The Role of the Mammalian Prion Protein in the Control of Sleep


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Sleep Dysfunction during Prion Disease Pathogenesis
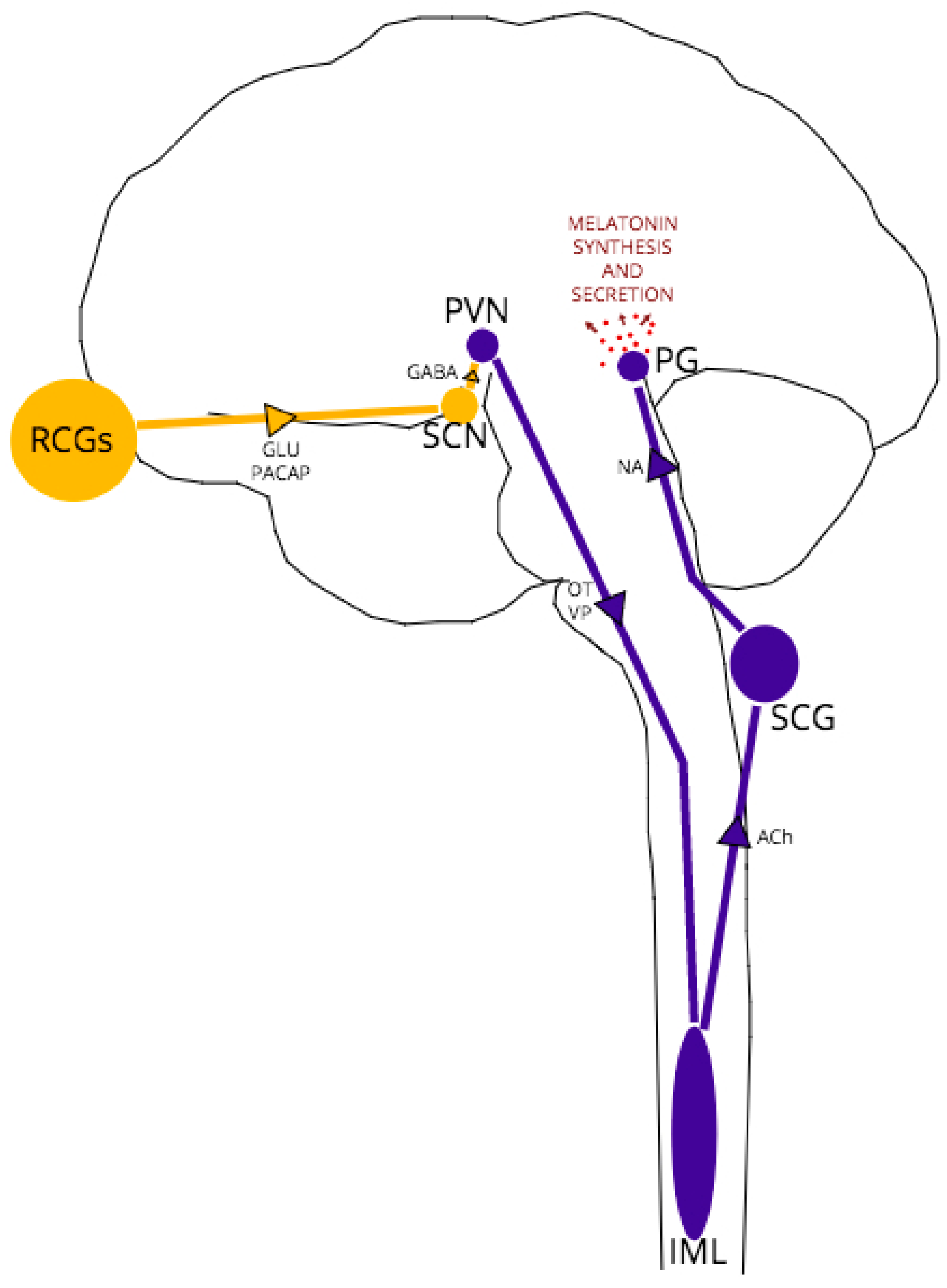
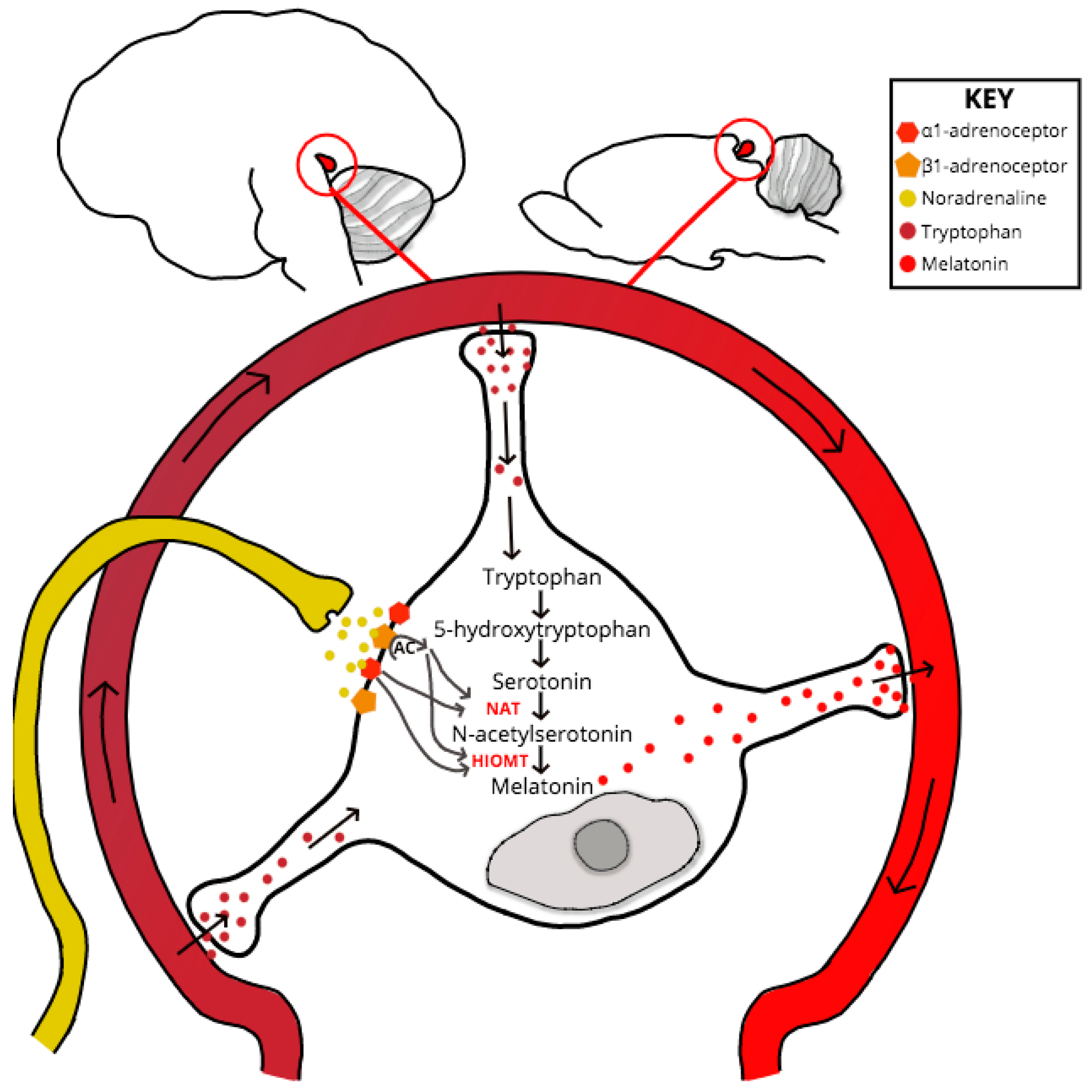
3. Circadian Rhythm, Homeostatic Sleep Pressure and Melatonin
4. A Role for PrPC in the Regulation of Melatonin Synthesis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sparkes, R.S.; Simon, M.; Cohn, V.H.; Fournier, R.E.; Lem, J.; Klisak, I.; Heinzmann, C.; Blatt, C.; Lucero, M.; Mohandas, T.; et al. Assignment of the human and mouse prion protein genes to homologous chromosomes. Proc. Natl. Acad. Sci. USA 1986, 83, 7358–7362. [Google Scholar] [CrossRef] [PubMed]
- Colby, D.W.; Prusiner, S.B. Prions. Cold Spring Harb. Perspect. Biol. 2011, 3, a006833. [Google Scholar] [CrossRef] [PubMed]
- Wulf, M.A.; Senatore, A.; Aguzzi, A. The biological function of the cellular prion protein: An update. BMC Biol. 2017, 15, 34. [Google Scholar] [CrossRef] [PubMed]
- Castle, A.R.; Gill, A.C. Physiological functions of the cellular prion protein. Front. Mol. Biosci. 2017, 4, 19. [Google Scholar] [CrossRef] [PubMed]
- Peralta, O.A.; Eyestone, W.H. Quantitative and qualitative analysis of cellular prion protein (prp(c)) expression in bovine somatic tissues. Prion 2009, 3, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Bueler, H.; Fischer, M.; Lang, Y.; Bluethmann, H.; Lipp, H.P.; DeArmond, S.J.; Prusiner, S.B.; Aguet, M.; Weissmann, C. Normal development and behaviour of mice lacking the neuronal cell-surface prp protein. Nature 1992, 356, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Bakkebo, M.K.; Mouillet-Richard, S.; Espenes, A.; Goldmann, W.; Tatzelt, J.; Tranulis, M.A. The cellular prion protein: A player in immunological quiescence. Front. Immunol. 2015, 6, 450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bremer, J.; Baumann, F.; Tiberi, C.; Wessig, C.; Fischer, H.; Schwarz, P.; Steele, A.D.; Toyka, K.V.; Nave, K.A.; Weis, J.; et al. Axonal prion protein is required for peripheral myelin maintenance. Nat. Neurosci. 2010, 13, U310–U319. [Google Scholar] [CrossRef] [PubMed]
