
Mechanisms of Innate Immune Sensing of HTLV-1 and Viral Immune Evasion

{kind=link}
{kind=link}
{kind=link}
Abstract
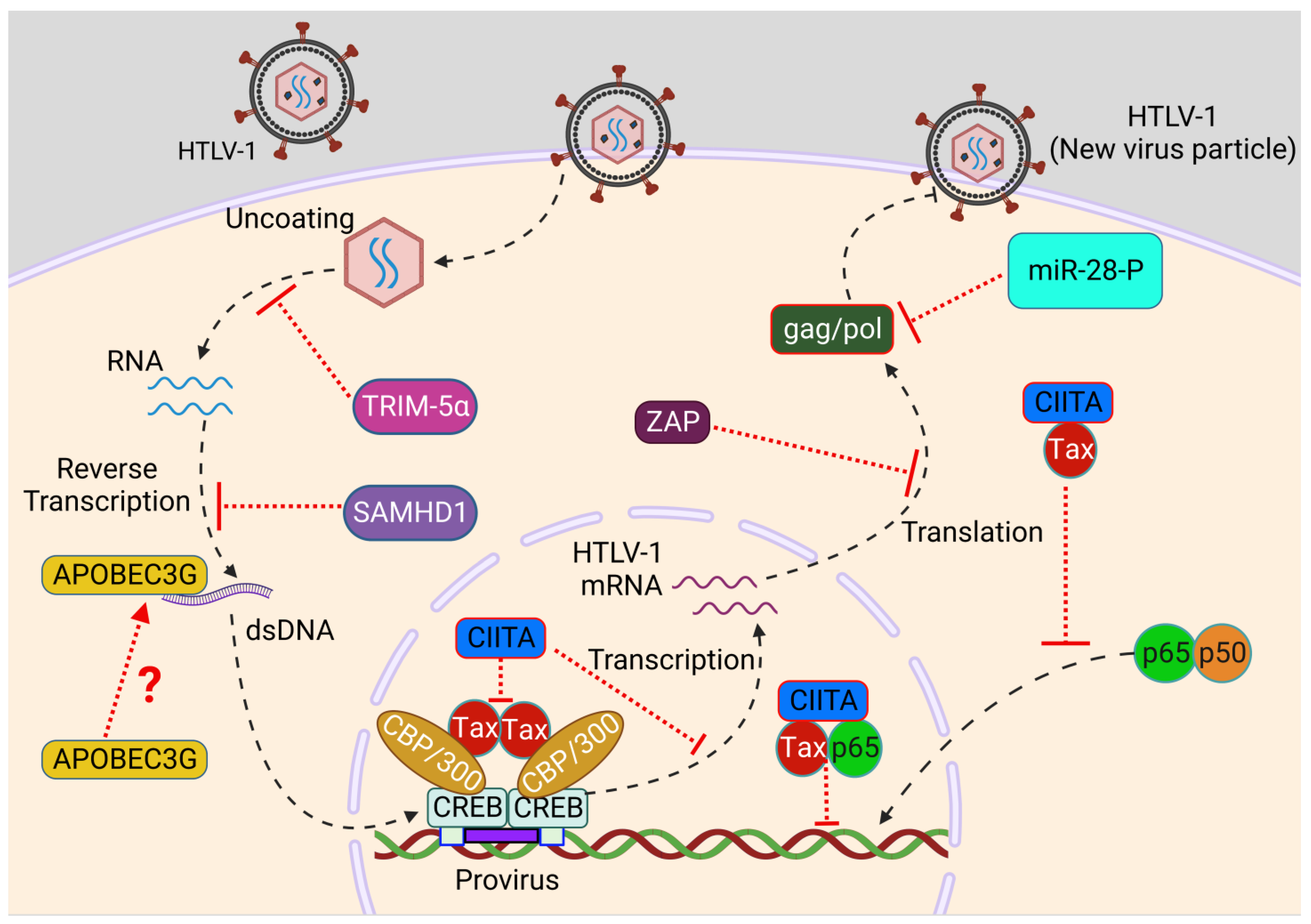
:1. Introduction
2. HTLV-1 Genomic Organization and Viral Life Cycle
3. Immune Sensors of HTLV-1
3.1. TLRs
3.2. IFI16
3.3. STING
3.4. Ku70
4. HTLV-1 Restriction Factors
4.1. APOBEC3
4.2. SAMHD1
4.3. Tetherin
4.4. CIITA
4.5. ZAP
4.6. TRIM Family
4.7. ADAR1
4.8. Micro RNAs
5. Innate Immunity and Inflammation
6. Innate Immune Evasion by HTLV-1
6.1. Tax
6.2. HBZ
6.3. p30
6.4. p12
7. Concluding Remarks and Future Prospective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Poiesz, B.J.; Ruscetti, F.W.; Gazdar, A.F.; Bunn, P.A.; Minna, J.D.; Gallo, R.C. Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc. Natl. Acad. Sci. USA 1980, 77, 7415–7419. [Google Scholar] [CrossRef]
- Hinuma, Y.; Nagata, K.; Hanaoka, M.; Nakai, M.; Matsumoto, T.; Kinoshita, K.I.; Shirakawa, S.; Miyoshi, I. Adult T-cell leukemia: Antigen in an ATL cell line and detection of antibodies to the antigen in human sera. Proc. Natl. Acad. Sci. USA 1981, 78, 6476–6480. [Google Scholar] [CrossRef]
- Mahieux, R.; Gessain, A. Adult T-cell leukemia/lymphoma and HTLV-1. Curr. Hematol. Malig. Rep. 2007, 2, 257–264. [Google Scholar] [CrossRef]
- Gessain, A.; Barin, F.; Vernant, J.C.; Gout, O.; Maurs, L.; Calender, A.; de The, G. Antibodies to human T-lymphotropic virus type-I in patients with tropical spastic paraparesis. Lancet 1985, 2, 407–410. [Google Scholar] [CrossRef]
- Osame, M.; Usuku, K.; Izumo, S.; Ijichi, N.; Amitani, H.; Igata, A.; Matsumoto, M.; Tara, M. HTLV-I associated myelopathy, a new clinical entity. Lancet 1986, 1, 1031–1032. [Google Scholar] [CrossRef] [PubMed]
- Einsiedel, L.J.; Pham, H.; Woodman, R.J.; Pepperill, C.; Taylor, K.A. The prevalence and clinical associations of HTLV-1 infection in a remote Indigenous community. Med. J. Aust. 2016, 205, 305–309. [Google Scholar] [CrossRef]
- Gessain, A.; Cassar, O. Epidemiological Aspects and World Distribution of HTLV-1 Infection. Front. Microbiol. 2012, 3, 388. [Google Scholar] [CrossRef]
- Abbaszadegan, M.R.; Gholamin, M.; Tabatabaee, A.; Farid, R.; Houshmand, M.; Abbaszadegan, M. Prevalence of human T-lymphotropic virus type 1 among blood donors from Mashhad, Iran. J. Clin. Microbiol. 2003, 41, 2593–2595. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, M.; Yamochi, T.; Watanabe, M.; Uchimaru, K.; Utsunomiya, A.; Higashihara, M.; Watanabe, T.; Horie, R. CD30 Characterizes Polylobated Lymphocytes and Disease Progression in HTLV-1-Infected Individuals. Clin. Cancer Res. 2018, 24, 5445–5457. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, E.; Nozuma, S.; Tashiro, Y.; Kubota, R.; Izumo, S.; Takashima, H. HTLV-1 associated myelopathy/tropical spastic paraparesis (HAM/TSP): A comparative study to identify factors that influence disease progression. J. Neurol. Sci. 2016, 371, 112–116. [Google Scholar] [CrossRef]
- Gout, O.; Baulac, M.; Gessain, A.; Semah, F.; Saal, F.; Peries, J.; Cabrol, C.; Foucault-Fretz, C.; Laplane, D.; Sigaux, F.; et al. Rapid development of myelopathy after HTLV-I infection acquired by transfusion during cardiac transplantation. N. Engl. J. Med. 1990, 322, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.J.; Zheng, X.Y.; Yang, X.Z.; Liu, T.B.; Yang, T.; Zheng, Z.H.; Gao, F.; Chen, C.X.; Li, J.G.; Zhang, C.Q.; et al. Clinical characteristics and prognosis in 12 patients with adult T cell leukemia/lymphoma confirmed by HTLV-1 provirus gene detection. Zhonghua Xue Ye Xue Za Zhi 2016, 37, 1027–1032. [Google Scholar] [CrossRef] [PubMed]
- Grigsby, I.F.; Zhang, W.; Johnson, J.L.; Fogarty, K.H.; Chen, Y.; Rawson, J.M.; Crosby, A.J.; Mueller, J.D.; Mansky, L.M. Biophysical analysis of HTLV-1 particles reveals novel insights into particle morphology and Gag stochiometry. Retrovirology 2010, 7, 75. [Google Scholar] [CrossRef] [PubMed]
- Kannian, P.; Green, P.L. Human T Lymphotropic Virus Type 1 (HTLV-1): Molecular Biology and Oncogenesis. Viruses 2010, 2, 2037–2077. [Google Scholar] [CrossRef]
- Majorovits, E.; Nejmeddine, M.; Tanaka, Y.; Taylor, G.P.; Fuller, S.D.; Bangham, C.R. Human T-lymphotropic virus-1 visualized at the virological synapse by electron tomography. PLoS ONE 2008, 3, e2251. [Google Scholar] [CrossRef]
- Igakura, T.; Stinchcombe, J.C.; Goon, P.K.; Taylor, G.P.; Weber, J.N.; Griffiths, G.M.; Tanaka, Y.; Osame, M.; Bangham, C.R. Spread of HTLV-I between lymphocytes by virus-induced polarization of the cytoskeleton. Science 2003, 299, 1713–1716. [Google Scholar] [CrossRef]
- Pais-Correia, A.M.; Sachse, M.; Guadagnini, S.; Robbiati, V.; Lasserre, R.; Gessain, A.; Gout, O.; Alcover, A.; Thoulouze, M.I. Biofilm-like extracellular viral assemblies mediate HTLV-1 cell-to-cell transmission at virological synapses. Nat. Med. 2010, 16, 83–89. [Google Scholar] [CrossRef]
- Van Prooyen, N.; Gold, H.; Andresen, V.; Schwartz, O.; Jones, K.; Ruscetti, F.; Lockett, S.; Gudla, P.; Venzon, D.; Franchini, G. Human T-cell leukemia virus type 1 p8 protein increases cellular conduits and virus transmission. Proc. Natl. Acad. Sci. USA 2010, 107, 20738–20743. [Google Scholar] [CrossRef]
- Ilinskaya, A.; Derse, D.; Hill, S.; Princler, G.; Heidecker, G. Cell-cell transmission allows human T-lymphotropic virus 1 to circumvent tetherin restriction. Virology 2013, 436, 201–209. [Google Scholar] [CrossRef]
- Ghez, D.; Lepelletier, Y.; Lambert, S.; Fourneau, J.M.; Blot, V.; Janvier, S.; Arnulf, B.; van Endert, P.M.; Heveker, N.; Pique, C.; et al. Neuropilin-1 is involved in human T-cell lymphotropic virus type 1 entry. J. Virol. 2006, 80, 6844–6854. [Google Scholar] [CrossRef]
- Pinto, D.O.; DeMarino, C.; Pleet, M.L.; Cowen, M.; Branscome, H.; Al Sharif, S.; Jones, J.; Dutartre, H.; Lepene, B.; Liotta, L.A.; et al. HTLV-1 Extracellular Vesicles Promote Cell-to-Cell Contact. Front. Microbiol. 2019, 10, 2147. [Google Scholar] [CrossRef]
