1. Introduction
Herpes simplex virus 1 (HSV-1) is the prototype member of the human
Herpesviridae family. Epidemiological surveys show that HSV-1 is a ubiquitous virus with a global seroprevalence rate of 50–90% in normal individuals [
1,
2,
3], which is the most common causative agent for cold sores, viral encephalitis, and infectious blindness [
4,
5,
6]. HSV-1 usually causes mild and sometimes asymptomatic infections in healthy or immunocompetent individuals but can lead to severe eyelid inflammation and encephalitis caused by severe central nervous system (CNS) infection in neonates or immune-compromised patients [
7].
HSV-1 can engage in lytic infection to produce viral progeny and establish a latent infection that can co-exist with the host during the lifetime of the host. HSV-1 is one of the most common causes of encephalitis in children and adults [
8,
9]. Neurotropic HSV-1 can infect the axon terminals of olfactory neurons, enter CNS via the retrograde axon transport of neurons, and finally reach the olfactory bulb in the brain. HSV-1 can also be reactivated from the neurons in the trigeminal ganglion and be transported anterogradely to the skin or CNS [
10]. Herpes simplex encephalitis (HSE) is one of the most serious manifestations of HSV-1 infection, with a mortality rate of 97%. HSV-1 infection in the cornea can cause chronic immune inflammatory disease, namely herpes simplex keratitis (HSK), which will lead to irreversible damage and blindness in the infected patient [
11]. Currently, there is no vaccine for preventing HSV infection [
3,
12]. Both primary viral infection and reactivation after latent infection may cause chronic inflammatory diseases of different degrees [
13]. Due to the widespread prevalence of HSV-1, the issue of latent infection is serious. The commonly used clinical therapeutic drugs are nucleoside analogues represented by acyclovir (ACV) [
3], which inhibit viral DNA synthesis and are highly effective in blocking viral replication and lytic infection [
14]. Although the use of antiviral drugs ACV can reduce the mortality rate of HSE patients, most patients have permanent neurological sequelae after recovery, including symptoms of cognitive, memory and behavioral disorders, and will increase the incidence rate of epilepsy [
10,
15]. Moreover, they have little effect on viral latency and cannot completely eliminate viral infection. These issues point to a strong need to develop improved and specific strategies for the prevention and treatment of HSV-1 infection, especially for latent viral infection.
HSV-1 is an enveloped double-stranded DNA virus. The genome of HSV-1 is large and complex, consisting of a unique long (UL) region and a unique short (US) region, with reverse repeats on its flanks. HSV genome is about 152 kb, encoding more than 80 different proteins [
16]. The diameter of HSV-1 virus particles is about 200 nm. Viral entry initiates with glycoprotein gD binding to one of its receptors: nectin-1, HVEM or modified heparan sulphates [
17,
18]. Therefore, gD is thought to be the major determinant of HSV tropism and also important in HSV-1 progeny replication and package [
19].
Following the viral entry step, viral genomes are released within the nuclei and replicated, and progeny viruses are assembled [
20]. During HSV-1 lytic infection, viral genes are expressed sequentially in a cascade fashion, and they are defined by immediate–early (IE), early (E), and late (L) phases [
2]. The entire lifecycle of HSV lytic infection is tightly regulated by a collection of viral genes (e.g.,
VP16,
ICP4, and
ICP27) [
21,
22,
23], which are essential for HSV-1 replication and lytic infection. VP16 (alpha-gene-transactivating factor, α-TIF) is a viral tegument protein that plays a crucial role in initiating the lytic cycle of HSV. Upon entry into the host cell, VP16 forms a complex with cellular factors Oct-1 and host cell factor 1 (HCF-1), enabling the activation of viral immediate–early (IE) genes. The expression of IE genes is the first step in the HSV lytic cycle, which subsequently leads to the expression of early and late viral genes, viral DNA replication, and the production of progeny viral particles [
24]. ICP4 (infected cell polypeptide 4) is an HSV immediate–early protein that functions as a viral transcriptional regulator. ICP4 is essential for the expression of early and late viral genes, as it binds to viral promoters and modulates gene expression. By regulating the transcription of viral genes, ICP4 plays a pivotal role in controlling the progression of the viral lytic cycle and ensuring efficient virus replication [
25,
26]. ICP27 (infected cell polypeptide 27) is another HSV immediate–early protein that has multiple roles in the viral life cycle, including the transcriptional and post-transcriptional regulation of viral gene expression. ICP27 influences the export of viral mRNA from the nucleus to the cytoplasm and has been shown to interact with cellular splicing factors, potentially affecting viral RNA processing. Additionally, ICP27 may play a role in inhibiting host cell gene expression, thereby promoting viral replication [
27].