- Miele, G.; Jeffrey, M.; Turnbull, D.; Manson, J.; Clinton, M. Ablation of cellular prion protein expression affects mitochondrial numbers and morphology. Biochem. Biophys. Res. Commun. 2002, 291, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Tobler, I.; Gaus, S.E.; Deboer, T.; Achermann, P.; Fischer, M.; Rulicke, T.; Moser, M.; Oesch, B.; McBride, P.A.; Manson, J.C. Altered circadian activity rhythms and sleep in mice devoid of prion protein. Nature 1996, 380, 639–642. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [PubMed]
- Annus, A.; Csati, A.; Vecsei, L. Prion diseases: New considerations. Clin. Neurol. Neurosurg. 2016, 150, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Brandner, S.; Jaunmuktane, Z. Prion disease: Experimental models and reality. Acta Neuropathol. 2017, 133, 197–222. [Google Scholar] [CrossRef] [PubMed]
- Fraser, J.R. What is the basis of transmissible spongiform encephalopathy induced neurodegeneration and can it be repaired? Neuropathol. Appl. Neurobiol. 2002, 28, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mays, C.E.; Kim, C.; Haldiman, T.; van der Merwe, J.; Lau, A.; Yang, J.; Grams, J.; Di Bari, M.A.; Nonno, R.; Telling, G.C.; et al. Prion disease tempo determined by host-dependent substrate reduction. J. Clin. Investig. 2014, 124, 847–858. [Google Scholar] [CrossRef] [PubMed]
- Llorens, F.; Ansoleaga, B.; Garcia-Esparcia, P.; Zafar, S.; Grau-Rivera, O.; Lopez-Gonzalez, I.; Blanco, R.; Carmona, M.; Yague, J.; Nos, C.; et al. Prp mrna and protein expression in brain and prp(c) in csf in creutzfeldt-jakob disease mm1 and vv2. Prion 2013, 7, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Llorens, F.; Zarranz, J.J.; Fischer, A.; Zerr, I.; Ferrer, I. Fatal familial insomnia: Clinical aspects and molecular alterations. Curr. Neurol. Neurosci. Rep. 2017, 17, 30. [Google Scholar] [CrossRef] [PubMed]
- Portaluppi, F.; Cortelli, P.; Avoni, P.; Vergnani, L.; Maltoni, P.; Pavani, A.; Sforza, E.; Degli Uberti, E.C.; Gambetti, P.; Lugaresi, E. Progressive disruption of the circadian rhythm of melatonin in fatal familial insomnia. J. Clin. Endocrinol. Metab. 1994, 78, 1075–1078. [Google Scholar] [PubMed]
- Kang, P.; de Bruin, G.S.; Wang, L.H.; Ward, B.A.; Ances, B.M.; Lim, M.M.; Bucelli, R.C. Sleep pathology in creutzfeldt-jakob disease. J. Clin. Sleep Med. 2016, 12, 1033–1039. [Google Scholar] [CrossRef] [PubMed]
- Givaty, G.; Maggio, N.; Cohen, O.S.; Blatt, I.; Chapman, J. Early pathology in sleep studies of patients with familial creutzfeldt-jakob disease. J. Sleep Res. 2016, 25, 571–575. [Google Scholar] [CrossRef] [PubMed]
