
Impact of Plasmodium relictum Infection on the Colonization Resistance of Bird Gut Microbiota: A Preliminary Study

 , , , , , ,
, , , , , ,  ,
,  and
and
Abstract
:1. Introduction
2. Methods
2.1. Data Source
2.2. Microbiota Diversity, Composition, and Abundance Analyses
2.3. Bacterial Co-Occurrence Networks
2.4. Comparative Network Analysis and Robustness
3. Results
3.1. Dynamics of Plasmodium relictum Infection
3.2. The Impact of Plasmodium relictum Infection on Host Microbiota Diversity and Composition
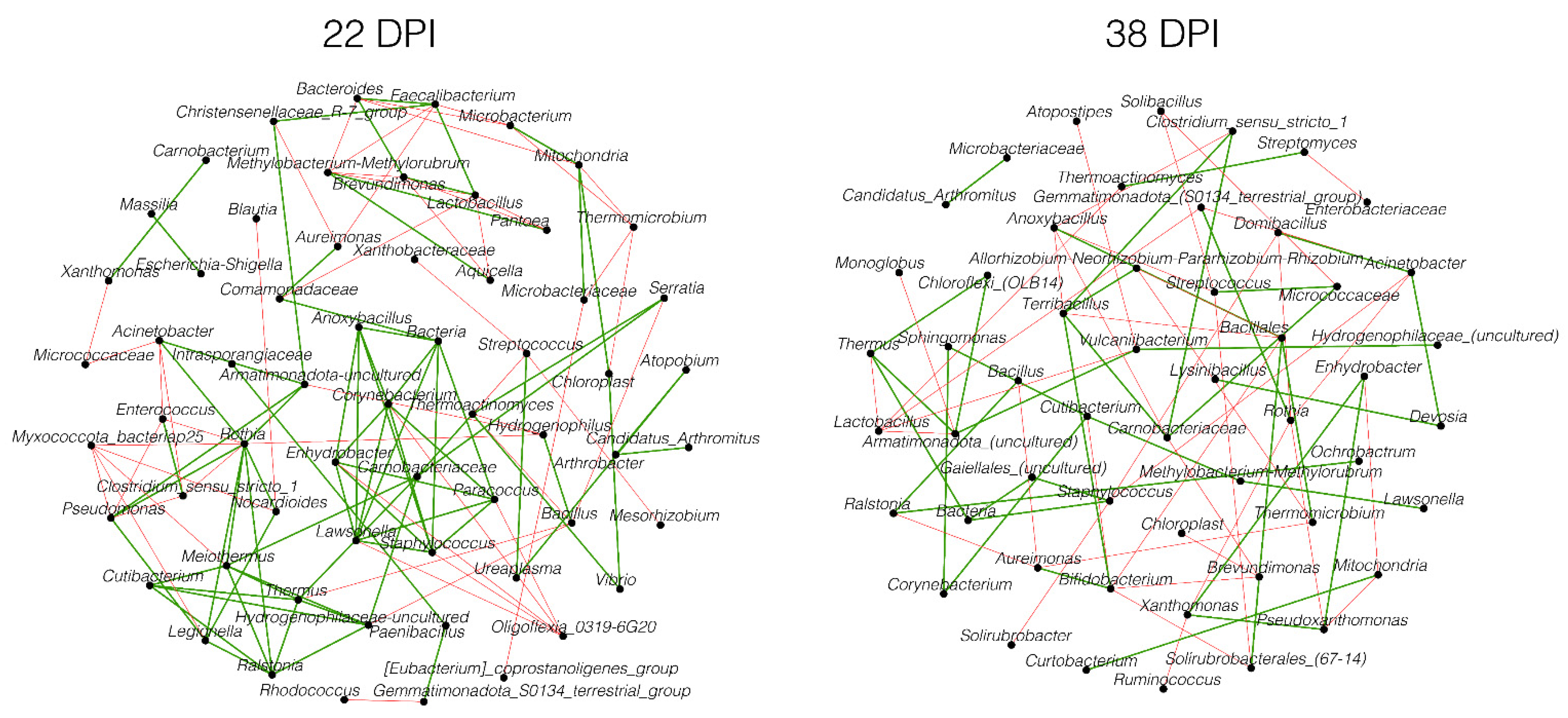
3.3. Changes in the Microbiota Assembly Due to Plasmodium relictum Infection
3.4. Network Robustness
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Valkiūnas, G. Avian Malaria Parasites and Other Haemosporidia; CRC Press: Boca Raton, FL, USA, 2005; ISBN 978-0-415-30097-1. [Google Scholar]
- Ducarmon, Q.R.; Zwittink, R.D.; Hornung, B.V.H.; van Schaik, W.; Young, V.B.; Kuijper, E.J. Gut Microbiota and Colonization Resistance against Bacterial Enteric Infection. Microbiol. Mol. Biol. Rev. 2019, 83, e00007-19. [Google Scholar] [CrossRef] [PubMed]
- Karita, Y.; Limmer, D.T.; Hallatschek, O. Scale-Dependent Tipping Points of Bacterial Colonization Resistance. Proc. Natl. Acad. Sci. USA 2022, 119, e2115496119. [Google Scholar] [CrossRef] [PubMed]
- Mullineaux-Sanders, C.; Suez, J.; Elinav, E.; Frankel, G. Sieving through Gut Models of Colonization Resistance. Nat. Microbiol. 2018, 3, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Stacy, A.; Andrade-Oliveira, V.; McCulloch, J.A.; Hild, B.; Oh, J.H.; Perez-Chaparro, P.J.; Sim, C.K.; Lim, A.I.; Link, V.M.; Enamorado, M.; et al. Infection Trains the Host for Microbiota-Enhanced Resistance to Pathogens. Cell 2021, 184, 615–627.e17. [Google Scholar] [CrossRef]
- Martínez-de la Puente, J.; Santiago-Alarcon, D.; Palinauskas, V.; Bensch, S. Plasmodium Relictum. Trends Parasitol. 2021, 37, 355–356. [Google Scholar] [CrossRef]
- Chen, Y.; Li, H. Avian Leukosis Virus Subgroup J Infection Influences the Gut Microbiota Composition in Huiyang Bearded Chickens. Lett. Appl. Microbiol. 2022, 74, 344–353. [Google Scholar] [CrossRef]
- Sun, F.; Chen, J.; Liu, K.; Tang, M.; Yang, Y. The Avian Gut Microbiota: Diversity, Influencing Factors, and Future Directions. Front. Microbiol. 2022, 13, 934272. [Google Scholar] [CrossRef]