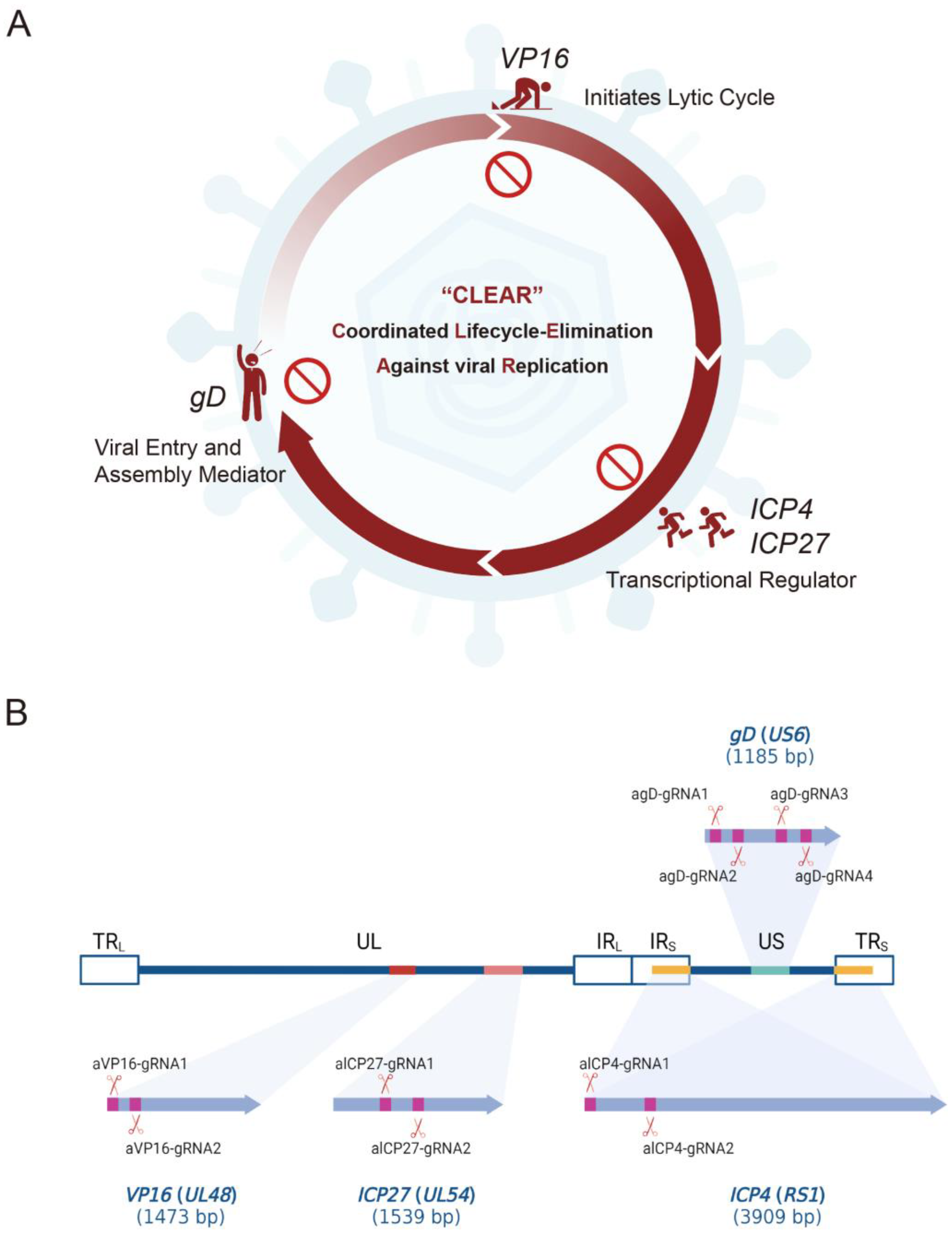
The genome editing strategy represents a promising approach against viral infection and replication by modifying or destroying the genetic material of viruses [
28,
29,
30,
31]. Several studies have used the CRISPR/Cas9 system against HSV-1 productive infection, and gRNAs were constructed to target HSV-1 essential genes, such as
ICP0,
ICP4,
ICP27,
UL8,
UL29,
US2, etc. [
32,
33,
34]. In a recent study, researchers presented evidence demonstrating the effective inhibition of HSV-1 infection
in vitro using CRISPR-Cas9 and CRISPR-CasX systems, highlighting a promising direction for developing antiviral therapies targeting UL30 [
35]. Here, we developed a multi-target genome editing approach against HSV-1 infection, targeting and eliminating HSV-1 genomes via a method named CLEAR (coordinated lifecycle elimination against viral replication). RNA-guided CRISPR/Cas9 genome editing was used to specifically target the DNA sequences of HSV-1 genes
VP16,
ICP27,
ICP4, or
gD, which express key viral proteins that stimulate HSV-1 gene expression, replication, or packaging [
36]. Our findings revealed that lentivirus delivered CRISPR-Cas9/gRNA-mediated targeting
VP16,
ICP27,
ICP4, and
gD genes reduced HSV-1 infectivity in cell culture models and protected mice against HSV-1 infection. These results support the promise of the CLEAR approach for developing potential anti-HSV-1 therapies that are capable of eliminating latent viral infections.
3. Discussion
Herpes cold sores caused by HSV-1 infection will occur repeatedly, which can be attributed to the lifelong persistent infection of the virus in the host [
37,
38]. This strategy is crucial for the survival of viruses because the entire life process of the host provides a repository for regular viral reactivation. When the host’s immunity declines, the virus recurs in the patient via reactivation. Relevant studies indicate that 70% of HSE cases in adults are attributable to previous HSV-1 infection and virus reactivation [
4].
HSV-1 therapy consists of nucleotide analogs such as acyclovir (ACV) and valacyclovir. Since the 1980s, ACV and its derivatives have become the first choice for the prevention and treatment of herpes virus infection [
3]. The continuous emergence of drug-resistant virus cases also poses higher requirements for antiviral treatment. Consequently, the issue of drug resistance warrants significant attention, underscoring the urgent need for research and development of novel therapeutic strategies. Increasing evidence shows that CRISPR technology can be used to effectively inhibit virus replication, including HSV, SARS-CoV-2 [
39], HBV [
40], HIV [
41], etc. Due to the large size of the Cas9 gene, a viral gene delivery vector with a large load capacity is required. Currently, AAV and lentivirus are the most commonly used viral vector candidates. Considering the limited gene-carrying capacity of AAV vectors, utilizing lentiviral vectors to deliver CRISPR elements may present a viable approach for herpes simplex keratitis (HSK) treatment. This prospective strategy could be applied through corneal therapy. Beyond merely eliminating HSV at the local corneal level, the lentivirus vector can retrogradely be transported along the axon and infect trigeminal ganglion neurons, and subsequently disrupting HSV reservoirs within the trigeminal ganglion [
32].
