A.; Popova, T.; Kainulainen, M.; Bailey, C.; Kashanchi, F.; Weber, F.; Kehn-Hall, K. Induction of DNA Damage Signaling upon Rift Valley Fever Virus Infection Results in Cell Cycle Arrest and Increased Viral Replication. J. Biol. Chem. 2012, 287, 7399–7410. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.-H.; Liao, C.-L.; Lin, Y.-L. Flavivirus induces interferon-beta gene expression through a pathway involving RIG-I-dependent IRF-3 and PI3K-dependent NF-κB activation. Microbes Infect. 2006, 8, 157–171. [Google Scholar] [CrossRef]
- Jin, R.; Zhu, W.; Cao, S.; Chen, R.; Jin, H.; Liu, Y.; Wang, S.; Wang, W.; Xiao, G. Japanese encephalitis virus activates autophagy as a viral immune evasion strategy. PLoS ONE 2013, 8, e52909. [Google Scholar] [CrossRef]
- Lin, R.J.; Chang, B.L.; Yu, H.P.; Liao, C.L.; Lin, Y.L. Blocking of interferon-induced Jak-Stat signaling by Japanese encephalitis virus NS5 through a protein tyrosine phosphatase-mediated mechanism. J. Virol. 2006, 80, 5908–5918. [Google Scholar] [CrossRef]
- Li, Q.; Zhou, D.; Jia, F.; Zhang, L.; Ashraf, U.; Li, Y.; Duan, H.; Song, Y.; Chen, H.; Cao, S.; et al. Japanese Encephalitis Virus NS1′ Protein Interacts with Host CDK1 Protein to Regulate Antiviral Response. Microbiol. Spectr. 2021, 9, e01661-21. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, N. Myeloid-Derived Suppressor Cells Inhibit T Follicular Helper Cell Immune Response in Japanese Encephalitis Virus Infection. J. Immunol. 2017, 199, 3094–3105. [Google Scholar] [CrossRef]
- Aleyas, A.G.; George, J.A.; Han, Y.W.; Rahman, M.M.; Kim, S.J.; Han, S.B.; Kim, B.S.; Kim, K.; Eo, S.K. Functional modulation of dendritic cells and macrophages by Japanese encephalitis virus through MyD88 adaptor molecule-dependent and -independent pathways. J. Immunol. 2009, 183, 2462–2474. [Google Scholar] [CrossRef]
- Kundu, K.; Dutta, K.; Nazmi, A.; Basu, A. Japanese encephalitis virus infection modulates the expression of suppressors of cytokine signaling (SOCS) in macrophages: Implications for the hosts’ innate immune response. Cell. Immunol. 2013, 285, 100–110. [Google Scholar] [CrossRef]
- Adhya, D.; Dutta, K.; Kundu, K.; Basu, A. Histone deacetylase inhibition by Japanese encephalitis virus in monocyte/macrophages: A novel viral immune evasion strategy. Immunobiology 2013, 218, 1235–1247. [Google Scholar] [CrossRef]
- Rastogi, M.; Singh, S.K. Japanese Encephalitis Virus exploits microRNA-155 to suppress the non-canonical NF-κB pathway in human microglial cells. Biochim. Biophys. Acta (BBA)—Gene Regul. Mech. 2020, 1863, 194639. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.; Jin, W.; Sun, S.C. Peli1 facilitates TRIF-dependent Toll-like receptor signaling and proinflammatory cytokine production. Nat. Immunol. 2009, 10, 1089–1095. [Google Scholar] [CrossRef] [PubMed]
- Hazra, B.; Chakraborty, S.; Basu, A. miR-301a mediated immune evasion by Japanese encephalitis virus. Oncotarget 2017, 8, 90620–90621. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.-Y.; Hsu, T.-W.; Chen, Y.-L.; Liu, S.-F.; Tsai, Y.-J.; Lin, Y.-T.; Chen, Y.-S.; Fan, Y.-H. Japanese encephalitis virus non-coding RNA inhibits activation of interferon by blocking nuclear translocation of interferon regulatory factor 3. Vet. Microbiol. 2013, 166, 11–21. [Google Scholar] [CrossRef]
- Han, N.; Adams, J.; Chen, P.; Guo, Z.Y.; Zhong, X.F.; Fang, W.; Li, N.; Wen, L.; Tao, X.Y.; Yuan, Z.M.; et al. Comparison of genotypes I and III in Japanese encephalitis virus reveals distinct differences in their genetic and host diversity. J. Virol. 2014, 88, 11469–11479. [Google Scholar] [CrossRef] [PubMed]
- Khare, B.; Kuhn, R.J. The Japanese Encephalitis Antigenic Complex Viruses: From Structure to Immunity. Viruses 2022, 14, 2213. [Google Scholar] [CrossRef]