- Bassant, M.H.; Cathala, F.; Court, L.; Gourmelon, P.; Hauw, J.J. Experimental scrapie in rats: First electrophysiological observations. Electroencephalogr. Clin. Neurophysiol. 1984, 57, 541–547. [Google Scholar] [CrossRef]
- Bert, J.; Vuillon-Cacciuttolo, G.; Balzamo, E.; De Micco, P.; Gambarelli, D.; Tamalet, J.; Gastaut, H. Experimental kuru in the rhesus monkey: A study of eeg modifications in the waking state and during sleep. Electroencephalogr. Clin. Neurophysiol. 1978, 45, 611–620. [Google Scholar] [CrossRef]
- Steele, A.D.; Jackson, W.S.; King, O.D.; Lindquist, S. The power of automated high-resolution behavior analysis revealed by its application to mouse models of huntington’s and prion diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 1983–1988. [Google Scholar] [CrossRef] [PubMed]
- Pierangeli, G.; Bono, F.; Aguglia, U.; Maltoni, P.; Montagna, P.; Lugaresi, E.; Quattrone, A.; Cortelli, P. Normal sleep-wake and circadian rhythms in a case of gerstmann-straussler-sheinker (gss) disease. Clin. Auton. Res. 2004, 14, 39–41. [Google Scholar] [CrossRef] [PubMed]
- Provini, F.; Vetrugno, R.; Pierangeli, G.; Cortelli, P.; Rizzo, G.; Filla, A.; Strisciuglio, C.; Gallassi, R.; Montagna, P. Sleep and temperature rhythms in two sisters with p102l gerstmann-straussler-scheinker (gss) disease. Sleep Med. 2009, 10, 374–377. [Google Scholar] [CrossRef] [PubMed]
- Amzica, F.; Steriade, M. Cellular substrates and laminar profile of sleep k-complex. Neuroscience 1998, 82, 671–686. [Google Scholar] [CrossRef]
- McCormick, D.A.; Bal, T. Sleep and arousal: Thalamocortical mechanisms. Annu. Rev. Neurosci. 1997, 20, 185–215. [Google Scholar] [CrossRef] [PubMed]
- Jahnke, K.; von Wegner, F.; Morzelewski, A.; Borisov, S.; Maischein, M.; Steinmetz, H.; Laufs, H. To wake or not to wake? The two-sided nature of the human k-complex. Neuroimage 2012, 59, 1631–1638. [Google Scholar] [CrossRef] [PubMed]
- Clawson, B.C.; Durkin, J.; Aton, S.J. Form and function of sleep spindles across the lifespan. Neural Plast. 2016, 2016, 6936381. [Google Scholar] [CrossRef] [PubMed]
- Cohen, O.S.; Chapman, J.; Korczyn, A.D.; Warman-Alaluf, N.; Orlev, Y.; Givaty, G.; Nitsan, Z.; Appel, S.; Rosenmann, H.; Kahana, E.; et al. Characterization of sleep disorders in patients with e200k familial creutzfeldt-jakob disease. J. Neurol. 2015, 262, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Ferrillo, F.; Plazzi, G.; Nobili, L.; Beelke, M.; De Carli, F.; Cortelli, P.; Tinuper, P.; Avoni, P.; Vandi, S.; Gambetti, P.; et al. Absence of sleep eeg markers in fatal familial insomnia healthy carriers: A spectral analysis study. Clin. Neurophysiol. 2001, 112, 1888–1892. [Google Scholar] [CrossRef]