In this study, we assessed the ability of CRISPR/Cas9 editing to eliminate HSV-1 replication by targeting specific DNA sequences essential to viral protein expression during the early and late phases of viral infection/reactivation. We designed specific gRNAs targeting the essential genes of HSV—
VP16 (2 gRNAs),
ICP27 (2 gRNAs),
ICP4 (2 gRNAs), and
gD (4 gRNAs)—and constructed 10 core plasmids of lentiviral vectors carrying SpCas9 and the corresponding gRNA (
Figure 1); then, we packaged and prepared lentiviruses for the gene delivery of the CRISPR editing system. First of all, we screened the inhibitory effect of the HSV replication of 10 gRNA-mediated
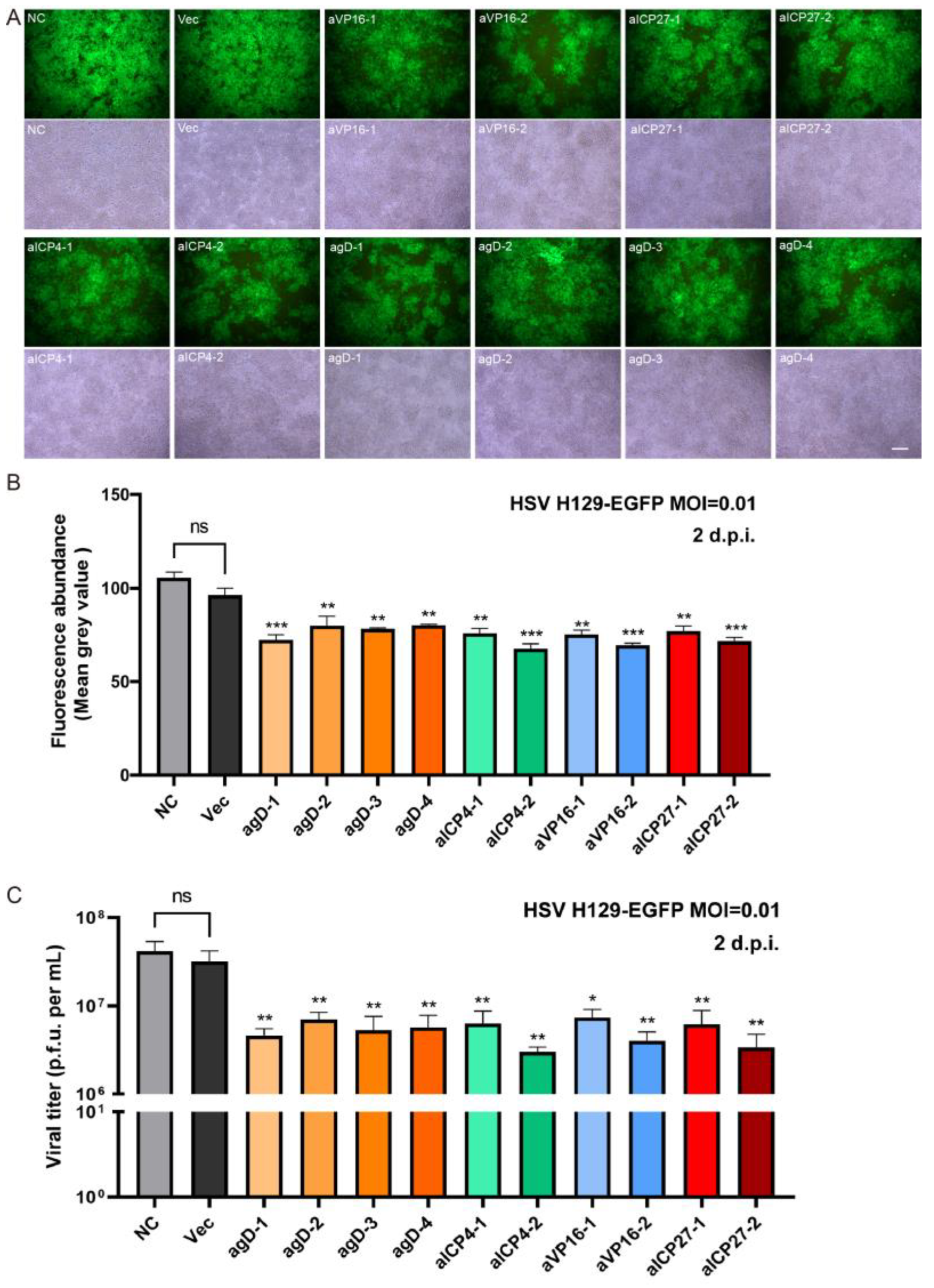
in vitro editing instances. Two days after infection with the H129-EGFP virus, the viruses in the control groups (NC and Vec) proliferated rapidly, and almost all cells expressed green fluorescent proteins, while the fluorescence ratio of single gRNA groups decreased to varying degrees (
Figure 3A). By determining the virus titers of the cell supernatant, all viral loads of the Cas9/gRNA groups were significantly lower than that of the control groups. Four types of gRNA—agD-1, aICP4-2, aVP16-2, and aICP27-2—were observed to have the most obvious inhibitory effect (
Figure 3B), thus serving as candidates for producing lentivirus vectors. At present, lentiviral vectors are widely used in CAR-T cell therapy to transduce patient-derived T cells
in vitro. For gene therapy, the viral genome of lentivirus will be integrated into the host genome, which is one of the main safety risks. However, the lentiviral vector also has certain advantages. As a pseudovirus vector, it is less toxic. The lentivirus itself does not proliferate, and the risk is controllable. Moreover, Cai et al. reported that HSV elimination in the TG reservoirs could be achieved by lentivirus-mediated CRISPR editing via the lentivirus retrogradely transport from the cornea to the trigeminal ganglion [