- Fadnis, P.R.; Ravi, V.; Desai, A.; Turtle, L.; Solomon, T. Innate immune mechanisms in Japanese encephalitis virus infection: Effect on transcription of pattern recognition receptors in mouse neuronal cells and brain tissue. Viral Immunol. 2013, 26, 366–377. [Google Scholar] [CrossRef]
- Fan, Y.C.; Chen, Y.Y.; Chen, J.M.; Huang, C.; Huang, M.; Chiou, S.S. Effectiveness of Live-Attenuated Genotype III Japanese Encephalitis Viral Vaccine against Circulating Genotype I Viruses in Swine. Viruses 2022, 14, 114. [Google Scholar] [CrossRef]
- Yun, S.I.; Lee, Y.M. Japanese encephalitis: The virus and vaccines. Hum. Vaccines Immunother. 2014, 10, 263–279. [Google Scholar] [CrossRef]
- Sharma, K.B.; Vrati, S.; Kalia, M. Pathobiology of Japanese encephalitis virus infection. Mol. Asp. Med. 2021, 81, 100994. [Google Scholar] [CrossRef]
- Getts, D.R.; Terry, R.L.; Getts, M.T.; Muüller, M.; Rana, S.; Shrestha, B.; Radford, J.; Van Rooijen, N.; Campbell, I.L.; King, N.J.C. Ly6c+ “inflammatory monocytes” are microglial precursors recruited in a pathogenic manner in West Nile virus encephalitis. J. Exp. Med. 2008, 205, 2319–2337. [Google Scholar] [CrossRef]
- Fredericksen, B.L.; Smith, M.; Katze, M.G.; Shi, P.-Y.; Gale, M. The Host Response to West Nile Virus Infection Limits Viral Spread through the Activation of the Interferon Regulatory Factor 3 Pathway. J. Virol. 2004, 78, 7737–7747. [Google Scholar] [CrossRef]
- Wilson, J.R.; Sessions, P.F.d.; Leon, M.A.; Scholle, F. West Nile Virus Nonstructural Protein 1 Inhibits TLR3 Signal Transduction. J. Virol. 2008, 82, 8262–8271. [Google Scholar] [CrossRef] [PubMed]
- Keller, B.C.; Fredericksen, B.L.; Samuel, M.A.; Mock, R.E.; Mason, P.W.; Diamond, M.S.; Gale, M. Resistance to Alpha/Beta Interferon Is a Determinant of West Nile Virus Replication Fitness and Virulence. J. Virol. 2006, 80, 9424–9434. [Google Scholar] [CrossRef]
- Evans, J.D.; Crown, R.A.; Sohn, J.A.; Seeger, C. West Nile Virus Infection Induces Depletion of IFNAR1 Protein Levels. Viral Immunol. 2011, 24, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Fensterl, V.; Sen, G.C. The ISG56/IFIT1 Gene Family. J. Interferon Cytokine Res. 2011, 31, 71–78. [Google Scholar] [CrossRef]
- Daffis, S.; Szretter, K.J.; Schriewer, J.; Li, J.; Youn, S.; Errett, J.; Lin, T.-Y.; Schneller, S.; Zust, R.; Dong, H.; et al. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 2010, 468, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Krishna, V.D.; Rangappa, M.; Satchidanandam, V. Virus-Specific Cytolytic Antibodies to Nonstructural Protein 1 of Japanese Encephalitis Virus Effect Reduction of Virus Output from Infected Cells. J. Virol. 2009, 83, 4766–4777. [Google Scholar] [CrossRef]
- Avirutnan, P.; Hauhart, R.E.; Somnuke, P.; Blom, A.M.; Diamond, M.S.; Atkinson, J.P. Binding of Flavivirus Nonstructural Protein NS1 to C4b Binding Protein Modulates Complement Activation. J. Immunol. 2011, 187, 424–433. [Google Scholar] [CrossRef]
- Cho, H.; Diamond, M.S. Immune responses to West Nile virus infection in the central nervous system. Viruses 2012, 4, 3812–3830. [Google Scholar] [CrossRef]
- Bakonyi, T.; Ivanics, E.; Erdelyi, K.; Ursu, K.; Ferenczi, E.; Weissenbock, H.; Nowotny, N. Lineage 1 and 2 strains of encephalitic West Nile virus, central Europe. Emerg. Infect. Dis. 2006, 12, 618–623. [Google Scholar] [CrossRef]
- Arjona, A.; Wang, P.; Montgomery, R.R.; Fikrig, E. Innate immune control of West Nile virus infection. Cell. Microbiol. 2011, 13, 1648–1658. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Wang, T. Recent advances in understanding West Nile virus host immunity and viral pathogenesis. F1000Research 2018, 7, 338. [Google Scholar] [CrossRef]
- Gerardin, P.; Couderc, T.; Bintner, M.; Tournebize, P.; Renouil, M.; Lemant, J.; Boisson, V.; Borgherini, G.; Staikowsky, F.; Schramm, F.; et al. Chikungunya virus-associated encephalitis: A cohort study on La Reunion Island, 2005–2009. Neurology 2016, 86, 94–102. [Google Scholar] [CrossRef]