- Barnard, A.L.; Igakura, T.; Tanaka, Y.; Taylor, G.P.; Bangham, C.R. Engagement of specific T-cell surface molecules regulates cytoskeletal polarization in HTLV-1-infected lymphocytes. Blood 2005, 106, 988–995. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, K.H.; Zhang, W.; Grigsby, I.F.; Johnson, J.L.; Chen, Y.; Mueller, J.D.; Mansky, L.M. New insights into HTLV-1 particle structure, assembly, and Gag-Gag interactions in living cells. Viruses 2011, 3, 770–793. [Google Scholar] [CrossRef]
- Konvalinka, J.; Krausslich, H.G.; Muller, B. Retroviral proteases and their roles in virion maturation. Virology 2015, 479–480, 403–417. [Google Scholar] [CrossRef]
- Sundquist, W.I.; Krausslich, H.G. HIV-1 assembly, budding, and maturation. Cold Spring Harb. Perspect. Med. 2012, 2, a006924. [Google Scholar] [CrossRef] [PubMed]
- Nagai, M.; Brennan, M.B.; Sakai, J.A.; Mora, C.A.; Jacobson, S. CD8(+) T cells are an in vivo reservoir for human T-cell lymphotropic virus type I. Blood 2001, 98, 1858–1861. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.S.; Petrow-Sadowski, C.; Huang, Y.K.; Bertolette, D.C.; Ruscetti, F.W. Cell-free HTLV-1 infects dendritic cells leading to transmission and transformation of CD4(+) T cells. Nat. Med. 2008, 14, 429–436. [Google Scholar] [CrossRef]
- Thompson, M.R.; Kaminski, J.J.; Kurt-Jones, E.A.; Fitzgerald, K.A. Pattern recognition receptors and the innate immune response to viral infection. Viruses 2011, 3, 920–940. [Google Scholar] [CrossRef]
- Orzalli, M.H.; Knipe, D.M. Cellular sensing of viral DNA and viral evasion mechanisms. Annu. Rev. Microbiol. 2014, 68, 477–492. [Google Scholar] [CrossRef]
- Wu, J.; Chen, Z.J. Innate immune sensing and signaling of cytosolic nucleic acids. Annu. Rev. Immunol. 2014, 32, 461–488. [Google Scholar] [CrossRef]
- van Montfoort, N.; Olagnier, D.; Hiscott, J. Unmasking immune sensing of retroviruses: Interplay between innate sensors and host effectors. Cytokine Growth Factor. Rev. 2014, 25, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Oshiumi, H.; Seya, T. Antiviral responses induced by the TLR3 pathway. Rev. Med. Virol. 2011, 21, 67–77. [Google Scholar] [CrossRef]
- Habibabadi, H.M.; Parsania, M.; Pourfathollah, A.A.; Haghighat, S.; Sharifi, Z. Association of TLR3 single nucleotide polymorphisms with susceptibility to HTLV-1 infection in Iranian asymptomatic blood donors. Rev. Soc. Bras. Med. Trop. 2020, 53, e20200026. [Google Scholar] [CrossRef] [PubMed]
- Beignon, A.S.; McKenna, K.; Skoberne, M.; Manches, O.; DaSilva, I.; Kavanagh, D.G.; Larsson, M.; Gorelick, R.J.; Lifson, J.D.; Bhardwaj, N. Endocytosis of HIV-1 activates plasmacytoid dendritic cells via Toll-like receptor-viral RNA interactions. J. Clin. Investig. 2005, 115, 3265–3275. [Google Scholar] [CrossRef] [PubMed]
- Diebold, S.S.; Kaisho, T.; Hemmi, H.; Akira, S.; Reis e Sousa, C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 2004, 303, 1529–1531. [Google Scholar] [CrossRef]
- Colisson, R.; Barblu, L.; Gras, C.; Raynaud, F.; Hadj-Slimane, R.; Pique, C.; Hermine, O.; Lepelletier, Y.; Herbeuval, J.P. Free HTLV-1 induces TLR7-dependent innate immune response and TRAIL relocalization in killer plasmacytoid dendritic cells. Blood 2010, 115, 2177–2185. [Google Scholar] [CrossRef]
- Demontis, M.A.; Sadiq, M.T.; Golz, S.; Taylor, G.P. HTLV-1 viral RNA is detected rarely in plasma of HTLV-1 infected subjects. J. Med. Virol. 2015, 87, 2130–2134. [Google Scholar] [CrossRef]
- Gross, C.; Thoma-Kress, A.K. Molecular Mechanisms of HTLV-1 Cell-to-Cell Transmission. Viruses 2016, 8, 74. [Google Scholar] [CrossRef]
- Hishizawa, M.; Imada, K.; Kitawaki, T.; Ueda, M.; Kadowaki, N.; Uchiyama, T. Depletion and impaired interferon-alpha-producing capacity of blood plasmacytoid dendritic cells in human T-cell leukaemia virus type I-infected individuals. Br. J. Haematol. 2004, 125, 568–575. [Google Scholar] [CrossRef]
- Broz, P.; Monack, D.M. Newly described pattern recognition receptors team up against intracellular pathogens. Nat. Rev. Immunol. 2013, 13, 551–565. [Google Scholar] [CrossRef]
- Ouchi, M.; Ouchi, T. Role of IFI16 in DNA damage and checkpoint. Front. Biosci. 2008, 13, 236–239. [Google Scholar] [CrossRef] [PubMed]
- Unterholzner, L.; Keating, S.E.; Baran, M.; Horan, K.A.; Jensen, S.B.; Sharma, S.; Sirois, C.M.; Jin, T.; Latz, E.; Xiao, T.S.; et al. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2010, 11, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Horan, K.A.; Hansen, K.; Jakobsen, M.R.; Holm, C.K.; Soby, S.; Unterholzner, L.; Thompson, M.; West, J.A.; Iversen, M.B.; Rasmussen, S.B.; et al. Proteasomal degradation of herpes simplex virus capsids in macrophages releases DNA to the cytosol for recognition by DNA sensors. J. Immunol. 2013, 190, 2311–2319. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Dutta, D.; Iqbal, J.; Pisano, G.; Gjyshi, O.; Ansari, M.A.; Kumar, B.; Chandran, B. Nuclear Innate Immune DNA Sensor IFI16 Is Degraded during Lytic Reactivation of Kaposi’s Sarcoma-Associated Herpesvirus (KSHV): Role of IFI16 in Maintenance of KSHV Latency. J. Virol. 2016, 90, 8822–8841. [Google Scholar] [CrossRef]
- Ansari, M.A.; Singh, V.V.; Dutta, S.; Veettil, M.V.; Dutta, D.; Chikoti, L.; Lu, J.; Everly, D.; Chandran, B. Constitutive interferon-inducible protein 16-inflammasome activation during Epstein-Barr virus latency I, II, and III in B and epithelial cells. J. Virol. 2013, 87, 8606–8623. [Google Scholar] [CrossRef]
- Morrone, S.R.; Wang, T.; Constantoulakis, L.M.; Hooy, R.M.; Delannoy, M.J.; Sohn, J. Cooperative assembly of IFI16 filaments on dsDNA provides insights into host defense strategy. Proc. Natl. Acad. Sci. USA 2014, 111, E62–E71. [Google Scholar] [CrossRef]
- Yang, B.; Song, D.; Liu, Y.; Cui, Y.; Lu, G.; Di, W.; Xing, H.; Ma, L.; Guo, Z.; Guan, Y.; et al. IFI16 regulates HTLV-1 replication through promoting HTLV-1 RTI-induced innate immune responses. FEBS Lett. 2018, 592, 1693–1704. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef]
- Hiller, B.; Hornung, V. STING Signaling the enERGIC Way. Cell. Host Microbe 2015, 18, 137–139. [Google Scholar] [CrossRef]
- Sze, A.; Belgnaoui, S.M.; Olagnier, D.; Lin, R.; Hiscott, J.; van Grevenynghe, J. Host restriction factor SAMHD1 limits human T cell leukemia virus type 1 infection of monocytes via STING-mediated apoptosis. Cell. Host Microbe 2013, 14, 422–434. [Google Scholar] [CrossRef]