32]. Of course, it is safer to choose the recombinant adeno-associated virus (rAAV) virus vector. However, the disadvantage is that the carrying capacity of rAAV is too small for delivering CRISPR system.
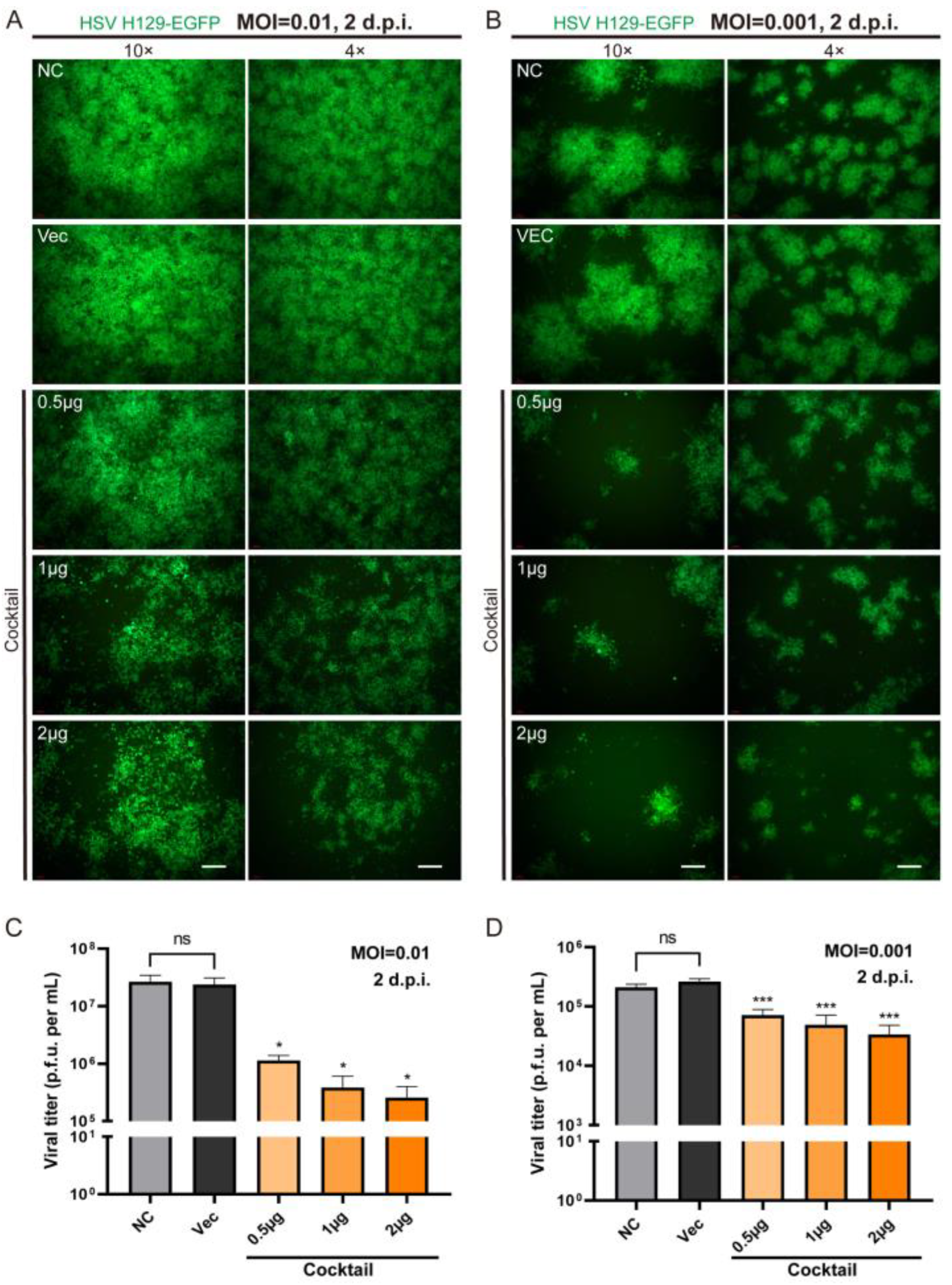
In order to more effectively block the life cycle of HSV replication by multi-point genome editing, we tested whether the combination of Cas9/gRNAs (CLEAR strategy) simultaneously targeting four genes could achieve improved inhibition effects against HSV replication. We applied different concentrations (0.5 μg, 1 μg, and 2 μg) of cocktail gRNA plasmids to transfect cells, and then the HSV1 H129-EGFP virus infected the transfected cells at MOI = 0.01 and MOI = 0.001 (
Figure 4). It was found that the cocktail groups showed significant inhibition on HSV replication, even at low doses. With the dose increase in Cas9/gRNA plasmids, the inhibition of viral proliferation was stronger (
Figure 4A,C). These results indicated that the combined utilization strategy of Cas9/gRNA targeting the four genes could effectively inhibit HSV replication and proliferation.
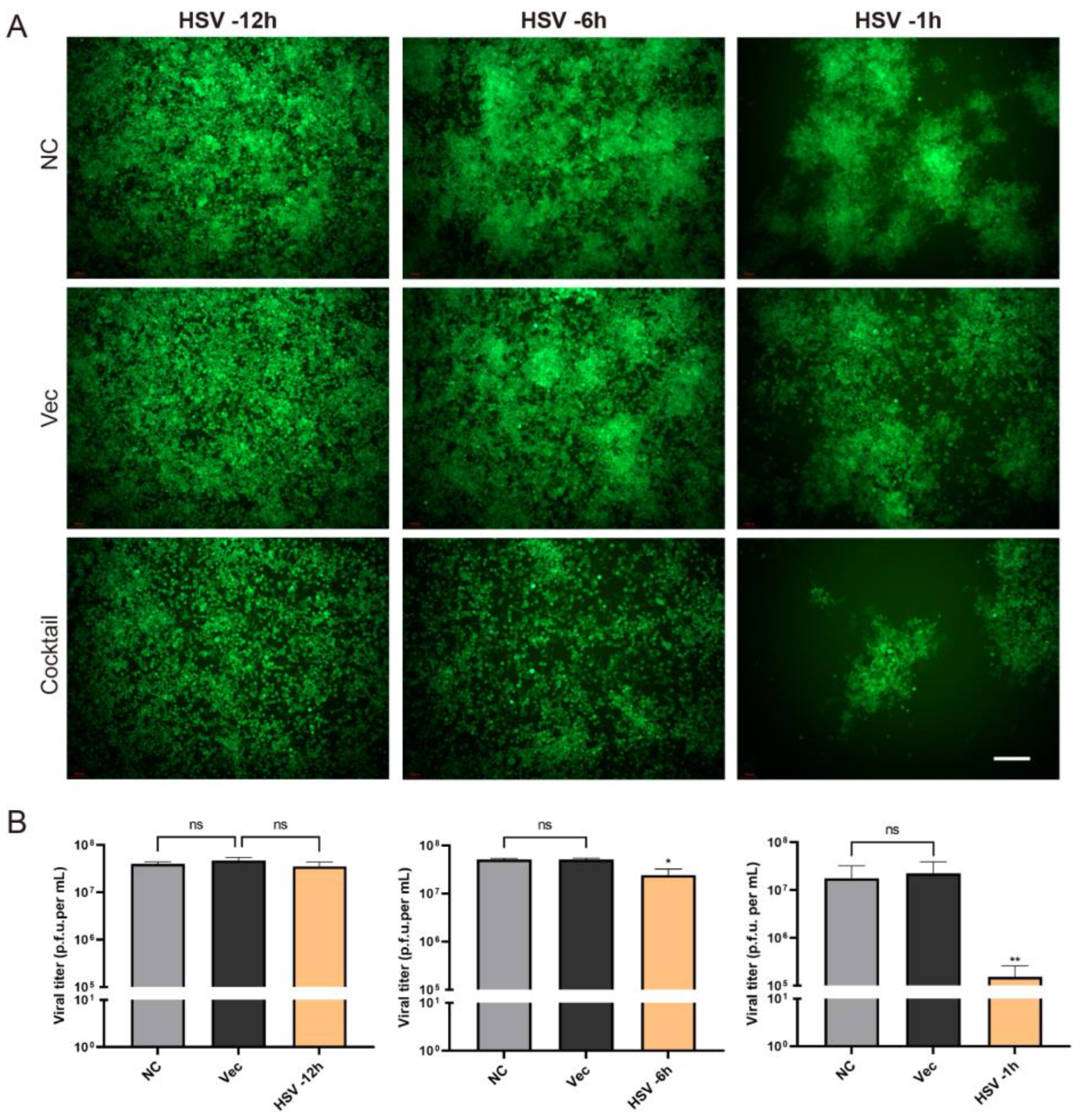
As described above, we tested the efficiency of the CRISPR editing system in preventing HSV infection. What was the therapeutic effect of the editing system? In order to solve this problem, we carried out an experiment in which the cells were first infected with HSV H129-EGFP and then treated the cells with CRISPR editing. H129-EGFP viruses were challenged 12 h, 6 h, and 1 h in advance and then transfected with Cas9/gRNA cocktail plasmids. It was found that the Cas9/gRNA intervention had a certain inhibition effect but was not obvious in the robust proliferation stage of HSV acute infection (−12 h) (
Figure 5). If CRISPR intervened earlier after HSV infection, the CRISPR editing treatment in the −6 h group and −1 h group could significantly reduce HSV replication; the earlier it intervenes, the better.