- Waite, D.W.; Taylor, M.W. Exploring the Avian Gut Microbiota: Current Trends and Future Directions. Front. Microbiol. 2015, 6, 673. [Google Scholar] [CrossRef]
- Palinauskas, V.; Mateos-Hernandez, L.; Wu-Chuang, A.; de la Fuente, J.; Aželytė, J.; Obregon, D.; Cabezas-Cruz, A. Exploring the Ecological Implications of Microbiota Diversity in Birds: Natural Barriers against Avian Malaria. Front. Immunol. 2022, 13, 807682. [Google Scholar] [CrossRef]
- Cisek, A.A.; Binek, M. Chicken Intestinal Microbiota Function with a Special Emphasis on the Role of Probiotic Bacteria. Pol. J. Vet. Sci. 2014, 17, 385–394. [Google Scholar] [CrossRef]
- Hird, S.M.; Sánchez, C.; Carstens, B.C.; Brumfield, R.T. Comparative Gut Microbiota of 59 Neotropical Bird Species. Front. Microbiol. 2015, 6, 1403. [Google Scholar] [CrossRef] [PubMed]
- Grond, K.; Santo Domingo, J.W.; Lanctot, R.B.; Jumpponen, A.; Bentzen, R.L.; Boldenow, M.L.; Brown, S.C.; Casler, B.; Cunningham, J.A.; Doll, A.C.; et al. Composition and Drivers of Gut Microbial Communities in Arctic-Breeding Shorebirds. Front. Microbiol. 2019, 10, 2258. [Google Scholar] [CrossRef] [PubMed]
- Mammeri, M.; Obregón, D.A.; Chevillot, A.; Polack, B.; Julien, C.; Pollet, T.; Cabezas-Cruz, A.; Adjou, K.T. Cryptosporidium Parvum Infection Depletes Butyrate Producer Bacteria in Goat Kid Microbiome. Front. Microbiol. 2020, 11, 548737. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, T.; Miyauchi, E.; Nakamura, S.; Hirai, M.; Suzue, K.; Imai, T.; Nomura, T.; Handa, T.; Okada, H.; Shimokawa, C.; et al. Plasmodium Berghei ANKA Causes Intestinal Malaria Associated with Dysbiosis. Sci. Rep. 2015, 5, 15699. [Google Scholar] [CrossRef] [PubMed]
- Stough, J.M.A.; Dearth, S.P.; Denny, J.E.; LeCleir, G.R.; Schmidt, N.W.; Campagna, S.R.; Wilhelm, S.W. Functional Characteristics of the Gut Microbiome in C57BL/6 Mice Differentially Susceptible to Plasmodium yoelii. Front. Microbiol. 2016, 7, 1520. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, B.; Portugal, S.; Tran, T.M.; Gozzelino, R.; Ramos, S.; Gomes, J.; Regalado, A.; Cowan, P.J.; d’Apice, A.J.F.; Chong, A.S.; et al. Gut Microbiota Elicits a Protective Immune Response against Malaria Transmission. Cell 2014, 159, 1277–1289. [Google Scholar] [CrossRef]