- Sissoko, D.; Malvy, D.; Giry, C.; Delmas, G.; Paquet, C.; Gabrie, P.; Pettinelli, F.; Sanquer, M.A.; Pierre, V. Outbreak of Chikungunya fever in Mayotte, Comoros archipelago, 2005–2006. Trans. R Soc. Trop. Med. Hyg. 2008, 102, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Robillard, P.Y.; Boumahni, B.; Gerardin, P.; Michault, A.; Fourmaintraux, A.; Schuffenecker, I.; Carbonnier, M.; Djemili, S.; Choker, G.; Roge-Wolter, M.; et al. Vertical maternal fetal transmission of the chikungunya virus. Ten cases among 84 pregnant women. Presse Med. 2006, 35, 785–788. [Google Scholar] [CrossRef]
- Shenoy, S.; Pradeep, G.C. Neurodevelopmental outcome of neonates with vertically transmitted Chikungunya fever with encephalopathy. Indian Pediatr. 2012, 49, 238–240. [Google Scholar] [PubMed]
- Villamil-Gomez, W.; Alba-Silvera, L.; Menco-Ramos, A.; Gonzalez-Vergara, A.; Molinares-Palacios, T.; Barrios-Corrales, M.; Rodriguez-Morales, A.J. Congenital Chikungunya Virus Infection in Sincelejo, Colombia: A Case Series. J. Trop. Pediatr. 2015, 61, 386–392. [Google Scholar] [CrossRef]
- Torres, J.R.; Falleiros-Arlant, L.H.; Duenas, L.; Pleitez-Navarrete, J.; Salgado, D.M.; Castillo, J.B. Congenital and perinatal complications of chikungunya fever: A Latin American experience. Int. J. Infect. Dis. 2016, 51, 85–88. [Google Scholar] [CrossRef]
- Bandeira, A.C.; Campos, G.S.; Sardi, S.I.; Rocha, V.F.; Rocha, G.C. Neonatal encephalitis due to Chikungunya vertical transmission: First report in Brazil. IDCases 2016, 5, 57–59. [Google Scholar] [CrossRef]
- Samra, J.A.; Hagood, N.L.; Summer, A.; Medina, M.T.; Holden, K.R. Clinical Features and Neurologic Complications of Children Hospitalized with Chikungunya Virus in Honduras. J. Child. Neurol. 2017, 32, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Karthiga, V.; Kommu, P.P.; Krishnan, L. Perinatal chikungunya in twins. J. Pediatr. Neurosci. 2016, 11, 223–224. [Google Scholar] [CrossRef] [PubMed]
- Ramos, R.; Viana, R.; Brainer-Lima, A.; Flore Ancio, T.; Carvalho, M.D.; van der Linden, V.; Amorim, A.; Rocha, M.A.; Medeiros, F. Perinatal Chikungunya Virus-associated Encephalitis Leading to Postnatal-Onset Microcephaly and Optic Atrophy. Pediatr. Infect. Dis. J. 2018, 37, 94–95. [Google Scholar] [CrossRef]
- Gerardin, P.; Barau, G.; Michault, A.; Bintner, M.; Randrianaivo, H.; Choker, G.; Lenglet, Y.; Touret, Y.; Bouveret, A.; Grivard, P.; et al. Multidisciplinary prospective study of mother-to-child chikungunya virus infections on the island of La Reunion. PLoS Med. 2008, 5, e60. [Google Scholar] [CrossRef]
- Ramful, D.; Carbonnier, M.; Pasquet, M.; Bouhmani, B.; Ghazouani, J.; Noormahomed, T.; Beullier, G.; Attali, T.; Samperiz, S.; Fourmaintraux, A.; et al. Mother-to-child transmission of Chikungunya virus infection. Pediatr. Infect. Dis. J. 2007, 26, 811–815. [Google Scholar] [CrossRef]
- Lyra, P.P.; Campos, G.S.; Bandeira, I.D.; Sardi, S.I.; Costa, L.F.; Santos, F.R.; Ribeiro, C.A.; Jardim, A.M.; Santiago, A.C.; de Oliveira, P.M.; et al. Congenital Chikungunya Virus Infection after an Outbreak in Salvador, Bahia, Brazil. AJP Rep. 2016, 6, e299–e300. [Google Scholar] [CrossRef]
- Simon, F.; Paule, P.; Oliver, M. Chikungunya Virus–Induced Myopericarditis: Toward an Increase of Dilated Cardiomyopathy in Countries with Epidemics? Am. J. Trop. Med. Hyg. 2008, 78, 212–213. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Vibha, D.; Srivastava, A.K.; Shukla, G.; Prasad, K. Guillain-Barre syndrome complicating chikungunya virus infection. J. Neurovirol. 2017, 23, 504–507. [Google Scholar] [CrossRef]
- Patwardhan, A.; Nalini, A.; Baishya, P.P.; Kulanthaivelu, K.; Krishnareddy, H.; Dutta, D.; Chawla, T.; Chowdary, R.M.; Yadav, R.; Vengalil, S. Case Report: Post-Chikungunya-Associated Myeloneuropathy. Am. J. Trop. Med. Hyg. 2021, 105, 942–945. [Google Scholar] [CrossRef]