Subsequently,
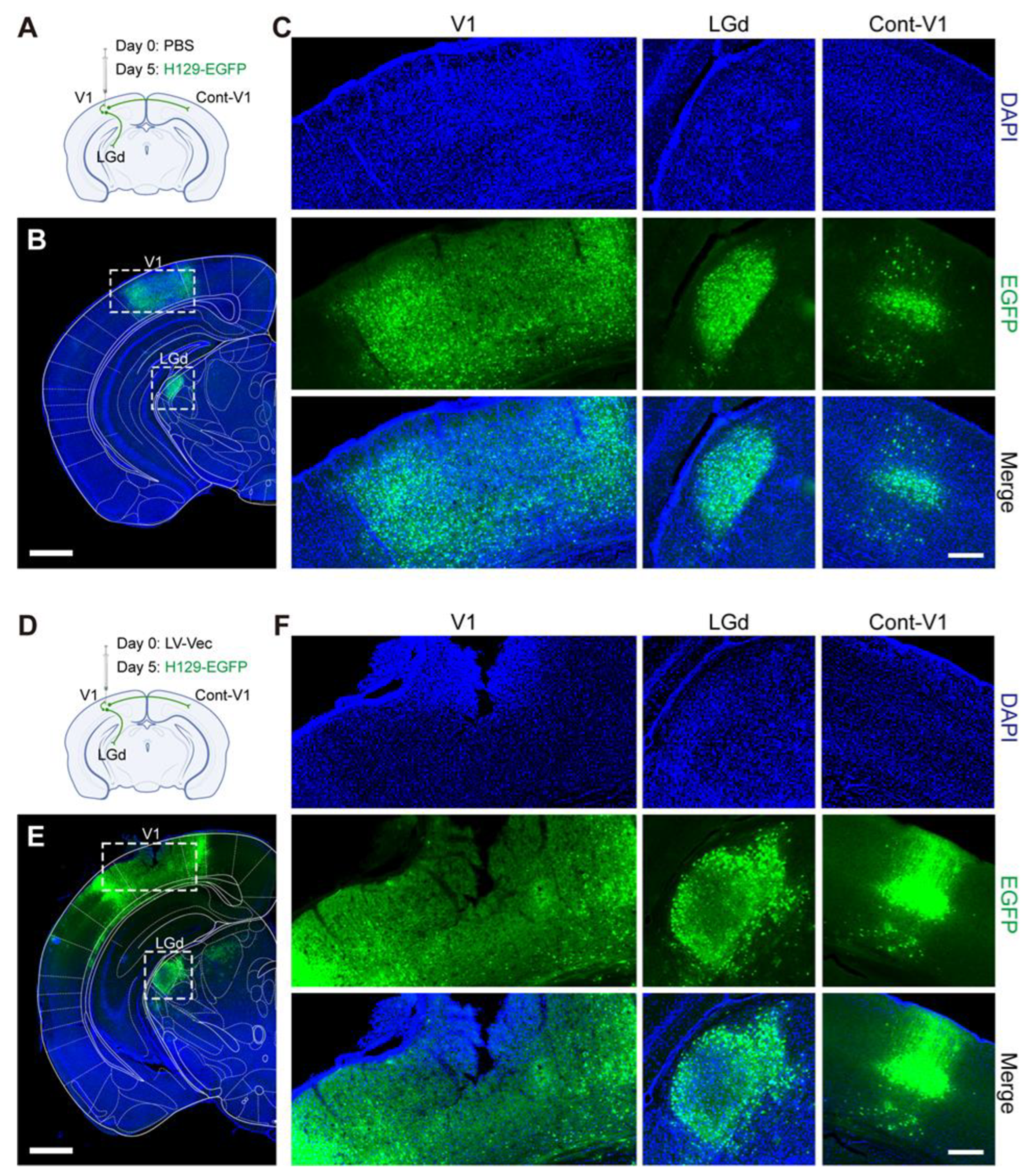
in vivo animal experiments with the produced lentiviruses were carried out. It was found that the Cas9/gRNA (Cocktail) strategy could effectively inhibit virus proliferation and transmission. In the control groups without any intervention (NC,
Figure 6A,B) and the LV-Vec control group (Vec,
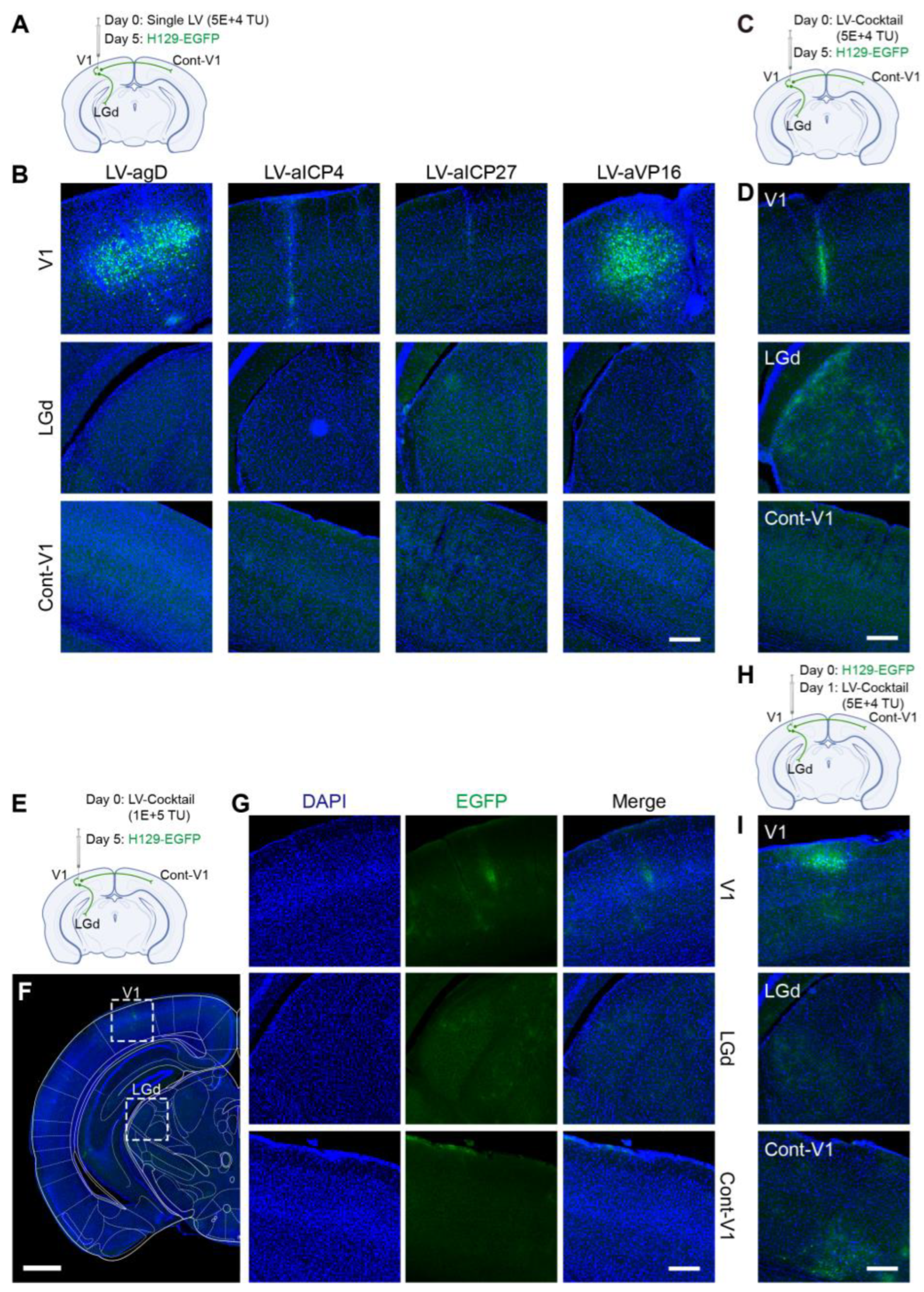
Figure 6D,E), HSV had effectively proliferated for 5 days after H129-EGFP was injected into the V1 brain region of mice, and it spread to LGd and other brain regions along the neural circuit. The infected neurons were labeled by a green fluorescent protein, and the LV-Vec treatment had no inhibitory effect. When the designed lentivirus carrying Cas9/gRNA was administrated alone (
Figure 7A,B), the inhibitory effect on HSV replication (few fluorescent-labeling cells at the injection site) and virus transmission (no fluorescent-labeling cells in the LGd region) were observed, which was better in the LV-aICP4 and LV-aICP27 groups compared with the LV-agD and LV-aVP16 groups. The treatment effect of the Cas9/gRNA combination (Cocktail) group was relatively the best. There were no or only a small number of fluorescent-labeling cells at injection site V1 (
Figure 7D,G). Furthermore, we also performed the therapeutic effect experiment. H129-EGFP was first injected in the V1 brain region of mice, and LV-cocktail was administrated 1-day post-infection; it was found that the cocktail treatment could effectively inhibit HSV replication (
Figure 7H,I), and the effect was better than that of a single lentivirus administration.
In this study, we developed an antiviral strategy comprising coordinated lifecycle elimination against viral replication (CLEAR). This strategy is different from targeting a key viral gene to inhibit viral genome replication. Instead, it starts with analyzing the characteristics of the viral life cycle and selects a multi-pronged approach to block various life cycle stages of the virus. In this way, it can effectively avoid the disability of single-point genome editing once the virus genome mutates. Multi-point genome editing can simultaneously target multiple key gene targets, thereby effectively inhibiting viral genome replication and producing a synergistic antiviral effect. This method is effective for both proliferative viruses and latent infected viruses, as it involves editing and cutting viral genomic materials. As mentioned above, the CLEAR strategy using CRISPR targeting the key HSV genes—
VP16,
ICP27,
ICP4, and