- Walker, J.R.; Corpina, R.A.; Goldberg, J. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature 2001, 412, 607–614. [Google Scholar] [CrossRef]
- Goodarzi, A.A.; Jeggo, P.A. The repair and signaling responses to DNA double-strand breaks. Adv. Genet. 2013, 82, 1–45. [Google Scholar] [CrossRef]
- Cohen, H.Y.; Lavu, S.; Bitterman, K.J.; Hekking, B.; Imahiyerobo, T.A.; Miller, C.; Frye, R.; Ploegh, H.; Kessler, B.M.; Sinclair, D.A. Acetylation of the C terminus of Ku70 by CBP and PCAF controls Bax-mediated apoptosis. Mol. Cell. 2004, 13, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Fu, Z.; Li, X.; Liang, Y.; Pei, S.; Hao, S.; Zhu, Q.; Yu, T.; Pei, Y.; Yuan, J.; et al. Cytoplasmic DNA sensing by KU complex in aged CD4(+) T cell potentiates T cell activation and aging-related autoimmune inflammation. Immunity 2021, 54, 632–647.e9. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Kang, L.; Song, D.; Liu, L.; Yang, S.; Ma, L.; Guo, Z.; Ding, H.; Wang, H.; Yang, B. Ku70 Senses HTLV-1 DNA and Modulates HTLV-1 Replication. J. Immunol. 2017, 199, 2475–2482. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Ao, Z.; Wang, B.; Jayappa, K.D.; Yao, X. Host protein Ku70 binds and protects HIV-1 integrase from proteasomal degradation and is required for HIV replication. J. Biol. Chem. 2011, 286, 17722–17735. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wu, Y.; Zheng, X.; Cong, J.; Liu, Y.; Li, J.; Sun, R.; Tian, Z.G.; Wei, H.M. Cytoplasm-Translocated Ku70/80 Complex Sensing of HBV DNA Induces Hepatitis-Associated Chemokine Secretion. Front. Immunol. 2016, 7, 569. [Google Scholar] [CrossRef]
- Sui, H.; Zhou, M.; Imamichi, H.; Jiao, X.; Sherman, B.T.; Lane, H.C.; Imamichi, T. STING is an essential mediator of the Ku70-mediated production of IFN-lambda1 in response to exogenous DNA. Sci. Signal. 2017, 10, eaah5054. [Google Scholar] [CrossRef]
- Chan, D.W.; Ye, R.; Veillette, C.J.; Lees-Miller, S.P. DNA-dependent protein kinase phosphorylation sites in Ku 70/80 heterodimer. Biochemistry 1999, 38, 1819–1828. [Google Scholar] [CrossRef]
- Ferguson, B.J.; Mansur, D.S.; Peters, N.E.; Ren, H.; Smith, G.L. DNA-PK is a DNA sensor for IRF-3-dependent innate immunity. eLife 2012, 1, e00047. [Google Scholar] [CrossRef]
- Bieniasz, P.D. Intrinsic immunity: A front-line defense against viral attack. Nat. Immunol. 2004, 5, 1109–1115. [Google Scholar] [CrossRef] [PubMed]
- Malim, M.H.; Bieniasz, P.D. HIV Restriction Factors and Mechanisms of Evasion. Cold Spring Harb. Perspect. Med. 2012, 2, a006940. [Google Scholar] [CrossRef]
- Colomer-Lluch, M.; Ruiz, A.; Moris, A.; Prado, J.G. Restriction Factors: From Intrinsic Viral Restriction to Shaping Cellular Immunity Against HIV-1. Front. Immunol. 2018, 9, 2876. [Google Scholar] [CrossRef] [PubMed]
- Carcone, A.; Journo, C.; Dutartre, H. Is the HTLV-1 Retrovirus Targeted by Host Restriction Factors? Viruses 2022, 14, 1611. [Google Scholar] [CrossRef] [PubMed]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.L.; Greene, W.C. The APOBEC3 cytidine deaminases: An innate defensive network opposing exogenous retroviruses and endogenous retroelements. Annu. Rev. Immunol. 2008, 26, 317–353. [Google Scholar] [CrossRef]
- Holmes, R.K.; Malim, M.H.; Bishop, K.N. APOBEC-mediated viral restriction: Not simply editing? Trends Biochem. Sci. 2007, 32, 118–128. [Google Scholar] [CrossRef]
- Baig, T.T.; Feng, Y.; Chelico, L. Determinants of efficient degradation of APOBEC3 restriction factors by HIV-1 Vif. J. Virol. 2014, 88, 14380–14395. [Google Scholar] [CrossRef]
- Ohsugi, T.; Koito, A. Human T cell leukemia virus type I is resistant to the antiviral effects of APOBEC3. J. Virol. Methods 2007, 139, 93–96. [Google Scholar] [CrossRef]
- Mahieux, R.; Suspene, R.; Delebecque, F.; Henry, M.; Schwartz, O.; Wain-Hobson, S.; Vartanian, J.P. Extensive editing of a small fraction of human T-cell leukemia virus type 1 genomes by four APOBEC3 cytidine deaminases. J. Gen. Virol. 2005, 86, 2489–2494. [Google Scholar] [CrossRef]
- Derse, D.; Hill, S.A.; Princler, G.; Lloyd, P.; Heidecker, G. Resistance of human T cell leukemia virus type 1 to APOBEC3G restriction is mediated by elements in nucleocapsid. Proc. Natl. Acad. Sci. USA 2007, 104, 2915–2920. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, K.; Nagata, Y.; Kitanaka, A.; Shiraishi, Y.; Shimamura, T.; Yasunaga, J.; Totoki, Y.; Chiba, K.; Sato-Otsubo, A.; Nagae, G.; et al. Integrated molecular analysis of adult T cell leukemia/lymphoma. Nat. Genet. 2015, 47, 1304–1315. [Google Scholar] [CrossRef]
- Yao, J.; Tanaka, M.; Takenouchi, N.; Ren, Y.; Lee, S.I.; Fujisawa, J.I. Induction of APOBEC3B cytidine deaminase in HTLV-1-infected humanized mice. Exp. Ther. Med. 2019, 17, 3701–3708. [Google Scholar] [CrossRef] [PubMed]
- Sasada, A.; Takaori-Kondo, A.; Shirakawa, K.; Kobayashi, M.; Abudu, A.; Hishizawa, M.; Imada, K.; Tanaka, Y.; Uchiyama, T. APOBEC3G targets human T-cell leukemia virus type 1. Retrovirology 2005, 2, 32. [Google Scholar] [CrossRef]
- Ooms, M.; Krikoni, A.; Kress, A.K.; Simon, V.; Munk, C. APOBEC3A, APOBEC3B, and APOBEC3H haplotype 2 restrict human T-lymphotropic virus type 1. J. Virol. 2012, 86, 6097–6108. [Google Scholar] [CrossRef]
- Alais, S.; Mahieux, R.; Dutartre, H. Viral Source-Independent High Susceptibility of Dendritic Cells to Human T-Cell Leukemia Virus Type 1 Infection Compared to That of T Lymphocytes. J. Virol. 2015, 89, 10580–10590. [Google Scholar] [CrossRef]
- de Castro-Amarante, M.F.; Pise-Masison, C.A.; McKinnon, K.; Washington Parks, R.; Galli, V.; Omsland, M.; Andresen, V.; Massoud, R.; Brunetto, G.; Caruso, B.; et al. Human T Cell Leukemia Virus Type 1 Infection of the Three Monocyte Subsets Contributes to Viral Burden in Humans. J. Virol. 2015, 90, 2195–2207. [Google Scholar] [CrossRef] [PubMed]
- Goldstone, D.C.; Ennis-Adeniran, V.; Hedden, J.J.; Groom, H.C.; Rice, G.I.; Christodoulou, E.; Walker, P.A.; Kelly, G.; Haire, L.F.; Yap, M.W.; et al. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature 2011, 480, 379–382. [Google Scholar] [CrossRef]