- Pancharoen, C.; Thisyakorn, U. Neurological manifestations in dengue patients. Southeast Asian J. Trop. Med. Public Health 2001, 32, 341–345. [Google Scholar]
- Bopeththa, B.; Ralapanawa, U. Post encephalitic parkinsonism following dengue viral infection. BMC Res. Notes 2017, 10, 655. [Google Scholar] [CrossRef] [PubMed]
- Datta, G.; Mitra, P. A Study on Cardiac Manifestations of Dengue Fever. J. Assoc. Physicians India 2019, 67, 14–16. [Google Scholar]
- Wali, J.P.; Biswas, A.; Chandra, S.; Malhotra, A.; Aggarwal, P.; Handa, R.; Wig, N.; Bahl, V.K. Cardiac involvement in Dengue Haemorrhagic Fever. Int. J. Cardiol. 1998, 64, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Sheetal, S.; Jacob, E. A Study on the Cardiac Manifestations of Dengue. J. Assoc Physicians India 2016, 64, 30–34. [Google Scholar] [PubMed]
- Kularatne, S.A.; Pathirage, M.M.; Kumarasiri, P.V.; Gunasena, S.; Mahindawanse, S.I. Cardiac complications of a dengue fever outbreak in Sri Lanka, 2005. Trans. R. Soc. Trop. Med. Hyg. 2007, 101, 804–808. [Google Scholar] [CrossRef] [PubMed]
- da Costa, P.S.; Ribeiro, G.M.; Junior, C.S.; da Costa Campos, L. Severe thrombotic events associated with dengue fever, Brazil. Am. J. Trop. Med. Hyg. 2012, 87, 741–742. [Google Scholar] [CrossRef]
- Guadalajara-Boo, J.F.; Ruiz-Esparza, M.E.; Aranda Frausto, A.; Soto Abraham, M.V.; Gaspar-Hernandez, J. Histologic and angiographic imaging of acute shock dengue myocarditis. Rev. Esp. Cardiol. 2014, 67, 226–227. [Google Scholar] [CrossRef]
- Kularatne, S.A.M.; Rajapakse, M.M.; Ralapanawa, U.; Waduge, R.; Pathirage, L.; Rajapakse, R. Heart and liver are infected in fatal cases of dengue: Three PCR based case studies. BMC Infect. Dis. 2018, 18, 681. [Google Scholar] [CrossRef]
- Marques, N.; Gan, V.C.; Leo, Y.S. Dengue myocarditis in Singapore: Two case reports. Infection 2013, 41, 709–714. [Google Scholar] [CrossRef]
- Bich, T.D.; Pham, O.K.; Hai, D.H.; Nguyen, N.M.; Van, H.N.; The, T.D.; Wills, B.; Yacoub, S. A pregnant woman with acute cardiorespiratory failure: Dengue myocarditis. Lancet 2015, 385, 1260. [Google Scholar] [CrossRef]
- Naresh, G.; Kulkarni, A.V.; Sinha, N.; Jhamb, N.; Gulati, S. Dengue hemorrhagic fever complicated with encephalopathy and myocarditis: A case report. J. Commun. Dis. 2008, 40, 223–224. [Google Scholar] [PubMed]
- Chou, M.T.; Yu, W.L. Takotsubo cardiomyopathy in a patient with dengue fever. J. Formos. Med. Assoc. Taiwan Yi Zhi 2016, 115, 818–819. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, K.; Teo, L.; Raymond, W.C.; MacLaren, G. Dengue Myopericarditis Mimicking Acute Myocardial Infarction. Circulation 2015, 131, e519–e522. [Google Scholar] [CrossRef]
- Tayeb, B.; Piot, C.; Roubille, F. Acute pericarditis after dengue fever. Ann. Cardiol. D’angeiologie 2011, 60, 240–242. [Google Scholar] [CrossRef] [PubMed]
- Dhariwal, A.K.; Sanzgiri, P.S.; Nagvekar, V. High degree atrioventricular block with ventricular asystole in a case of dengue fever. Indian Heart J. 2016, 68 (Suppl. S2), S194–S197. [Google Scholar] [CrossRef] [PubMed]
- Navinan, M.R.; Yudhishdran, J.; Herath, S.; Liyanage, I.; Kugadas, T.; Kumara, D.; Kulatunga, A. Complete heart block in dengue complicating management of shock due to both bleeding and leakage: A case report. BMC Res. Notes 2015, 8, 68. [Google Scholar] [CrossRef]
- Sharda, M.; Gupt, A.; Nagar, D.; Soni, A.K. Dengue fever: An additional cause for bradycardia. J. Assoc. Physicians India 2014, 62, 362–363. [Google Scholar]
- Khongphatthallayothin, A.; Chotivitayatarakorn, P.; Somchit, S.; Mitprasart, A.; Sakolsattayadorn, S.; Thisyakorn, C. Morbitz type I second degree AV block during recovery from dengue hemorrhagic fever. Southeast Asian J. Trop. Med. Public Health 2000, 31, 642–645. [Google Scholar]
- Kaushik, J.S.; Gupta, P.; Rajpal, S.; Bhatt, S. Spontaneous resolution of sinoatrial exit block and atrioventricular dissociation in a child with dengue fever. Singap. Med. J. 2010, 51, e146–e148. [Google Scholar]