gD—could effectively inhibit HSV replication and proliferation, and the combination of Cas9/gRNAs strategy was more efficient. In order to effectively target latent HSV 1-containing neurons, the administration strategies include a local administration method or LV administration at the axonal terminals of neurons. Lentivirus delivers the CRISPR system to enter the cell body of HSV1 latently infected neurons in order to carry out the gene-cutting function in the nucleus, which can effectively prevent viral genome replication in latent cells and achieve the fundamental elimination of the latent virus. In addition, it is worth noting that there was no significant difference in the effect of agD, aICP4, aVP16, and aICP27 editing on inhibiting HSV replication
in vitro. However, in
in vivo experiments, the effect of the targeted editing of ICP4 or ICP27 alone was better than that of editing gD and VP16 (
Figure 7B). The effectiveness of editing ICP4 or ICP27 in inhibiting HSV replication was consistent with previous reports [
14,
31,
32,
34].
In the present study, we assessed the efficiency of Cas9/gRNA genome editing to inhibit HSV replication at the preventive and therapeutic levels. In order to explore the accessibility of gRNAs to their target sequences and determine the time window for CRISPR gene editing, we designed the experiments involving the “prevention strategy” and “treatment strategy” and compared their effects. The results showed that the CRISPR editing system could inhibit HSV replication more effectively by expressing gRNAs in the cells before infection. In the productive life cycle of HSV viruses, when the viral genome is in a naked DNA state, it can certainly be easily recognized, captured, and cut by the Cas9/gRNA complex. When the viral genome is in the form of chromatin, it will increase the difficulty of editing to a certain extent. On one level, the cocktail of gRNA editing and cutting (multiple targeting effects of CLEAR strategy) could provide enhanced efficacy in preventing viral genome replication. Moreover, multi-target blockades could effectively prevent the occurrence of ineffective single-target editing interventions during viral gene mutations. The determination and optimization of the CRISPR gene editing time window need to be further studied.
In conclusion, the study demonstrated that the CRISPR gene editing strategy had a good inhibitory effect in the experiments of both “prevention strategy” and “treatment strategy”, especially in vivo experiments. Compared with the control groups, Cas9/gRNA treatments, whether administered alone or combined, significantly inhibited HSV proliferation and spread. The demonstrated dose–response relationship and the applicability of the CLEAR strategy across different MOIs further highlight its potential for developing novel anti-HSV therapies.