- Laguette, N.; Sobhian, B.; Casartelli, N.; Ringeard, M.; Chable-Bessia, C.; Segeral, E.; Yatim, A.; Emiliani, S.; Schwartz, O.; Benkirane, M. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 2011, 474, 654–657. [Google Scholar] [CrossRef]
- Hrecka, K.; Hao, C.; Gierszewska, M.; Swanson, S.K.; Kesik-Brodacka, M.; Srivastava, S.; Florens, L.; Washburn, M.P.; Skowronski, J. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 2011, 474, 658–661. [Google Scholar] [CrossRef]
- Gramberg, T.; Kahle, T.; Bloch, N.; Wittmann, S.; Mullers, E.; Daddacha, W.; Hofmann, H.; Kim, B.; Lindemann, D.; Landau, N.R. Restriction of diverse retroviruses by SAMHD1. Retrovirology 2013, 10, 26. [Google Scholar] [CrossRef]
- Van Damme, N.; Goff, D.; Katsura, C.; Jorgenson, R.L.; Mitchell, R.; Johnson, M.C.; Stephens, E.B.; Guatelli, J. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell. Host Microbe 2008, 3, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Neil, S.J.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Miyagi, E.; Andrew, A.J.; Kao, S.; Strebel, K. Vpu enhances HIV-1 virus release in the absence of Bst-2 cell surface down-modulation and intracellular depletion. Proc. Natl. Acad. Sci. USA 2009, 106, 2868–2873. [Google Scholar] [CrossRef] [PubMed]
- Maali, Y.; Journo, C.; Mahieux, R.; Dutartre, H. Microbial Biofilms: Human T-cell Leukemia Virus Type 1 First in Line for Viral Biofilm but Far Behind Bacterial Biofilms. Front. Microbiol. 2020, 11, 2041. [Google Scholar] [CrossRef]
- Pai, R.K.; Askew, D.; Boom, W.H.; Harding, C.V. Regulation of class II MHC expression in APCs: Roles of types I, III, and IV class II transactivator. J. Immunol. 2002, 169, 1326–1333. [Google Scholar] [CrossRef]
- Accolla, R.S.; Mazza, S.; De Lerma Barbaro, A.; De Maria, A.; Tosi, G. The HLA class II transcriptional activator blocks the function of HIV-1 Tat and inhibits viral replication. Eur. J. Immunol. 2002, 32, 2783–2791. [Google Scholar] [CrossRef]
- Accolla, R.S.; De Lerma Barbaro, A.; Mazza, S.; Casoli, C.; De Maria, A.; Tosi, G. The MHC class II transactivator: Prey and hunter in infectious diseases. Trends Immunol. 2001, 22, 560–563. [Google Scholar] [CrossRef]
- Tosi, G.; Pilotti, E.; Mortara, L.; De Lerma Barbaro, A.; Casoli, C.; Accolla, R.S. Inhibition of human T cell leukemia virus type 2 replication by the suppressive action of class II transactivator and nuclear factor Y. Proc. Natl. Acad. Sci. USA 2006, 103, 12861–12866. [Google Scholar] [CrossRef]
- Tosi, G.; Forlani, G.; Andresen, V.; Turci, M.; Bertazzoni, U.; Franchini, G.; Poli, G.; Accolla, R.S. Major histocompatibility complex class II transactivator CIITA is a viral restriction factor that targets human T-cell lymphotropic virus type 1 Tax-1 function and inhibits viral replication. J. Virol. 2011, 85, 10719–10729. [Google Scholar] [CrossRef]
- Forlani, G.; Abdallah, R.; Accolla, R.S.; Tosi, G. The Major Histocompatibility Complex Class II Transactivator CIITA Inhibits the Persistent Activation of NF-kappaB by the Human T Cell Lymphotropic Virus Type 1 Tax-1 Oncoprotein. J. Virol. 2016, 90, 3708–3721. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Guo, X.; Goff, S.P. Inhibition of retroviral RNA production by ZAP, a CCCH-type zinc finger protein. Science 2002, 297, 1703–1706. [Google Scholar] [CrossRef]
- Zhu, M.; Ma, X.; Cui, X.; Zhou, J.; Li, C.; Huang, L.; Shang, Y.; Cheng, Z. Inhibition of avian tumor virus replication by CCCH-type zinc finger antiviral protein. Oncotarget 2017, 8, 58865–58871. [Google Scholar] [CrossRef]
- Mao, R.; Nie, H.; Cai, D.; Zhang, J.; Liu, H.; Yan, R.; Cuconati, A.; Block, T.M.; Guo, J.T.; Guo, H. Inhibition of hepatitis B virus replication by the host zinc finger antiviral protein. PLoS Pathog. 2013, 9, e1003494. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Wang, X.; Gao, G. The Short Form of the Zinc Finger Antiviral Protein Inhibits Influenza A Virus Protein Expression and Is Antagonized by the Virus-Encoded NS1. J. Virol. 2017, 91, e01909-16. [Google Scholar] [CrossRef] [PubMed]
- Chiu, H.P.; Chiu, H.; Yang, C.F.; Lee, Y.L.; Chiu, F.L.; Kuo, H.C.; Lin, R.J.; Lin, Y.L. Inhibition of Japanese encephalitis virus infection by the host zinc-finger antiviral protein. PLoS Pathog. 2018, 14, e1007166. [Google Scholar] [CrossRef]
- Takata, M.A.; Goncalves-Carneiro, D.; Zang, T.M.; Soll, S.J.; York, A.; Blanco-Melo, D.; Bieniasz, P.D. CG dinucleotide suppression enables antiviral defence targeting non-self RNA. Nature 2017, 550, 124–127. [Google Scholar] [CrossRef]
- Miyazato, P.; Matsuo, M.; Tan, B.J.Y.; Tokunaga, M.; Katsuya, H.; Islam, S.; Ito, J.; Murakawa, Y.; Satou, Y. HTLV-1 contains a high CG dinucleotide content and is susceptible to the host antiviral protein ZAP. Retrovirology 2019, 16, 38. [Google Scholar] [CrossRef]
- van Gent, M.; Sparrer, K.M.J.; Gack, M.U. TRIM Proteins and Their Roles in Antiviral Host Defenses. Annu. Rev. Virol. 2018, 5, 385–405. [Google Scholar] [CrossRef]
- Skorupka, K.A.; Roganowicz, M.D.; Christensen, D.E.; Wan, Y.; Pornillos, O.; Ganser-Pornillos, B.K. Hierarchical assembly governs TRIM5alpha recognition of HIV-1 and retroviral capsids. Sci. Adv. 2019, 5, eaaw3631. [Google Scholar] [CrossRef]
- Imam, S.; Komurlu, S.; Mattick, J.; Selyutina, A.; Talley, S.; Eddins, A.; Diaz-Griffero, F.; Campbell, E.M. K63-Linked Ubiquitin Is Required for Restriction of HIV-1 Reverse Transcription and Capsid Destabilization by Rhesus TRIM5alpha. J. Virol. 2019, 93, e00558-19. [Google Scholar] [CrossRef] [PubMed]
- Nozuma, S.; Matsuura, E.; Kodama, D.; Tashiro, Y.; Matsuzaki, T.; Kubota, R.; Izumo, S.; Takashima, H. Effects of host restriction factors and the HTLV-1 subtype on susceptibility to HTLV-1-associated myelopathy/tropical spastic paraparesis. Retrovirology 2017, 14, 26. [Google Scholar] [CrossRef] [PubMed]
- Leal, F.E.; Menezes, S.M.; Costa, E.A.S.; Brailey, P.M.; Gama, L.; Segurado, A.C.; Kallas, E.G.; Nixon, D.F.; Dierckx, T.; Khouri, R.; et al. Comprehensive Antiretroviral Restriction Factor Profiling Reveals the Evolutionary Imprint of the ex Vivo and in Vivo IFN-beta Response in HTLV-1-Associated Neuroinflammation. Front. Microbiol. 2018, 9, 985. [Google Scholar] [CrossRef]