- Mahmod, M.; Darul, N.D.; Mokhtar, I.; Nor, N.M.; Anshar, F.M.; Maskon, O. Atrial fibrillation as a complication of dengue hemorrhagic fever: Non-self-limiting manifestation. Int. J. Infect. Dis. 2009, 13, e316–e318. [Google Scholar] [CrossRef]
- Horta Veloso, H.; Ferreira Junior, J.A.; Braga de Paiva, J.M.; Faria Honorio, J.; Junqueira Bellei, N.C.; Vicenzo de Paola, A.A. Acute atrial fibrillation during dengue hemorrhagic fever. Braz. J. Infect. Dis. 2003, 7, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.A.; Azhar, E.I.; El-Fiky, S.; Madani, H.H.; Abuljadial, M.A.; Ashshi, A.M.; Turkistani, A.M.; Hamouh, E.A. Clinical profile and outcome of hospitalized patients during first outbreak of dengue in Makkah, Saudi Arabia. Acta Trop. 2008, 105, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.C.; Lu, P.L.; Chang, J.M.; Lin, M.Y.; Tsai, J.J.; Chen, Y.H.; Chang, K.; Chen, H.C.; Hwang, S.J. Impact of renal failure on the outcome of dengue viral infection. Clin. J. Am. Soc. Nephrol. CJASN 2008, 3, 1350–1356. [Google Scholar] [CrossRef]
- Lee, I.-K.; Liu, J.-W.; Yang, K.D. Clinical Characteristics, Risk Factors, and Outcomes in Adults Experiencing Dengue Hemorrhagic Fever Complicated with Acute Renal Failure. Am. J. Trop. Med. Hyg. 2009, 80, 651–655. [Google Scholar] [CrossRef] [PubMed]
- Laoprasopwattana, K.; Pruekprasert, P.; Dissaneewate, P.; Geater, A.; Vachvanichsanong, P. Outcome of dengue hemorrhagic fever-caused acute kidney injury in Thai children. J. Pediatr. 2010, 157, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Basu, G.; Chrispal, A.; Boorugu, H.; Gopinath, K.G.; Chandy, S.; Prakash, J.A.; Thomas, K.; Abraham, A.M.; John, G.T. Acute kidney injury in tropical acute febrile illness in a tertiary care centre—RIFLE criteria validation. Nephrol. Dial. Transplant. 2011, 26, 524–531. [Google Scholar] [CrossRef]
- Khalil, M.A.; Sarwar, S.; Chaudry, M.A.; Maqbool, B.; Khalil, Z.; Tan, J.; Yaqub, S.; Hussain, S.A. Acute kidney injury in dengue virus infection. Clin. Kidney J. 2012, 5, 390–394. [Google Scholar] [CrossRef]
- Lee, I.K.; Liu, J.W.; Yang, K.D. Fatal dengue hemorrhagic fever in adults: Emphasizing the evolutionary pre-fatal clinical and laboratory manifestations. PLoS Negl. Trop. Dis. 2012, 6, e1532. [Google Scholar] [CrossRef]
- Mehra, N.; Patel, A.; Abraham, G.; Reddy, Y.N.; Reddy, Y.N. Acute kidney injury in dengue fever using Acute Kidney Injury Network criteria: Incidence and risk factors. Trop. Dr. 2012, 42, 160–162. [Google Scholar] [CrossRef]
- Huang, S.Y.; Lee, I.K.; Liu, J.W.; Kung, C.T.; Wang, L. Clinical features of and risk factors for rhabdomyolysis among adult patients with dengue virus infection. Am. J. Trop. Med. Hyg. 2015, 92, 75–81. [Google Scholar] [CrossRef]
- Mallhi, T.H.; Khan, A.H.; Adnan, A.S.; Sarriff, A.; Khan, Y.H.; Jummaat, F. Incidence, Characteristics and Risk Factors of Acute Kidney Injury among Dengue Patients: A Retrospective Analysis. PLoS ONE 2015, 10, e0138465. [Google Scholar] [CrossRef]
- Huang, H.S.; Hsu, C.C.; Ye, J.C.; Su, S.B.; Huang, C.C.; Lin, H.J. Predicting the mortality in geriatric patients with dengue fever. Medicine 2017, 96, e7878. [Google Scholar] [CrossRef] [PubMed]
- Vakrani, G.P.; Subramanyam, N.T. Acute Renal Failure in Dengue Infection. J. Clin. Diagn. Res. 2017, 11, OC10–OC13. [Google Scholar] [CrossRef]
- Basu, B.; Roy, B. Acute Renal Failure Adversely Affects Survival in Pediatric Dengue Infection. Indian J. Crit. Care Med. 2018, 22, 30–33. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.K.; Huang, C.H.; Huang, W.C.; Chen, Y.C.; Tsai, C.Y.; Chang, K.; Chen, Y.H. Prognostic Factors in Adult Patients with Dengue: Developing Risk Scoring Models and Emphasizing Factors Associated with Death ≤7 Days after Illness Onset and ≤3 Days after Presentation. J. Clin. Med. 2018, 7, 396. [Google Scholar] [CrossRef]