- Gemignani, A.; Laurino, M.; Provini, F.; Piarulli, A.; Barletta, G.; d’Ascanio, P.; Bedini, R.; Lodi, R.; Manners, D.N.; Allegrini, P.; et al. Thalamic contribution to sleep slow oscillation features in humans: A single case cross sectional EEG study in fatal familial insomnia. Sleep Med. 2012, 13, 946–952. [Google Scholar] [CrossRef] [PubMed]
- Piao, Y.S.; Kakita, A.; Watanabe, H.; Kitamoto, T.; Takahashi, H. Sporadic fatal insomnia with spongiform degeneration in the thalamus and widespread prpsc deposits in the brain. Neuropathology 2005, 25, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Bouybayoune, I.; Mantovani, S.; Del Gallo, F.; Bertani, I.; Restelli, E.; Comerio, L.; Tapella, L.; Baracchi, F.; Fernandez-Borges, N.; Mangieri, M.; et al. Transgenic fatal familial insomnia mice indicate prion infectivity-independent mechanisms of pathogenesis and phenotypic expression of disease. PLoS Pathog. 2015, 11, e1004796. [Google Scholar] [CrossRef] [PubMed]
- Jackson, W.S.; Borkowski, A.W.; Faas, H.; Steele, A.D.; King, O.D.; Watson, N.; Jasanoff, A.; Lindquist, S. Spontaneous generation of prion infectivity in fatal familial insomnia knockin mice. Neuron 2009, 63, 438–450. [Google Scholar] [CrossRef] [PubMed]
- Bumb, J.M.; Schilling, C.; Enning, F.; Haddad, L.; Paul, F.; Lederbogen, F.; Deuschle, M.; Schredl, M.; Nolte, I. Pineal gland volume in primary insomnia and healthy controls: A magnetic resonance imaging study. J. Sleep Res. 2014, 23, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Hajak, G.; Rodenbeck, A.; Staedt, J.; Bandelow, B.; Huether, G.; Ruther, E. Nocturnal plasma melatonin levels in patients suffering from chronic primary insomnia. J. Pineal Res. 1995, 19, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Borbely, A.A. A two process model of sleep regulation. Hum. Neurobiol. 1982, 1, 195–204. [Google Scholar] [PubMed]
- Carter, M.D.; Juurlink, D.N. Melatonin. Can. Med. Assoc. J. 2012, 184, 1923. [Google Scholar] [CrossRef] [PubMed]
- Bjorness, T.E.; Dale, N.; Mettlach, G.; Sonneborn, A.; Sahin, B.; Fienberg, A.A.; Yanagisawa, M.; Bibb, J.A.; Greene, R.W. An adenosine-mediated glial-neuronal circuit for homeostatic sleep. J. Neurosci. 2016, 36, 3709–3721. [Google Scholar] [CrossRef] [PubMed]