- Dassouki, Z.; Sahin, U.; El Hajj, H.; Jollivet, F.; Kfoury, Y.; Lallemand-Breitenbach, V.; Hermine, O.; de The, H.; Bazarbachi, A. ATL response to arsenic/interferon therapy is triggered by SUMO/PML/RNF4-dependent Tax degradation. Blood 2015, 125, 474–482. [Google Scholar] [CrossRef]
- Hogg, M.; Paro, S.; Keegan, L.P.; O’Connell, M.A. RNA editing by mammalian ADARs. Adv. Genet. 2011, 73, 87–120. [Google Scholar] [CrossRef] [PubMed]
- Samuel, C.E. Adenosine deaminases acting on RNA (ADARs) are both antiviral and proviral. Virology 2011, 411, 180–193. [Google Scholar] [CrossRef]
- Taylor, D.R.; Puig, M.; Darnell, M.E.; Mihalik, K.; Feinstone, S.M. New antiviral pathway that mediates hepatitis C virus replicon interferon sensitivity through ADAR1. J. Virol. 2005, 79, 6291–6298. [Google Scholar] [CrossRef]
- Zhang, H.; Ni, G.; Damania, B. ADAR1 Facilitates KSHV Lytic Reactivation by Modulating the RLR-Dependent Signaling Pathway. Cell. Rep. 2020, 31, 107564. [Google Scholar] [CrossRef]
- Radetskyy, R.; Daher, A.; Gatignol, A. ADAR1 and PKR, interferon stimulated genes with clashing effects on HIV-1 replication. Cytokine Growth Factor. Rev. 2018, 40, 48–58. [Google Scholar] [CrossRef]
- Clerzius, G.; Gelinas, J.F.; Daher, A.; Bonnet, M.; Meurs, E.F.; Gatignol, A. ADAR1 interacts with PKR during human immunodeficiency virus infection of lymphocytes and contributes to viral replication. J. Virol. 2009, 83, 10119–10128. [Google Scholar] [CrossRef]
- Cachat, A.; Alais, S.; Chevalier, S.A.; Journo, C.; Fusil, F.; Dutartre, H.; Boniface, A.; Ko, N.L.; Gessain, A.; Cosset, F.L.; et al. ADAR1 enhances HTLV-1 and HTLV-2 replication through inhibition of PKR activity. Retrovirology 2014, 11, 93. [Google Scholar] [CrossRef] [PubMed]
- Cachat, A.; Chevalier, S.A.; Alais, S.; Ko, N.L.; Ratner, L.; Journo, C.; Dutartre, H.; Mahieux, R. Alpha interferon restricts human T-lymphotropic virus type 1 and 2 de novo infection through PKR activation. J. Virol. 2013, 87, 13386–13396. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, G.; Navas-Martin, S.; Martin-Garcia, J. MicroRNAs and HIV-1 infection: Antiviral activities and beyond. J. Mol. Biol. 2014, 426, 1178–1197. [Google Scholar] [CrossRef]
- Ahluwalia, J.K.; Khan, S.Z.; Soni, K.; Rawat, P.; Gupta, A.; Hariharan, M.; Scaria, V.; Lalwani, M.; Pillai, B.; Mitra, D.; et al. Human cellular microRNA hsa-miR-29a interferes with viral nef protein expression and HIV-1 replication. Retrovirology 2008, 5, 117. [Google Scholar] [CrossRef] [PubMed]
- Fochi, S.; Ciminale, V.; Trabetti, E.; Bertazzoni, U.; D’Agostino, D.M.; Zipeto, D.; Romanelli, M.G. NF-kappaB and MicroRNA Deregulation Mediated by HTLV-1 Tax and HBZ. Pathogens 2019, 8, 290. [Google Scholar] [CrossRef] [PubMed]
- Cobos Jimenez, V.; Booiman, T.; de Taeye, S.W.; van Dort, K.A.; Rits, M.A.; Hamann, J.; Kootstra, N.A. Differential expression of HIV-1 interfering factors in monocyte-derived macrophages stimulated with polarizing cytokines or interferons. Sci. Rep. 2012, 2, 763. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.T.; Nicot, C. miR-28-3p is a cellular restriction factor that inhibits human T cell leukemia virus, type 1 (HTLV-1) replication and virus infection. J. Biol. Chem. 2015, 290, 5381–5390. [Google Scholar] [CrossRef]
- Pichler, K.; Schneider, G.; Grassmann, R. MicroRNA miR-146a and further oncogenesis-related cellular microRNAs are dysregulated in HTLV-1-transformed T lymphocytes. Retrovirology 2008, 5, 100. [Google Scholar] [CrossRef]
- Tili, E.; Michaille, J.J.; Cimino, A.; Costinean, S.; Dumitru, C.D.; Adair, B.; Fabbri, M.; Alder, H.; Liu, C.G.; Calin, G.A.; et al. Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-alpha stimulation and their possible roles in regulating the response to endotoxin shock. J. Immunol. 2007, 179, 5082–5089. [Google Scholar] [CrossRef]
- Gottwein, E.; Mukherjee, N.; Sachse, C.; Frenzel, C.; Majoros, W.H.; Chi, J.T.; Braich, R.; Manoharan, M.; Soutschek, J.; Ohler, U.; et al. A viral microRNA functions as an orthologue of cellular miR-155. Nature 2007, 450, 1096–1099. [Google Scholar] [CrossRef]
- Sharma, S.; tenOever, B.R.; Grandvaux, N.; Zhou, G.P.; Lin, R.; Hiscott, J. Triggering the interferon antiviral response through an IKK-related pathway. Science 2003, 300, 1148–1151. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Weidmer, A.; Liu, C.G.; Volinia, S.; Croce, C.M.; Lieberman, P.M. Epstein-Barr virus-induced miR-155 attenuates NF-kappaB signaling and stabilizes latent virus persistence. J. Virol. 2008, 82, 10436–10443. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Wang, P.; Lin, L.; Liu, X.; Ma, F.; An, H.; Wang, Z.; Cao, X. MicroRNA-146a feedback inhibits RIG-I-dependent Type I IFN production in macrophages by targeting TRAF6, IRAK1, and IRAK2. J. Immunol. 2009, 183, 2150–2158. [Google Scholar] [CrossRef] [PubMed]
- Taganov, K.D.; Boldin, M.P.; Chang, K.J.; Baltimore, D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12481–12486. [Google Scholar] [CrossRef]
- Oliere, S.; Douville, R.; Sze, A.; Belgnaoui, S.M.; Hiscott, J. Modulation of innate immune responses during human T-cell leukemia virus (HTLV-1) pathogenesis. Cytokine Growth Factor. Rev. 2011, 22, 197–210. [Google Scholar] [CrossRef]
- Leal, F.E.; Ndhlovu, L.C.; Hasenkrug, A.M.; Bruno, F.R.; Carvalho, K.I.; Wynn-Williams, H.; Neto, W.K.; Sanabani, S.S.; Segurado, A.C.; Nixon, D.F.; et al. Expansion in CD39(+) CD4(+) immunoregulatory t cells and rarity of Th17 cells in HTLV-1 infected patients is associated with neurological complications. PLoS Negl. Trop. Dis. 2013, 7, e2028. [Google Scholar] [CrossRef]
- Mariathasan, S.; Newton, K.; Monack, D.M.; Vucic, D.; French, D.M.; Lee, W.P.; Roose-Girma, M.; Erickson, S.; Dixit, V.M. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 2004, 430, 213–218. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell. 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Zhao, C.; Zhao, W. NLRP3 Inflammasome-A Key Player in Antiviral Responses. Front. Immunol. 2020, 11, 211. [Google Scholar] [CrossRef]