- Mallhi, T.H.; Khan, A.H.; Adnan, A.S.; Sarriff, A.; Khan, Y.H.; Gan, S.H. Short-term renal outcomes following acute kidney injury among dengue patients: A follow-up analysis from large prospective cohort. PLoS ONE 2018, 13, e0192510. [Google Scholar] [CrossRef]
- Mehta, K.; Pajai, A.; Bhurke, S.; Shirkande, A.; Bhadade, R.; D’Souza, R. Acute Kidney Injury of Infectious Etiology in Monsoon Season: A Prospective Study Using Acute Kidney Injury Network Criteria. Indian J. Nephrol. 2018, 28, 143–152. [Google Scholar] [CrossRef]
- Diptyanusa, A.; Phumratanaprapin, W.; Phonrat, B.; Poovorawan, K.; Hanboonkunupakarn, B.; Sriboonvorakul, N.; Thisyakorn, U. Characteristics and associated factors of acute kidney injury among adult dengue patients: A retrospective single-center study. PLoS ONE 2019, 14, e0210360. [Google Scholar] [CrossRef]
- Padyana, M.; Karanth, S.; Vaidya, S.; Gopaldas, J.A. Clinical Profile and Outcome of Dengue Fever in Multidisciplinary Intensive Care Unit of a Tertiary Level Hospital in India. Indian J. Crit. Care Med. 2019, 23, 270–273. [Google Scholar] [CrossRef]
- Patel, M.L.; Himanshu, D.; Chaudhary, S.C.; Atam, V.; Sachan, R.; Misra, R.; Mohapatra, S.D. Clinical Characteristic and Risk Factors of Acute Kidney Injury among Dengue Viral Infections in Adults: A Retrospective Analysis. Indian J. Nephrol. 2019, 29, 15–21. [Google Scholar] [CrossRef]
- Xiang, J.Y.; Zhang, Y.H.; Tan, Z.R.; Huang, J.; Zhao, Y.W. Guillain-Barre syndrome associated with Japanese encephalitis virus infection in China. Viral Immunol. 2014, 27, 418–420. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.; Praharaj, H.N.; Patil, T.B.; Giri, P. Acute transverse myelitis following Japanese encephalitis viral infection: An uncommon complication of a common disease. BMJ Case Rep. 2012, 2012, bcr2012007094. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.B.; Kumar, H.; Meena, B.L.; Chandra, S.; Agrawal, J.; Kanogiya, N. Neuropsychiatric Profile in Malaria: An Overview. J. Clin. Diagn. Res. 2016, 10, OC24–OC28. [Google Scholar] [CrossRef]
- Pace, A.A.; Edwards, S.; Weatherby, S. A new clinical variant of the post-malaria neurological syndrome. J. Neurol. Sci. 2013, 334, 183–185. [Google Scholar] [CrossRef]
- Derkach, A.; Otim, I.; Pfeiffer, R.M.; Onabajo, O.O.; Legason, I.D.; Nabalende, H.; Ogwang, M.D.; Kerchan, P.; Talisuna, A.O.; Ayers, L.W.; et al. Associations between IgG reactivity to Plasmodium falciparum erythrocyte membrane protein 1 (PfEMP1) antigens and Burkitt lymphoma in Ghana and Uganda case-control studies. EBioMedicine 2019, 39, 358–368. [Google Scholar] [CrossRef]
- Johnston, W.T.; Mutalima, N.; Sun, D.; Emmanuel, B.; Bhatia, K.; Aka, P.; Wu, X.; Borgstein, E.; Liomba, G.N.; Kamiza, S.; et al. Relationship between Plasmodium falciparum malaria prevalence, genetic diversity and endemic Burkitt lymphoma in Malawi. Sci. Rep. 2014, 4, 3741. [Google Scholar] [CrossRef]
- Peprah, S.; Ogwang, M.D.; Kerchan, P.; Reynolds, S.J.; Tenge, C.N.; Were, P.A.; Kuremu, R.T.; Wekesa, W.N.; Sumba, P.O.; Masalu, N.; et al. Risk factors for Burkitt lymphoma in East African children and minors: A case-control study in malaria-endemic regions in Uganda, Tanzania and Kenya. Int. J. Cancer 2020, 146, 953–969. [Google Scholar] [CrossRef]
- Wyss, K.; Granath, F.; Wangdahl, A.; Djarv, T.; Fored, M.; Naucler, P.; Farnert, A. Malaria and risk of lymphoid neoplasms and other cancer: A nationwide population-based cohort study. BMC Med. 2020, 18, 296. [Google Scholar] [CrossRef]
- Park, J.E. Parkinsonism and Tremor in a Patient with Plasmodium vivax Malaria. Tremor Other Hyperkinetic Mov. 2017, 7, 471. [Google Scholar] [CrossRef]
- Mittal, S.; Misri, Z.K.; Rakshith, K.C.; Mittal, S. Post Malarial Neurological Syndrome: An Uncommon Cause. J. Neuroinfect. Dis. 2015, 1, 1–4. [Google Scholar] [CrossRef]