- Mooney, J.P.; Lokken, K.L.; Byndloss, M.X.; George, M.D.; Velazquez, E.M.; Faber, F.; Butler, B.P.; Walker, G.T.; Ali, M.M.; Potts, R.; et al. Inflammation-Associated Alterations to the Intestinal Microbiota Reduce Colonization Resistance against Non-Typhoidal Salmonella during Concurrent Malaria Parasite Infection. Sci. Rep. 2015, 5, 14603. [Google Scholar] [CrossRef]
- Navine, A.K.; Paxton, K.L.; Paxton, E.H.; Hart, P.J.; Foster, J.T.; McInerney, N.; Fleischer, R.C.; Videvall, E. Microbiomes Associated with Avian Malaria Survival Differ between Susceptible Hawaiian Honeycreepers and Sympatric Malaria-resistant Introduced Birds. Mol. Ecol. 2023, 32, 6659–6670. [Google Scholar] [CrossRef]
- Videvall, E.; Marzal, A.; Magallanes, S.; Fleischer, R.C.; Espinoza, K.; García-Longoria, L. The Uropygial Gland Microbiome of House Sparrows with Malaria Infection. J. Avian Biol. 2021, 52, jav.02686. [Google Scholar] [CrossRef]
- Rohrer, S.D.; Robertson, B.Q.; Chubiz, L.M.; Parker, P.G. Gut Microbiome Composition Associated with Plasmodium Infection in the Eurasian Tree Sparrow. J. Avian Biol. 2023, 2023, e03027. [Google Scholar] [CrossRef]
- Aželytė, J.; Wu-Chuang, A.; Maitre, A.; Žiegytė, R.; Mateos-Hernández, L.; Obregón, D.; Palinauskas, V.; Cabezas-Cruz, A. Avian Malaria Parasites Modulate Gut Microbiome Assembly in Canaries. Microorganisms 2023, 11, 563. [Google Scholar] [CrossRef] [PubMed]
- Faust, K.; Raes, J. Microbial Interactions: From Networks to Models. Nat. Rev. Microbiol. 2012, 10, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Röttjers, L.; Faust, K. From Hairballs to Hypotheses–Biological Insights from Microbial Networks. FEMS Microbiol. Rev. 2018, 42, 761–780. [Google Scholar] [CrossRef] [PubMed]
- Shade, A.; Peter, H.; Allison, S.D.; Baho, D.L.; Berga, M.; Bürgmann, H.; Huber, D.H.; Langenheder, S.; Lennon, J.T.; Martiny, J.B.H.; et al. Fundamentals of Microbial Community Resistance and Resilience. Front. Microbio 2012, 3, 417. [Google Scholar] [CrossRef] [PubMed]