- Banerjee, P.; Rochford, R.; Antel, J.; Canute, G.; Wrzesinski, S.; Sieburg, M.; Feuer, G. Proinflammatory cytokine gene induction by human T-cell leukemia virus type 1 (HTLV-1) and HTLV-2 Tax in primary human glial cells. J. Virol. 2007, 81, 1690–1700. [Google Scholar] [CrossRef]
- Kamada, A.J.; Pontillo, A.; Guimaraes, R.L.; Loureiro, P.; Crovella, S.; Brandao, L.A. NLRP3 polymorphism is associated with protection against human T-lymphotropic virus 1 infection. Mem. Inst. Oswaldo Cruz 2014, 109, 960–963. [Google Scholar] [CrossRef] [PubMed]
- Moles, R.; Sarkis, S.; Galli, V.; Omsland, M.; Artesi, M.; Bissa, M.; McKinnon, K.; Brown, S.; Hahaut, V.; Washington-Parks, R.; et al. NK cells and monocytes modulate primary HTLV-1 infection. PLoS Pathog. 2022, 18, e1010416. [Google Scholar] [CrossRef] [PubMed]
- Tsukasaki, K.; Miller, C.W.; Kubota, T.; Takeuchi, S.; Fujimoto, T.; Ikeda, S.; Tomonaga, M.; Koeffler, H.P. Tumor necrosis factor alpha polymorphism associated with increased susceptibility to development of adult T-cell leukemia/lymphoma in human T-lymphotropic virus type 1 carriers. Cancer Res. 2001, 61, 3770–3774. [Google Scholar] [PubMed]
- Aggarwal, B.B.; Shishodia, S.; Sandur, S.K.; Pandey, M.K.; Sethi, G. Inflammation and cancer: How hot is the link? Biochem. Pharmacol. 2006, 72, 1605–1621. [Google Scholar] [CrossRef]
- Quaresma, J.A.; Yoshikawa, G.T.; Koyama, R.V.; Dias, G.A.; Fujihara, S.; Fuzii, H.T. HTLV-1, Immune Response and Autoimmunity. Viruses 2015, 8, 5. [Google Scholar] [CrossRef]
- Horiuchi, S.; Yamamoto, N.; Dewan, M.Z.; Takahashi, Y.; Yamashita, A.; Yoshida, T.; Nowell, M.A.; Richards, P.J.; Jones, S.A.; Yamamoto, N. Human T-cell leukemia virus type-I Tax induces expression of interleukin-6 receptor (IL-6R): Shedding of soluble IL-6R and activation of STAT3 signaling. Int. J. Cancer 2006, 119, 823–830. [Google Scholar] [CrossRef]
- Mohanty, S.; Harhaj, E.W. Mechanisms of Oncogenesis by HTLV-1 Tax. Pathogens 2020, 9, 543. [Google Scholar] [CrossRef]
- Harhaj, E.W.; Giam, C.Z. NF-kappaB signaling mechanisms in HTLV-1-induced adult T-cell leukemia/lymphoma. FEBS J. 2018, 285, 3324–3336. [Google Scholar] [CrossRef]
- Zhao, T. The Role of HBZ in HTLV-1-Induced Oncogenesis. Viruses 2016, 8, 34. [Google Scholar] [CrossRef]
- Satou, Y.; Yasunaga, J.; Zhao, T.; Yoshida, M.; Miyazato, P.; Takai, K.; Shimizu, K.; Ohshima, K.; Green, P.L.; Ohkura, N.; et al. HTLV-1 bZIP factor induces T-cell lymphoma and systemic inflammation in vivo. PLoS Pathog. 2011, 7, e1001274. [Google Scholar] [CrossRef]
- Higuchi, Y.; Yasunaga, J.I.; Mitagami, Y.; Tsukamoto, H.; Nakashima, K.; Ohshima, K.; Matsuoka, M. HTLV-1 induces T cell malignancy and inflammation by viral antisense factor-mediated modulation of the cytokine signaling. Proc. Natl. Acad. Sci. USA 2020, 117, 13740–13749. [Google Scholar] [CrossRef]
- Nyborg, J.K.; Egan, D.; Sharma, N. The HTLV-1 Tax protein: Revealing mechanisms of transcriptional activation through histone acetylation and nucleosome disassembly. Biochim. Biophys. Acta 2010, 1799, 266–274. [Google Scholar] [CrossRef]
- Mohanty, S.; Han, T.; Choi, Y.B.; Lavorgna, A.; Zhang, J.; Harhaj, E.W. The E3/E4 ubiquitin conjugation factor UBE4B interacts with and ubiquitinates the HTLV-1 Tax oncoprotein to promote NF-kappaB activation. PLoS Pathog. 2020, 16, e1008504. [Google Scholar] [CrossRef]
- Kfoury, Y.; Setterblad, N.; El-Sabban, M.; Zamborlini, A.; Dassouki, Z.; El Hajj, H.; Hermine, O.; Pique, C.; de The, H.; Saib, A.; et al. Tax ubiquitylation and SUMOylation control the dynamic shuttling of Tax and NEMO between Ubc9 nuclear bodies and the centrosome. Blood 2011, 117, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Shembade, N.; Harhaj, E.W. Role of post-translational modifications of HTLV-1 Tax in NF-kappaB activation. World J. Biol. Chem. 2010, 1, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Lavorgna, A.; Harhaj, E.W. Regulation of HTLV-1 tax stability, cellular trafficking and NF-kappaB activation by the ubiquitin-proteasome pathway. Viruses 2014, 6, 3925–3943. [Google Scholar] [CrossRef] [PubMed]
- Durkin, S.S.; Ward, M.D.; Fryrear, K.A.; Semmes, O.J. Site-specific phosphorylation differentiates active from inactive forms of the human T-cell leukemia virus type 1 Tax oncoprotein. J. Biol. Chem. 2006, 281, 31705–31712. [Google Scholar] [CrossRef] [PubMed]
- Gatza, M.L.; Dayaram, T.; Marriott, S.J. Ubiquitination of HTLV-I Tax in response to DNA damage regulates nuclear complex formation and nuclear export. Retrovirology 2007, 4, 95. [Google Scholar] [CrossRef]
- Taniguchi, Y.; Nosaka, K.; Yasunaga, J.; Maeda, M.; Mueller, N.; Okayama, A.; Matsuoka, M. Silencing of human T-cell leukemia virus type I gene transcription by epigenetic mechanisms. Retrovirology 2005, 2, 64. [Google Scholar] [CrossRef]
- Mitre, E.; Thompson, R.W.; Carvalho, E.M.; Nutman, T.B.; Neva, F.A. Majority of interferon-gamma-producing CD4+ cells in patients infected with human T cell lymphotrophic virus do not express tax protein. J. Infect. Dis. 2003, 188, 428–432. [Google Scholar] [CrossRef]
- Masaki, A.; Ishida, T.; Suzuki, S.; Ito, A.; Narita, T.; Kinoshita, S.; Ri, M.; Kusumoto, S.; Komatsu, H.; Inagaki, H.; et al. Human T-cell lymphotropic/leukemia virus type 1 (HTLV-1) Tax-specific T-cell exhaustion in HTLV-1-infected individuals. Cancer Sci. 2018, 109, 2383–2390. [Google Scholar] [CrossRef] [PubMed]
- Mahgoub, M.; Yasunaga, J.I.; Iwami, S.; Nakaoka, S.; Koizumi, Y.; Shimura, K.; Matsuoka, M. Sporadic on/off switching of HTLV-1 Tax expression is crucial to maintain the whole population of virus-induced leukemic cells. Proc. Natl. Acad. Sci. USA 2018, 115, E1269–E1278. [Google Scholar] [CrossRef] [PubMed]
- Hyun, J.; Ramos, J.C.; Toomey, N.; Balachandran, S.; Lavorgna, A.; Harhaj, E.; Barber, G.N. Oncogenic human T-cell lymphotropic virus type 1 tax suppression of primary innate immune signaling pathways. J. Virol. 2015, 89, 4880–4893. [Google Scholar] [CrossRef]