4. Materials and Methods
4.1. Animals
All husbandry and experimental procedures in this study were approved by the Animal Care and Use Committees at the Shenzhen Institute of Advanced Technology or Innovation Academy for Precision Measurement Science and Technology, Chinese Academy of Sciences (approval No. SIAT-IACUC-200902-NS-WHD-A1438). Adult male C57BL/6 mice were purchased from Hunan SJA Laboratory Animal Company. Adult (8–10 weeks old) C57BL/6 mice (male, n = 27) were used for in vivo experiments. Mice were randomly assigned to groups of predetermined sample size. No mice were excluded from these analyses. All animals were housed in a dedicated housing room with a 12/12 h light/dark cycle, and food and water were available ad libitum. All the experiments with viruses were performed in biosafety level 2 (BSL-2) laboratory and animal facilities.
4.2. Cell and Virus
The wildtype HSV-1 H129 clinical strain was generously provided by Professor Lynn Enquist (Princeton University, Princeton, NJ, USA). Viral stocks were grown on BHK-21 cells (American Type Culture Collection) maintained in Dulbecco’s minimum essential media (DMEM) with 10% fetal bovine serum (FBS). Standard plaque assays were performed by serially diluting virus in DMEM supplemented with 2% FBS (2% DMEM) and overlaying infected cells with medium containing 2% FBS, antibiotics, and 1% agarose. Plaques were identified using neutral red staining and/or fluorescence microscopy where appropriate. Aliquots of viral stocks were stored frozen at −80 °C.
HSV1 H129-EGFP recombinant virus was constructed and maintained in our lab, in which an EGFP expression cassette (containing hUbC promoter, EGFP gene, WPRE and bGH ploy (A) signal) was inserted into the intergenic region between the UL37 and UL38 genes of H129 viral genome. The recombinant H129-EGFP viruses were mass-produced by infecting BHK-21 cells grown in T75 tissue culture flasks. After infected cells showed a prominent cytopathic effect (~2 days), medium containing the viruses was collected, centrifuged to remove cell debris (7000× g for 10 min), the supernatant passed through a 0.22 μm filter, and finally centrifuged at 50,000× g/3 h using Beckman Avanti J-26SXP Ultracentrifuge. Dissolved viruses were aliquoted into 3 μL and stored at −80 °C. The titer of viral stocks was determined using standard plaque assay and titers were expressed as plaque-forming units (PFU) per milliliter. A fresh aliquot of stock virus was thawed and used for each experiment. The titers of HSV1 H129-EGFP stocks used in these studies were ~2 × 109 PFU/mL.
4.3. gRNA and Lentiviral Vector Construction
Lentiviral delivery of both SpCas9 and sgRNA (lenti-CRISPR) for targeted gene editing was constructed. sgRNA was designed based on the target site sequence of selected genes and screened by CRISPR DESIGN system (
http://crispr.mit.edu/ (accessed on 15 October 2021)). sgRNA oligonucleotides sequences were as
Table 1. The empty vector lentiCRISPR v2 vector (addgene, #52961) was digested using BsmBI, and the annealed oligos were cloned into the single guide RNA scaffold. Then, lentiCRISPR-sgRNA virus was produced in HEK293T cells with the packaging plasmids as previously described methods [
42]. In brief, the above lentivirus constructs were co-transfected with three lentivirus packaging vectors (pGAG/POL, pREV and pMD2.G) into 293T cells, and the targeting viruses were obtained by collecting supernatants after 2 to 3 days. The titers of produced five lentiCRISPR-sgRNA (LV-vec, LV-agD-1, LV-aVP16-2, LV-aICP4-2, LV-aICP27-2) viral stocks were all ~1 × 10
8 PFU/mL.
4.4. Infection and Transduction of Cells
BHK-21 cells and 293T cells were seeded in 6-well plates at density of 1 × 106 cells per dish 24 h before 2 μg lentiCRISPR-sgRNA transfection. Six hours after transfection, the medium was refreshed with 2 mL DMEM supplemented with 2% FBS (Gibco, Thermo Fisher, Sydney, Australia) and 1% penicillin/streptomycin (BasalMedia, Shanghai, China), then BHK-21 cells were infected with H129-EGFP at MOI = 0.01 after 18 h. The supernatants were harvested 48 h after infection.
Next, we selected agD-1, aICP4-2, aICP27-2 and aVP16-2 sgRNA in combination of 0.5 μg, 1 μg and 2 μg to transfection BHK-21 cells. Six hours after transfection, the medium was refreshed with 2% DMEM, then BHK-21 cells were infected with various MOI (0.01 or 0.001) of H129-EGFP after 18 h. The supernatants were harvested 24 h and 48 h after infection.
For treatment strategy experiment, BHK-21 cells were infected with MOI at 0.01 of H129-EGFP. After 1 h, 6 h and 12 h infection, BHK-21 cells were transfected with four sgRNAs (agD-1, aICP4-2, aICP27-2 and aVP16-2) in combination of 1 μg. Six hours after transfection, the medium was refreshed with 2% DMEM, then the supernatants were harvested 42 h.