- Kanjalkar, M.; Karnad, D.R.; Narayana, R.V.; Shah, P.U. Guillain-Barre syndrome following malaria. J. Infect. 1999, 38, 48–50. [Google Scholar] [CrossRef] [PubMed]
- Lei, K.; Ji, W.; Bhaya, B.; Ahsan, C. A Rare Case of Cardiac Recovery after Acute Myocarditis from West Nile Virus Infection: A Review of the Current Literature. Case Rep. Cardiol. 2022, 2022, 8517728. [Google Scholar] [CrossRef]
- Gao, A.R.; Nichols, L.; Mannuru, D. A Rare Case of West Nile Virus-Associated Cardiomyopathy. Cureus 2022, 14, e28473. [Google Scholar] [CrossRef] [PubMed]
- Hasbun, R.; Garcia, M.N.; Kellaway, J.; Baker, L.; Salazar, L.; Woods, S.P.; Murray, K.O. West Nile Virus Retinopathy and Associations with Long Term Neurological and Neurocognitive Sequelae. PLoS ONE 2016, 11, e0148898. [Google Scholar] [CrossRef]
- Weatherhead, J.E.; Miller, V.E.; Garcia, M.N.; Hasbun, R.; Salazar, L.; Dimachkie, M.M.; Murray, K.O. Long-term neurological outcomes in West Nile virus-infected patients: An observational study. Am. J. Trop. Med. Hyg. 2015, 92, 1006–1012. [Google Scholar] [CrossRef]
- Beirao, P.; Pereira, P.; Nunes, A.; Antunes, P. Yellow fever vaccine-associated neurological disease, a suspicious case. BMJ Case Rep. 2017, 2017, bcr2016218706. [Google Scholar] [CrossRef] [PubMed]
- Bailey, A.L.; Kang, L.-I.; Zanella, L.G.F.D.A.B.D.; Silveira, C.G.T.; Ho, Y.-L.; Foquet, L.; Bial, G.; McCune, B.T.; Duarte-Neto, A.N.; Thomas, A.; et al. Consumptive coagulopathy of severe yellow fever occurs independently of hepatocellular tropism and massive hepatic injury. Proc. Natl. Acad. Sci. USA 2020, 117, 32648–32656. [Google Scholar] [CrossRef]
- Mecharles, S.; Herrmann, C.; Poullain, P.; Tran, T.H.; Deschamps, N.; Mathon, G.; Landais, A.; Breurec, S.; Lannuzel, A. Acute myelitis due to Zika virus infection. Lancet 2016, 387, 1481. [Google Scholar] [CrossRef]
- Schwartzmann, P.V.; Ramalho, L.N.; Neder, L.; Vilar, F.C.; Ayub-Ferreira, S.M.; Romeiro, M.F.; Takayanagui, O.M.; Dos Santos, A.C.; Schmidt, A.; Figueiredo, L.T.; et al. Zika Virus Meningoencephalitis in an Immunocompromised Patient. Mayo Clin. Proc. 2017, 92, 460–466. [Google Scholar] [CrossRef]
- Parra, B.; Lizarazo, J.; Jimenez-Arango, J.A.; Zea-Vera, A.F.; Gonzalez-Manrique, G.; Vargas, J.; Angarita, J.A.; Zuniga, G.; Lopez-Gonzalez, R.; Beltran, C.L.; et al. Guillain-Barre Syndrome Associated with Zika Virus Infection in Colombia. N. Engl. J. Med. 2016, 375, 1513–1523. [Google Scholar] [CrossRef]
- Anaya, J.M.; Rodriguez, Y.; Monsalve, D.M.; Vega, D.; Ojeda, E.; Gonzalez-Bravo, D.; Rodriguez-Jimenez, M.; Pinto-Diaz, C.A.; Chaparro, P.; Gunturiz, M.L.; et al. A comprehensive analysis and immunobiology of autoimmune neurological syndromes during the Zika virus outbreak in Cucuta, Colombia. J. Autoimmun. 2017, 77, 123–138. [Google Scholar] [CrossRef]
- Nunes, M.L.; Esper, N.B.; Franco, A.R.; Radaelli, G.; Soder, R.B.; Bomfim, R.; Kalil Neto, F.; Gameleira, F.T.; Portuguez, M.W.; da Costa, J.C. Epilepsy after congenital zika virus infection: EEG and neuroimaging features. Seizure 2021, 84, 14–22. [Google Scholar] [CrossRef]
- Riley, E.M.; Stewart, V.A. Immune mechanisms in malaria: New insights in vaccine development. Nat. Med. 2013, 19, 168–178. [Google Scholar] [CrossRef]
- Takala, S.L.; Plowe, C.V. Genetic diversity and malaria vaccine design, testing and efficacy: Preventing and overcoming ‘vaccine resistant malaria’. Parasite Immunol. 2009, 31, 560–573. [Google Scholar] [CrossRef] [PubMed]
- Draper, S.J.; Sack, B.K.; King, C.R.; Nielsen, C.M.; Rayner, J.C.; Higgins, M.K.; Long, C.A.; Seder, R.A. Malaria Vaccines: Recent Advances and New Horizons. Cell Host Microbe 2018, 24, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Waide, M.L.; Schmidt, N.W. The gut microbiome, immunity, and Plasmodium severity. Curr. Opin. Microbiol. 2020, 58, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Datoo, M.S.; Natama, M.H.; Some, A.; Traore, O.; Rouamba, T.; Bellamy, D.; Yameogo, P.; Valia, D.; Tegneri, M.; Ouedraogo, F.; et al. Efficacy of a low-dose candidate malaria vaccine, R21 in adjuvant Matrix-M, with seasonal administration to children in Burkina Faso: A randomised controlled trial. Lancet 2021, 397, 1809–1818. [Google Scholar] [CrossRef] [PubMed]