- Maitre, A.; Wu-Chuang, A.; Mateos-Hernández, L.; Piloto-Sardiñas, E.; Foucault-Simonin, A.; Cicculli, V.; Moutailler, S.; Paoli, J.; Falchi, A.; Obregón, D.; et al. Rickettsial Pathogens Drive Microbiota Assembly in Hyalomma Marginatum and Rhipicephalus Bursa Ticks. Mol. Ecol. 2023, 32, 4660–4676. [Google Scholar] [CrossRef] [PubMed]
- Maitre, A.; Wu-Chuang, A.; Mateos-Hernández, L.; Foucault-Simonin, A.; Moutailler, S.; Paoli, J.-C.; Falchi, A.; Díaz-Sánchez, A.A.; Banović, P.; Obregón, D.; et al. Rickettsia Helvetica Infection Is Associated with Microbiome Modulation in Ixodes Ricinus Collected from Humans in Serbia. Sci. Rep. 2022, 12, 11464. [Google Scholar] [CrossRef]
- Mateos-Hernández, L.; Obregón, D.; Wu-Chuang, A.; Maye, J.; Bornères, J.; Versillé, N.; de la Fuente, J.; Díaz-Sánchez, S.; Bermúdez-Humarán, L.G.; Torres-Maravilla, E.; et al. Anti-Microbiota Vaccines Modulate the Tick Microbiome in a Taxon-Specific Manner. Front. Immunol. 2021, 12, 704621. [Google Scholar] [CrossRef]
- Estrada-Peña, A.; Cabezas-Cruz, A.; Obregón, D. Resistance of Tick Gut Microbiome to Anti-Tick Vaccines, Pathogen Infection and Antimicrobial Peptides. Pathogens 2020, 9, 309. [Google Scholar] [CrossRef]
- Aželytė, J.; Wu-Chuang, A.; Žiegytė, R.; Platonova, E.; Mateos-Hernandez, L.; Maye, J.; Obregon, D.; Palinauskas, V.; Cabezas-Cruz, A. Anti-Microbiota Vaccine Reduces Avian Malaria Infection within Mosquito Vectors. Front. Immunol. 2022, 13, 841835. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Yarza, P.; Yilmaz, P.; Pruesse, E.; Glöckner, F.O.; Ludwig, W.; Schleifer, K.-H.; Whitman, W.B.; Euzéby, J.; Amann, R.; Rosselló-Móra, R. Uniting the Classification of Cultured and Uncultured Bacteria and Archaea Using 16S rRNA Gene Sequences. Nat. Rev. Microbiol. 2014, 12, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Faith, D.P. Conservation Evaluation and Phylogenetic Diversity. Biol. Conserv. 1992, 61, 1–10. [Google Scholar] [CrossRef]
- Bray, J.R.; Curtis, J.T. An Ordination of the Upland Forest Communities of Southern Wisconsin. Ecol. Monogr. 1957, 27, 325–349. [Google Scholar] [CrossRef]
- Pielou, E.C. The Measurement of Diversity in Different Types of Biological Collections. J. Theor. Biol. 1966, 13, 131–144. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021; Available online: https://www.r-project.org/ (accessed on 10 May 2023).