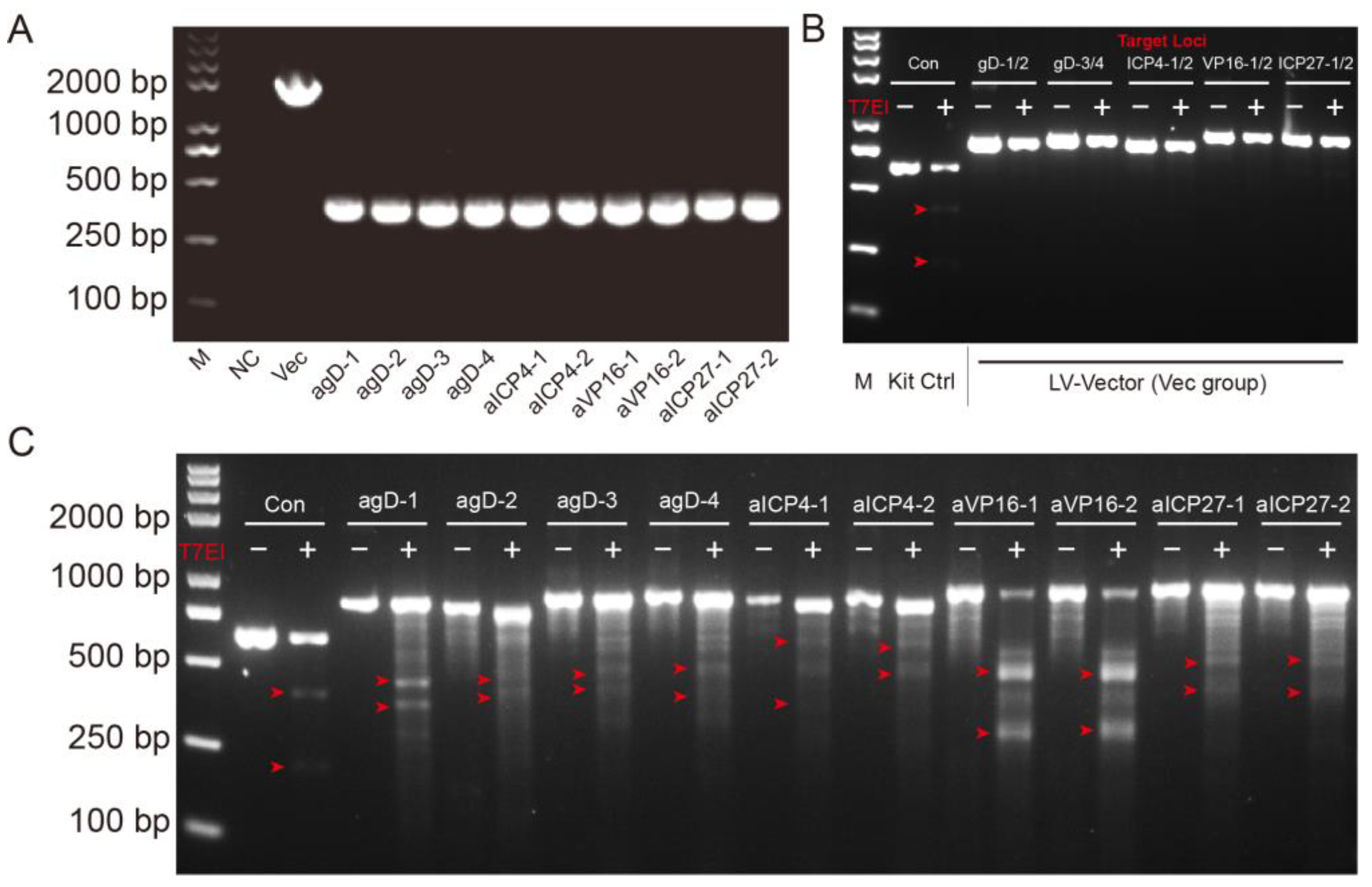
4.5. T7 Endonuclease Mutation Detection Assay
The EnGen Mutation Detection Kit (New England Biolabs, Ipswich, MA, USA, M0302) was used for detection of on-target genome editing events. BHK-21 cells were transfected with 2 μg Cas9-gRNA plasmids. Then, 24 h later, the cells were infected with H129-EGFP at MOI = 0.01. The cells were harvested 48 h after infection and lysed with 100 μL QuickExtract™ (Cambio/Epicentre, QE09050) per well and mix by pipetting. A 1 μL volume of lysates was used for the amplification of the target site. Put on PCR plate with 2 μL NEB Buffer and add ddH2O to a final volume of 19 μL. Run Cross-Hybridisation PCR program. After products had cross-hybridized add 1 μL T7 endonuclease I in each sample. Incubate at 37 °C for 15 min. Stop T7 endonuclease reaction straight after the end of the incubation period by adding 2 μL 0.25 M EDTA. Add loading buffer to all samples and load them on the 1.5% agarose gel. Run at 100 V and check the gel regularly under the UV, taking images at different time points.
4.6. Viral Titer Assay
H129-EGFP titer was performed in triplicate for each biological sample. BHK-21 (2 × 105) cell was seeded in 6-well plate, then infected BHK-21 with various dilution of virus supernatant on the following day. After 1 h of infection, 0.5% agarose (Biowest agarose, Baygene, Shanghai, China) in DMEM supplemented with 2% FBS (Gibco, Thermo Fisher, Australia) and 1% penicillin/streptomycin (BasalMedia, Shanghai, China, S110JV) was added. The plaques were counted after 48 h incubation in 35 °C, 5% CO2 Cell Culture Chamber.
4.7. Stereotactic Surgery
All procedures on animals were performed in Biosafety level 2 (BSL-2) animal facilities [
43]. Animals were anesthetized with pentobarbital sodium by intraperitoneal injection (80 mg/kg, i.p.), and placed in a stereotaxic apparatus (RWD, Shenzhen, China, 68030). During surgery and virus injection, all animals were kept anesthetized with isoflurane (1–1.5%). The skull above the targeted areas was thinned with a dental drill and removed carefully. Injections were conducted with a syringe pump (Stoelting, Wood Dale, IL, USA, 53311) connected to a glass micropipette with a tip diameter of 10–15 mm. The glass micropipette was held for an extra 10 min after the completion of the injection and then slowly retreated. After the surgery, the incisions were stitched and lincomycin hydrochloride and lidocaine hydrochloride gel was applied to prevent inflammation and alleviate pain for the animals. 5 × 10
4 TU lentiCRISPR-gRNA viruses and 500 PFU H129-EGFP viruses was injected into the adult male C57BL/6 mice (n = 3) at 0 d.p.i. and 5 d.p.i. respectively, with the following coordinates: V1 (AP, −2.80 mm; ML, −2.40 mm; and DV, −0.90 mm).
4.8. Slice Preparation and Confocal Imaging
Five days post injection with HSV H129-EGFP, the mice were anesthetized with pentobarbital sodium (100 mg/kg body weight, i.p.), and perfused transcardially with PBS (5 min), followed by ice-cold 4% paraformaldehyde (PFA, 158127 MSDS, sigma) dissolved in PBS (5 min). The brain tissues were carefully removed and post-fixed in PBS containing 4% PFA at 4 °C overnight, and then equilibrated in PBS containing 25% sucrose at 4 °C for 72 h. The 40-μm-thick coronal slices of the whole brain were obtained using the cryostat microtome and stored at −20 °C.
For all samples, every sixth section of the brain slices were selected, stained with DAPI, washed with PBS, mounted with 70% glycerol (in PBS) and sealed with nail polish. All of the images were captured with the Olympus VS120 virtual microscopy slide scanning system (Olympus, Tokyo, Japan).
4.9. Data Analysis
SPSS (version 13.0) and Origin 9.0 were used for data analysis (student’s t-tests) and statistical graphs, respectively. All data were presented as means ± SEM. Statistical significance was set as *, p < 0.05, **, p < 0.01 and ***, p < 0.001.


 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}