- Achermann, P.; Borbely, A.A. Sleep homeostasis and models of sleep regulation. In Principles and Practice of Sleep Medicine, 5th ed.; Kryger, M.H., Roth, T., Dement, W.C., Eds.; Elsevier: Saint Louis, MO, USA, 2011; pp. 431–444. [Google Scholar]
- Bersagliere, A.; Achermann, P. Slow oscillations in human non-rapid eye movement sleep electroencephalogram: Effects of increased sleep pressure. J. Sleep Res. 2010, 19, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Patel, V.R.; Ceglia, N.; Zeller, M.; Eckel-Mahan, K.; Sassone-Corsi, P.; Baldi, P. The pervasiveness and plasticity of circadian oscillations: The coupled circadian-oscillators framework. Bioinformatics 2015, 31, 3181–3188. [Google Scholar] [CrossRef] [PubMed]
- Shibata, S.; Sasaki, H.; Ikeda, Y. Chrono-nutrition and chrono-exercise. Nihon Rinsho 2013, 71, 2194–2199. [Google Scholar] [PubMed]
- Baker, F.C.; Driver, H.S. Circadian rhythms, sleep, and the menstrual cycle. Sleep Med. 2007, 8, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Acuna-Castroviejo, D.; Escames, G.; Venegas, C.; Diaz-Casado, M.E.; Lima-Cabello, E.; Lopez, L.C.; Rosales-Corral, S.; Tan, D.X.; Reiter, R.J. Extrapineal melatonin: Sources, regulation, and potential functions. Cell. Mol. Life Sci. 2014, 71, 2997–3025. [Google Scholar] [CrossRef] [PubMed]
- Simonneaux, V.; Ribelayga, C. Generation of the melatonin endocrine message in mammals: A review of the complex regulation of melatonin synthesis by norepinephrine, peptides, and other pineal transmitters. Pharmacol. Rev. 2003, 55, 325–395. [Google Scholar] [CrossRef] [PubMed]
- Hastings, M.H.; Brancaccio, M.; Maywood, E.S. Circadian pacemaking in cells and circuits of the suprachiasmatic nucleus. J. Neuroendocrinol. 2014, 26, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Bonmati-Carrion, M.A.; Arguelles-Prieto, R.; Martinez-Madrid, M.J.; Reiter, R.; Hardeland, R.; Rol, M.A.; Madrid, J.A. Protecting the melatonin rhythm through circadian healthy light exposure. Int. J. Mol. Sci. 2014, 15, 23448–23500. [Google Scholar] [CrossRef] [PubMed]
- Gupta, B.B.; Spessert, R.; Vollrath, L. Molecular components and mechanism of adrenergic signal transduction in mammalian pineal gland: Regulation of melatonin synthesis. Indian J. Exp. Biol. 2005, 43, 115–149. [Google Scholar] [PubMed]