- Yuen, C.K.; Chan, C.P.; Fung, S.Y.; Wang, P.H.; Wong, W.M.; Tang, H.V.; Yuen, K.S.; Chan, C.P.; Jin, D.Y.; Kok, K.H. Suppression of Type I Interferon Production by Human T-Cell Leukemia Virus Type 1 Oncoprotein Tax through Inhibition of IRF3 Phosphorylation. J. Virol. 2016, 90, 3902–3912. [Google Scholar] [CrossRef]
- Wang, J.; Yang, S.; Liu, L.; Wang, H.; Yang, B. HTLV-1 Tax impairs K63-linked ubiquitination of STING to evade host innate immunity. Virus Res. 2017, 232, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yamada, O.; Kawagishi, K.; Araki, H.; Yamaoka, S.; Hattori, T.; Shimotohno, K. Human T-cell leukemia virus type 1 Tax modulates interferon-alpha signal transduction through competitive usage of the coactivator CBP/p300. Virology 2008, 379, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Oliere, S.; Hernandez, E.; Lezin, A.; Arguello, M.; Douville, R.; Nguyen, T.L.; Olindo, S.; Panelatti, G.; Kazanji, M.; Wilkinson, P.; et al. HTLV-1 evades type I interferon antiviral signaling by inducing the suppressor of cytokine signaling 1 (SOCS1). PLoS Pathog. 2010, 6, e1001177. [Google Scholar] [CrossRef]
- Charoenthongtrakul, S.; Zhou, Q.; Shembade, N.; Harhaj, N.S.; Harhaj, E.W. Human T cell leukemia virus type 1 Tax inhibits innate antiviral signaling via NF-kappaB-dependent induction of SOCS1. J. Virol. 2011, 85, 6955–6962. [Google Scholar] [CrossRef]
- Liu, J.; Qian, C.; Cao, X. Post-Translational Modification Control of Innate Immunity. Immunity 2016, 45, 15–30. [Google Scholar] [CrossRef]
- Feng, X.; Heyden, N.V.; Ratner, L. Alpha interferon inhibits human T-cell leukemia virus type 1 assembly by preventing Gag interaction with rafts. J. Virol. 2003, 77, 13389–13395. [Google Scholar] [CrossRef]
- Kinpara, S.; Kijiyama, M.; Takamori, A.; Hasegawa, A.; Sasada, A.; Masuda, T.; Tanaka, Y.; Utsunomiya, A.; Kannagi, M. Interferon-alpha (IFN-alpha) suppresses HTLV-1 gene expression and cell cycling, while IFN-alpha combined with zidovudine induces p53 signaling and apoptosis in HTLV-1-infected cells. Retrovirology 2013, 10, 52. [Google Scholar] [CrossRef] [PubMed]
- Macchi, B.; Balestrieri, E.; Frezza, C.; Grelli, S.; Valletta, E.; Marcais, A.; Marino-Merlo, F.; Turpin, J.; Bangham, C.R.; Hermine, O.; et al. Quantification of HTLV-1 reverse transcriptase activity in ATL patients treated with zidovudine and interferon-alpha. Blood Adv. 2017, 1, 748–752. [Google Scholar] [CrossRef] [PubMed]
- Kannagi, M.; Hasegawa, A.; Nagano, Y.; Kimpara, S.; Suehiro, Y. Impact of host immunity on HTLV-1 pathogenesis: Potential of Tax-targeted immunotherapy against ATL. Retrovirology 2019, 16, 23. [Google Scholar] [CrossRef] [PubMed]
- Satou, Y.; Yasunaga, J.; Yoshida, M.; Matsuoka, M. HTLV-I basic leucine zipper factor gene mRNA supports proliferation of adult T cell leukemia cells. Proc. Natl. Acad. Sci. USA 2006, 103, 720–725. [Google Scholar] [CrossRef]
- Forlani, G.; Shallak, M.; Tedeschi, A.; Cavallari, I.; Marcais, A.; Hermine, O.; Accolla, R.S. Dual cytoplasmic and nuclear localization of HTLV-1-encoded HBZ protein is a unique feature of adult T-cell leukemia. Haematologica 2021, 106, 2076–2085. [Google Scholar] [CrossRef]
- Lemasson, I.; Lewis, M.R.; Polakowski, N.; Hivin, P.; Cavanagh, M.H.; Thebault, S.; Barbeau, B.; Nyborg, J.K.; Mesnard, J.M. Human T-cell leukemia virus type 1 (HTLV-1) bZIP protein interacts with the cellular transcription factor CREB to inhibit HTLV-1 transcription. J. Virol. 2007, 81, 1543–1553. [Google Scholar] [CrossRef]
- Arnold, J.; Zimmerman, B.; Li, M.; Lairmore, M.D.; Green, P.L. Human T-cell leukemia virus type-1 antisense-encoded gene, Hbz, promotes T-lymphocyte proliferation. Blood 2008, 112, 3788–3797. [Google Scholar] [CrossRef]
- Ma, G.; Yasunaga, J.; Fan, J.; Yanagawa, S.; Matsuoka, M. HTLV-1 bZIP factor dysregulates the Wnt pathways to support proliferation and migration of adult T-cell leukemia cells. Oncogene 2013, 32, 4222–4230. [Google Scholar] [CrossRef]
- Kuhlmann, A.S.; Villaudy, J.; Gazzolo, L.; Castellazzi, M.; Mesnard, J.M.; Duc Dodon, M. HTLV-1 HBZ cooperates with JunD to enhance transcription of the human telomerase reverse transcriptase gene (hTERT). Retrovirology 2007, 4, 92. [Google Scholar] [CrossRef]
- Shiohama, Y.; Naito, T.; Matsuzaki, T.; Tanaka, R.; Tomoyose, T.; Takashima, H.; Fukushima, T.; Tanaka, Y.; Saito, M. Absolute quantification of HTLV-1 basic leucine zipper factor (HBZ) protein and its plasma antibody in HTLV-1 infected individuals with different clinical status. Retrovirology 2016, 13, 29. [Google Scholar] [CrossRef]
- Mitagami, Y.; Yasunaga, J.; Kinosada, H.; Ohshima, K.; Matsuoka, M. Interferon-gamma Promotes Inflammation and Development of T-Cell Lymphoma in HTLV-1 bZIP Factor Transgenic Mice. PLoS Pathog. 2015, 11, e1005120. [Google Scholar] [CrossRef]
- Yamamoto-Taguchi, N.; Satou, Y.; Miyazato, P.; Ohshima, K.; Nakagawa, M.; Katagiri, K.; Kinashi, T.; Matsuoka, M. HTLV-1 bZIP factor induces inflammation through labile Foxp3 expression. PLoS Pathog. 2013, 9, e1003630. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Yasunaga, J.; Satou, Y.; Nakao, M.; Takahashi, M.; Fujii, M.; Matsuoka, M. Human T-cell leukemia virus type 1 bZIP factor selectively suppresses the classical pathway of NF-kappaB. Blood 2009, 113, 2755–2764. [Google Scholar] [CrossRef] [PubMed]
- Zhi, H.; Yang, L.; Kuo, Y.L.; Ho, Y.K.; Shih, H.M.; Giam, C.Z. NF-kappaB hyper-activation by HTLV-1 tax induces cellular senescence, but can be alleviated by the viral anti-sense protein HBZ. PLoS Pathog. 2011, 7, e1002025. [Google Scholar] [CrossRef] [PubMed]
- Narulla, M.S.; Alsairi, A.; Charmier, L.; Noonan, S.; Conroy, D.; Hall, W.W.; Sheehy, N. Positive and Negative Regulation of Type I Interferons by the Human T Cell Leukemia Virus Antisense Protein HBZ. J. Virol. 2017, 91, e00853-17. [Google Scholar] [CrossRef]
- Nicot, C.; Dundr, M.; Johnson, J.M.; Fullen, J.R.; Alonzo, N.; Fukumoto, R.; Princler, G.L.; Derse, D.; Misteli, T.; Franchini, G. HTLV-1-encoded p30II is a post-transcriptional negative regulator of viral replication. Nat. Med. 2004, 10, 197–201. [Google Scholar] [CrossRef]