- Gloor, G.B.; Macklaim, J.M.; Fernandes, A.D. Displaying Variation in Large Datasets: Plotting a Visual Summary of Effect Sizes. J. Comput. Graph. Stat. 2016, 25, 971–979. [Google Scholar] [CrossRef]
- Aitchison, J. The Statistical Analysis of Compositional Data; Springer: Dordrecht, The Netherlands, 1986; ISBN 978-94-010-8324-9. [Google Scholar]
- Friedman, J.; Alm, E.J. Inferring Correlation Networks from Genomic Survey Data. PLoS Comput. Biol. 2012, 8, e1002687. [Google Scholar] [CrossRef]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An Open Source Software for Exploring and Manipulating Networks. ICWSM 2009, 3, 361–362. [Google Scholar] [CrossRef]
- Peschel, S.; Müller, C.L.; von Mutius, E.; Boulesteix, A.-L.; Depner, M. NetCoMi: Network Construction and Comparison for Microbiome Data in R. Brief. Bioinform. 2021, 22, bbaa290. [Google Scholar] [CrossRef]
- Röttjers, L.; Vandeputte, D.; Raes, J.; Faust, K. Null-Model-Based Network Comparison Reveals Core Associations. ISME Commun. 2021, 1, 36. [Google Scholar] [CrossRef]
- Anaconda Software Distribution. Anaconda Documentation. Anaconda Inc. 2023. Available online: https://docs.anaconda.com/ (accessed on 10 June 2023).
- Lhomme, S. Analyse Spatiale de La Structure Des Réseaux Techniques Dans Un Contexte de Risques. Cybergeo 2015. [Google Scholar] [CrossRef]
- Csárdi, G.; Nepusz, T. The Igraph Software Package for Complex Network Research. InterJournal Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Csárdi, G.; Nepusz, T.; Müller, K.; Horvát, S.; Traag, V.; Zanini, F.; Noom, D. Igraph for R: R Interface of the Igraph Library for Graph Theory and Network Analysis 2023. Zenodo. Available online: https://CRAN.R-project.org/package=igraph (accessed on 30 April 2023).
- Freitas, S.; Yang, D.; Kumar, S.; Tong, H.; Chau, D.H. Evaluating Graph Vulnerability and Robustness Using TIGER. arXiv 2020. [Google Scholar] [CrossRef]
- Farinella, D.N.; Kaur, S.; Tran, V.; Cabrera-Mora, M.; Joyner, C.J.; Lapp, S.A.; Pakala, S.B.; Nural, M.V.; DeBarry, J.D.; Kissinger, J.C.; et al. Malaria Disrupts the Rhesus Macaque Gut Microbiome. Front. Cell. Infect. Microbiol. 2023, 12, 1058926. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.J.; Schloss, P.; Ramos, Y.; Raffa, K.; Handelsman, J. Robustness of the Bacterial Community in the Cabbage White Butterfly Larval Midgut. Microb. Ecol. 2010, 59, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Shea, K. Community Ecology Theory as a Framework for Biological Invasions. Trends Ecol. Evol. 2002, 17, 170–176. [Google Scholar] [CrossRef]
- Spragge, F.; Bakkeren, E.; Jahn, M.T.; Araujo, E.B.N.; Pearson, C.F.; Wang, X.; Pankhurst, L.; Cunrath, O.; Foster, K.R. Microbiome Diversity Protects against Pathogens by Nutrient Blocking. Science 2023, 382, eadj3502. [Google Scholar] [CrossRef] [PubMed]
- Schokker, D.; Veninga, G.; Vastenhouw, S.A.; Bossers, A.; De Bree, F.M.; Kaal-Lansbergen, L.M.T.E.; Rebel, J.M.J.; Smits, M.A. Early Life Microbial Colonization of the Gut and Intestinal Development Differ between Genetically Divergent Broiler Lines. BMC Genom. 2015, 16, 418. [Google Scholar] [CrossRef] [PubMed]
- Boisseau, M.; Dhorne-Pollet, S.; Bars-Cortina, D.; Courtot, É.; Serreau, D.; Annonay, G.; Lluch, J.; Gesbert, A.; Reigner, F.; Sallé, G.; et al. Species Interactions, Stability, and Resilience of the Gut Microbiota—Helminth Assemblage in Horses. iScience 2023, 26, 106044. [Google Scholar] [CrossRef]