- Leston, J.; Harthe, C.; Mottolese, C.; Mertens, P.; Sindou, M.; Claustrat, B. Is pineal melatonin released in the third ventricle in humans? A study in movement disorders. Neurochirurgie 2015, 61, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Clough, S.J.; Hutchinson, A.J.; Adamah-Biassi, E.B.; Popovska-Gorevski, M.; Dubocovich, M.L. Mt1 and mt2 melatonin receptors: A therapeutic perspective. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 361–383. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Q.; Fichna, J.; Bashashati, M.; Li, Y.Y.; Storr, M. Distribution, function and physiological role of melatonin in the lower gut. World J. Gastroenterol. 2011, 17, 3888–3898. [Google Scholar] [CrossRef] [PubMed]
- Gitto, E.; Reiter, R.J.; Cordaro, S.P.; La Rosa, M.; Chiurazzi, P.; Trimarchi, G.; Gitto, P.; Calabro, M.P.; Barberi, I. Oxidative and inflammatory parameters in respiratory distress syndrome of preterm newborns: Beneficial effects of melatonin. Am. J. Perinatol. 2004, 21, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Gallucci, M.; Flores-Obando, R.; Mazzuco, S.; Ongaro, F.; Di Giorgi, E.; Boldrini, P.; Durante, E.; Frigato, A.; Albani, D.; Forloni, G.; et al. Melatonin and the charlson comorbidity index (cci): The treviso longeva (trelong) study. Int. J. Biol. Markers 2014, 29, e253–e260. [Google Scholar] [CrossRef] [PubMed]
- Ozler, M.; Simsek, K.; Ozkan, C.; Akgul, E.O.; Topal, T.; Oter, S.; Korkmaz, A. Comparison of the effect of topical and systemic melatonin administration on delayed wound healing in rats that underwent pinealectomy. Scand. J. Clin. Lab. Investig. 2010, 70, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.D.; Rusak, B.; Piggins, H.D. Regulation of melatonin-sensitivity and firing-rate rhythms of hamster suprachiasmatic nucleus neurons: Constant light effects. Brain Res. 1993, 602, 191–199. [Google Scholar] [CrossRef]
- De Farias Tda, S.; de Oliveira, A.C.; Andreotti, S.; do Amaral, F.G.; Chimin, P.; de Proenca, A.R.; Leal, F.L.; Sertie, R.A.; Campana, A.B.; Lopes, A.B.; et al. Pinealectomy interferes with the circadian clock genes expression in white adipose tissue. J. Pineal Res. 2015, 58, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Egermann, M.; Gerhardt, C.; Barth, A.; Maestroni, G.J.; Schneider, E.; Alini, M. Pinealectomy affects bone mineral density and structure—An experimental study in sheep. BMC Musculoskelet. Disord. 2011, 12, 271. [Google Scholar] [CrossRef] [PubMed]