- Long, C.A.; Zavala, F. Immune Responses in Malaria. Cold Spring Harb. Perspect. Med. 2017, 7, a025577. [Google Scholar] [CrossRef]
- Pang, E.L.; Loh, H.S. Towards development of a universal dengue vaccine—How close are we? Asian Pac. J. Trop. Med. 2017, 10, 220–228. [Google Scholar] [CrossRef]
- Metz, S.W.; Geertsema, C.; Martina, B.E.; Andrade, P.; Heldens, J.G.; van Oers, M.M.; Goldbach, R.W.; Vlak, J.M.; Pijlman, G.P. Functional processing and secretion of Chikungunya virus E1 and E2 glycoproteins in insect cells. Virol. J. 2011, 8, 353. [Google Scholar] [CrossRef]
- Schlesinger, J.J.; Brandriss, M.W.; Walsh, E.E. Protection against 17D yellow fever encephalitis in mice by passive transfer of monoclonal antibodies to the nonstructural glycoprotein gp48 and by active immunization with gp48. J. Immunol. 1985, 135, 2805–2809. [Google Scholar] [CrossRef]
- Abbink, P.; Larocca, R.A.; De La Barrera, R.A.; Bricault, C.A.; Moseley, E.T.; Boyd, M.; Kirilova, M.; Li, Z.; Ng’ang’a, D.; Nanayakkara, O.; et al. Protective efficacy of multiple vaccine platforms against Zika virus challenge in rhesus monkeys. Science 2016, 353, 1129–1132. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [PubMed]
- Jagger, B.W.; Dowd, K.A.; Chen, R.E.; Desai, P.; Foreman, B.; Burgomaster, K.E.; Himansu, S.; Kong, W.P.; Graham, B.S.; Pierson, T.C.; et al. Protective Efficacy of Nucleic Acid Vaccines Against Transmission of Zika Virus During Pregnancy in Mice. J. Infect. Dis. 2019, 220, 1577–1588. [Google Scholar] [CrossRef] [PubMed]
- Spik, K.; Shurtleff, A.; McElroy, A.K.; Guttieri, M.C.; Hooper, J.W.; SchmalJohn, C. Immunogenicity of combination DNA vaccines for Rift Valley fever virus, tick-borne encephalitis virus, Hantaan virus, and Crimean Congo hemorrhagic fever virus. Vaccine 2006, 24, 4657–4666. [Google Scholar] [CrossRef]






| Plasmodium Species | Severity | Distribution | Innate Immunity | Humoral Immunity | Major Involved Pathway | References |
|---|---|---|---|---|---|---|
| P. falciparum | High | Africa, Asia, and Latin America | Neutrophils, monocytes, and NK cells | Long-lived plasma B cells and memory B cells; antibodies | Toll, immune deficiency (Imd), Janus kinase (JNK), and signal transducers and activators of transcription (STAT) | [52,53] |
| P. vivax | Moderate | Asia, Latin America, Africa | Cytokines, dendritic cells | B cells and antibodies | IFN-α/IFN-γ and TCR pathway, MAPK6/MAPK4 signalling | [46,54] |
| P. ovale | Low–moderate | Africa, Asia | NA | NA | NA | |
| P. malariae | Low | Africa, Asia, and Latin America | NA | NA | NA | |
| P. knowlesi | Moderate–high | Southeast Asia | NA | NA | NA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhattacharjee, S.; Ghosh, D.; Saha, R.; Sarkar, R.; Kumar, S.; Khokhar, M.; Pandey, R.K. Mechanism of Immune Evasion in Mosquito-Borne Diseases. Pathogens 2023, 12, 635. https://doi.org/10.3390/pathogens12050635
Bhattacharjee S, Ghosh D, Saha R, Sarkar R, Kumar S, Khokhar M, Pandey RK. Mechanism of Immune Evasion in Mosquito-Borne Diseases. Pathogens. 2023; 12(5):635. https://doi.org/10.3390/pathogens12050635
Chicago/Turabian StyleBhattacharjee, Swagato, Debanjan Ghosh, Rounak Saha, Rima Sarkar, Saurav Kumar, Manoj Khokhar, and Rajan Kumar Pandey. 2023. "Mechanism of Immune Evasion in Mosquito-Borne Diseases" Pathogens 12, no. 5: 635. https://doi.org/10.3390/pathogens12050635
APA StyleBhattacharjee, S., Ghosh, D., Saha, R., Sarkar, R., Kumar, S., Khokhar, M., & Pandey, R. K. (2023). Mechanism of Immune Evasion in Mosquito-Borne Diseases. Pathogens, 12(5), 635. https://doi.org/10.3390/pathogens12050635