- Britton, R.A.; Young, V.B. Interaction between the Intestinal Microbiota and Host in Clostridium Difficile Colonization Resistance. Trends Microbiol. 2012, 20, 313–319. [Google Scholar] [CrossRef]
- Kamdar, K.; Khakpour, S.; Chen, J.; Leone, V.; Brulc, J.; Mangatu, T.; Antonopoulos, D.A.; Chang, E.B.; Kahn, S.A.; Kirschner, B.S.; et al. Genetic and Metabolic Signals during Acute Enteric Bacterial Infection Alter the Microbiota and Drive Progression to Chronic Inflammatory Disease. Cell Host Microbe 2016, 19, 21–31. [Google Scholar] [CrossRef]
- King, K.C.; Brockhurst, M.A.; Vasieva, O.; Paterson, S.; Betts, A.; Ford, S.A.; Frost, C.L.; Horsburgh, M.J.; Haldenby, S.; Hurst, G.D. Rapid Evolution of Microbe-Mediated Protection against Pathogens in a Worm Host. ISME J. 2016, 10, 1915–1924. [Google Scholar] [CrossRef] [PubMed]
- Wu-Chuang, A.; Bates, K.A.; Obregon, D.; Estrada-Peña, A.; King, K.C.; Cabezas-Cruz, A. Rapid Evolution of a Novel Protective Symbiont into Keystone Taxon in Caenorhabditis Elegans Microbiota. Sci. Rep. 2022, 12, 14045. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Network Features | Uninfected | P. relictum-Infected | ||
|---|---|---|---|---|
| 22 DPI | 38 DPI | 22 DPI | 38 DPI | |
| Nodes | 109 | 82 | 109 | 139 |
| Edges | 2943 | 3235 | 1201 | 904 |
| Positive | 1482 (50.36%) | 1558 (48.16%) | 624 (51.96%) | 461 (51%) |
| Negative | 1461 (49.64%) | 1677 (51.84%) | 577 (48.04%) | 443 (49%) |
| Network diameter | 3 | 2 | 4 | 5 |
| Average degree | 54 | 78.902 | 22.037 | 13.007 |
| Weighted degree | 0.638 | −2.286 | 0.877 | 0.257 |
| Average path length | 1.509 | 1.026 | 2.238 | 2.673 |
| Modularity | 19.062 | −20.16 | 9.353 | 17.473 |
| Number of modules | 3 | 2 | 3 | 7 |
| Average clustering coefficient | 0.759 | 0.978 | 0.667 | 0.539 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aželytė, J.; Maitre, A.; Abuin-Denis, L.; Piloto-Sardiñas, E.; Wu-Chuang, A.; Žiegytė, R.; Mateos-Hernández, L.; Obregón, D.; Cabezas-Cruz, A.; Palinauskas, V. Impact of Plasmodium relictum Infection on the Colonization Resistance of Bird Gut Microbiota: A Preliminary Study. Pathogens 2024, 13, 91. https://doi.org/10.3390/pathogens13010091
Aželytė J, Maitre A, Abuin-Denis L, Piloto-Sardiñas E, Wu-Chuang A, Žiegytė R, Mateos-Hernández L, Obregón D, Cabezas-Cruz A, Palinauskas V. Impact of Plasmodium relictum Infection on the Colonization Resistance of Bird Gut Microbiota: A Preliminary Study. Pathogens. 2024; 13(1):91. https://doi.org/10.3390/pathogens13010091
Chicago/Turabian StyleAželytė, Justė, Apolline Maitre, Lianet Abuin-Denis, Elianne Piloto-Sardiñas, Alejandra Wu-Chuang, Rita Žiegytė, Lourdes Mateos-Hernández, Dasiel Obregón, Alejandro Cabezas-Cruz, and Vaidas Palinauskas. 2024. "Impact of Plasmodium relictum Infection on the Colonization Resistance of Bird Gut Microbiota: A Preliminary Study" Pathogens 13, no. 1: 91. https://doi.org/10.3390/pathogens13010091
APA StyleAželytė, J., Maitre, A., Abuin-Denis, L., Piloto-Sardiñas, E., Wu-Chuang, A., Žiegytė, R., Mateos-Hernández, L., Obregón, D., Cabezas-Cruz, A., & Palinauskas, V. (2024). Impact of Plasmodium relictum Infection on the Colonization Resistance of Bird Gut Microbiota: A Preliminary Study. Pathogens, 13(1), 91. https://doi.org/10.3390/pathogens13010091