- Slawik, H.; Stoffel, M.; Riedl, L.; Vesely, Z.; Behr, M.; Lehmberg, J.; Pohl, C.; Meyer, B.; Wiegand, M.; Krieg, S.M. Prospective study on salivary evening melatonin and sleep before and after pinealectomy in humans. J. Biol. Rhythm. 2016, 31, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Fisher, S.P.; Sugden, D. Endogenous melatonin is not obligatory for the regulation of the rat sleep-wake cycle. Sleep 2010, 33, 833–840. [Google Scholar] [CrossRef] [PubMed]
- De Butte, M.; Pappas, B.A. Pinealectomy causes hippocampal ca1 and ca3 cell loss: Reversal by melatonin supplementation. Neurobiol. Aging 2007, 28, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Lacoste, B.; Angeloni, D.; Dominguez-Lopez, S.; Calderoni, S.; Mauro, A.; Fraschini, F.; Descarries, L.; Gobbi, G. Anatomical and cellular localization of melatonin mt1 and mt2 receptors in the adult rat brain. J. Pineal Res. 2015, 58, 397–417. [Google Scholar] [CrossRef] [PubMed]
- Cagampang, F.R.; Whatley, S.A.; Mitchell, A.L.; Powell, J.F.; Campbell, I.C.; Coen, C.W. Circadian regulation of prion protein messenger rna in the rat forebrain: A widespread and synchronous rhythm. Neuroscience 1999, 91, 1201–1204. [Google Scholar] [CrossRef]
- Nakahara, D.; Nakamura, M.; Iigo, M.; Okamura, H. Bimodal circadian secretion of melatonin from the pineal gland in a living cba mouse. Proc. Natl. Acad. Sci. USA 2003, 100, 9584–9589. [Google Scholar] [CrossRef] [PubMed]
- Micic, G.; de Bruyn, A.; Lovato, N.; Wright, H.; Gradisar, M.; Ferguson, S.; Burgess, H.J.; Lack, L. The endogenous circadian temperature period length (tau) in delayed sleep phase disorder compared to good sleepers. J. Sleep Res. 2013, 22, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Verwey, M.; Robinson, B.; Amir, S. Recording and analysis of circadian rhythms in running-wheel activity in rodents. J. Vis. Exp. 2013, 113, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.R.; Nicholas, R.S.; Canevari, L. Lack of prion protein expression results in a neuronal phenotype sensitive to stress. J. Neurosci. Res. 2002, 67, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Dauvilliers, Y.; Cervena, K.; Carlander, B.; Espa, F.; Bassetti, C.; Claustrat, B.; Laplanche, J.L.; Billiard, M.; Touchon, J. Dissociation in circadian rhythms in a pseudohypersomnia form of fatal familial insomnia. Neurology 2004, 63, 2416–2418. [Google Scholar] [CrossRef] [PubMed]