- Zhang, W.; Nisbet, J.W.; Albrecht, B.; Ding, W.; Kashanchi, F.; Bartoe, J.T.; Lairmore, M.D. Human T-lymphotropic virus type 1 p30(II) regulates gene transcription by binding CREB binding protein/p300. J. Virol. 2001, 75, 9885–9895. [Google Scholar] [CrossRef]
- Malu, A.; Hutchison, T.; Yapindi, L.; Smith, K.; Nelson, K.; Bergeson, R.; Pope, J.; Romeo, M.; Harrod, C.; Ratner, L.; et al. The human T-cell leukemia virus type-1 tax oncoprotein dissociates NF-kappaB p65(RelA)-Stathmin complexes and causes catastrophic mitotic spindle damage and genomic instability. Virology 2019, 535, 83–101. [Google Scholar] [CrossRef]
- Datta, A.; Sinha-Datta, U.; Dhillon, N.K.; Buch, S.; Nicot, C. The HTLV-I p30 interferes with TLR4 signaling and modulates the release of pro- and anti-inflammatory cytokines from human macrophages. J. Biol. Chem. 2006, 281, 23414–23424. [Google Scholar] [CrossRef]
- Mori, N.; Prager, D. Interleukin-10 gene expression and adult T-cell leukemia. Leuk. Lymphoma 1998, 29, 239–248. [Google Scholar] [CrossRef]
- Mori, N.; Gill, P.S.; Mougdil, T.; Murakami, S.; Eto, S.; Prager, D. Interleukin-10 gene expression in adult T-cell leukemia. Blood 1996, 88, 1035–1045. [Google Scholar] [CrossRef] [PubMed]
- Moles, R.; Sarkis, S.; Galli, V.; Omsland, M.; Purcell, D.F.J.; Yurick, D.; Khoury, G.; Pise-Masison, C.A.; Franchini, G. p30 protein: A critical regulator of HTLV-1 viral latency and host immunity. Retrovirology 2019, 16, 42. [Google Scholar] [CrossRef] [PubMed]
- Datta, A.; Silverman, L.; Phipps, A.J.; Hiraragi, H.; Ratner, L.; Lairmore, M.D. Human T-lymphotropic virus type-1 p30 alters cell cycle G2 regulation of T lymphocytes to enhance cell survival. Retrovirology 2007, 4, 49. [Google Scholar] [CrossRef] [PubMed]
- Baydoun, H.H.; Pancewicz, J.; Bai, X.; Nicot, C. HTLV-I p30 inhibits multiple S phase entry checkpoints, decreases cyclin E-CDK2 interactions and delays cell cycle progression. Mol. Cancer 2010, 9, 302. [Google Scholar] [CrossRef]
- Nicot, C.; Mulloy, J.C.; Ferrari, M.G.; Johnson, J.M.; Fu, K.; Fukumoto, R.; Trovato, R.; Fullen, J.; Leonard, W.J.; Franchini, G. HTLV-1 p12(I) protein enhances STAT5 activation and decreases the interleukin-2 requirement for proliferation of primary human peripheral blood mononuclear cells. Blood 2001, 98, 823–829. [Google Scholar] [CrossRef]
- Johnson, J.M.; Nicot, C.; Fullen, J.; Ciminale, V.; Casareto, L.; Mulloy, J.C.; Jacobson, S.; Franchini, G. Free major histocompatibility complex class I heavy chain is preferentially targeted for degradation by human T-cell leukemia/lymphotropic virus type 1 p12(I) protein. J. Virol. 2001, 75, 6086–6094. [Google Scholar] [CrossRef]
- Ohashi, T.; Hanabuchi, S.; Suzuki, R.; Kato, H.; Masuda, T.; Kannagi, M. Correlation of major histocompatibility complex class I downregulation with resistance of human T-cell leukemia virus type 1-infected T cells to cytotoxic T-lymphocyte killing in a rat model. J. Virol. 2002, 76, 7010–7019. [Google Scholar] [CrossRef]
- Koralnik, I.J.; Gessain, A.; Klotman, M.E.; Lo Monico, A.; Berneman, Z.N.; Franchini, G. Protein isoforms encoded by the pX region of human T-cell leukemia/lymphotropic virus type I. Proc. Natl. Acad. Sci. USA 1992, 89, 8813–8817. [Google Scholar] [CrossRef]
- Collins, N.D.; Newbound, G.C.; Albrecht, B.; Beard, J.L.; Ratner, L.; Lairmore, M.D. Selective ablation of human T-cell lymphotropic virus type 1 p12I reduces viral infectivity in vivo. Blood 1998, 91, 4701–4707. [Google Scholar] [CrossRef]
- Banerjee, P.; Feuer, G.; Barker, E. Human T-cell leukemia virus type 1 (HTLV-1) p12I down-modulates ICAM-1 and -2 and reduces adherence of natural killer cells, thereby protecting HTLV-1-infected primary CD4+ T cells from autologous natural killer cell-mediated cytotoxicity despite the reduction of major histocompatibility complex class I molecules on infected cells. J. Virol. 2007, 81, 9707–9717. [Google Scholar] [CrossRef]
- Fukudome, K.; Furuse, M.; Fukuhara, N.; Orita, S.; Imai, T.; Takagi, S.; Nagira, M.; Hinuma, Y.; Yoshie, O. Strong induction of ICAM-1 in human T cells transformed by human T-cell-leukemia virus type 1 and depression of ICAM-1 or LFA-1 in adult T-cell-leukemia-derived cell lines. Int. J. Cancer 1992, 52, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Cheng, Q.; Wang, Z.; Yuan, P.; Li, Z.; Sun, Y.; Han, S.; Yin, J.; Peng, B.; He, X.; et al. Human Pirh2 is a novel inhibitor of prototype foamy virus replication. Viruses 2015, 7, 1668–1684. [Google Scholar] [CrossRef] [PubMed]
- Yuan, P.; Yan, J.; Wang, S.; Guo, Y.; Xi, X.; Han, S.; Yin, J.; Peng, B.; He, X.; Bodem, J.; et al. Trim28 acts as restriction factor of prototype foamy virus replication by modulating H3K9me3 marks and destabilizing the viral transactivator Tas. Retrovirology 2021, 18, 38. [Google Scholar] [CrossRef]
- Schnell, A.P.; Kohrt, S.; Aristodemou, A.; Taylor, G.P.; Bangham, C.R.M.; Thoma-Kress, A.K. HDAC inhibitors Panobinostat and Romidepsin enhance tax transcription in HTLV-1-infected cell lines and freshly isolated patients’ T-cells. Front. Immunol. 2022, 13, 978800. [Google Scholar] [CrossRef] [PubMed]
- Lezin, A.; Gillet, N.; Olindo, S.; Signate, A.; Grandvaux, N.; Verlaeten, O.; Belrose, G.; de Carvalho Bittencourt, M.; Hiscott, J.; Asquith, B.; et al. Histone deacetylase mediated transcriptional activation reduces proviral loads in HTLV-1 associated myelopathy/tropical spastic paraparesis patients. Blood 2007, 110, 3722–3728. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohanty, S.; Harhaj, E.W. Mechanisms of Innate Immune Sensing of HTLV-1 and Viral Immune Evasion. Pathogens 2023, 12, 735. https://doi.org/10.3390/pathogens12050735
Mohanty S, Harhaj EW. Mechanisms of Innate Immune Sensing of HTLV-1 and Viral Immune Evasion. Pathogens. 2023; 12(5):735. https://doi.org/10.3390/pathogens12050735
Chicago/Turabian StyleMohanty, Suchitra, and Edward W. Harhaj. 2023. "Mechanisms of Innate Immune Sensing of HTLV-1 and Viral Immune Evasion" Pathogens 12, no. 5: 735. https://doi.org/10.3390/pathogens12050735
APA StyleMohanty, S., & Harhaj, E. W. (2023). Mechanisms of Innate Immune Sensing of HTLV-1 and Viral Immune Evasion. Pathogens, 12(5), 735. https://doi.org/10.3390/pathogens12050735