- Beekes, M.; Baldauf, E.; Diringer, H. Sequential appearance and accumulation of pathognomonic markers in the central nervous system of hamsters orally infected with scrapie. J. Gen. Virol. 1996, 77 Pt 8, 1925–1934. [Google Scholar] [CrossRef] [PubMed]
- Pollera, C.; Bondiolotti, G.; Formentin, E.; Puricelli, M.; Mantegazza, P.; Bareggi, S.; Poli, G.; Ponti, W. Plasma noradrenalin as marker of neuroinvasion in prion diseases. Vet. Res. Commun. 2007, 31 (Suppl. 1), 249–252. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.W.; Choi, E.K.; Ju, W.K.; Ahn, M.S.; Carp, R.I.; Wisniewski, H.M.; Kim, Y.S. Extensive degeneration of catecholaminergic neurons to scrapie agent 87v in the brains of im mice. Mol. Chem. Neuropathol. 1998, 34, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Bassant, M.H.; Fage, D.; Dedek, J.; Cathala, F.; Court, L.; Scatton, B. Monoamine abnormalities in the brain of scrapie-infected rats. Brain Res. 1984, 308, 182–185. [Google Scholar] [CrossRef]
- Bondiolotti, G.; Rossoni, G.; Puricelli, M.; Formentin, E.; Lucchini, B.; Poli, G.; Ponti, W.; Bareggi, S.R. Changes in sympathetic activity in prion neuroinvasion. Neurobiol. Dis. 2010, 37, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Su, A.I.; Wiltshire, T.; Batalov, S.; Lapp, H.; Ching, K.A.; Block, D.; Zhang, J.; Soden, R.; Hayakawa, M.; Kreiman, G.; et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. USA 2004, 101, 6062–6067. [Google Scholar] [CrossRef] [PubMed]
- Hohjoh, H.; Takasu, M.; Shishikura, K.; Takahashi, Y.; Honda, Y.; Tokunaga, K. Significant association of the arylalkylamine n-acetyltransferase (aa-nat) gene with delayed sleep phase syndrome. Neurogenetics 2003, 4, 151–153. [Google Scholar] [PubMed]
- Baler, R.; Covington, S.; Klein, D.C. The rat arylalkylamine n-acetyltransferase gene promoter. Camp activation via a camp-responsive element-ccaat complex. J. Biol. Chem. 1997, 272, 6979–6985. [Google Scholar] [CrossRef] [PubMed]
- Beckman, D.; Santos, L.E.; Americo, T.A.; Ledo, J.H.; de Mello, F.G.; Linden, R. Prion protein modulates monoaminergic systems and depressive-like behavior in mice. J. Biol. Chem. 2015, 290, 20488–20498. [Google Scholar] [CrossRef] [PubMed]
- Gauer, F.; Poirel, V.J.; Garidou, M.L.; Simonneaux, V.; Pevet, P. Molecular cloning of the arylalkylamine-n-acetyltransferase and daily variations of its mrna expression in the syrian hamster pineal gland. Brain Res. Mol. Brain Res. 1999, 71, 87–95. [Google Scholar] [CrossRef]
- Green, S.A.; Holt, B.D.; Liggett, S.B. Beta 1- and beta 2-adrenergic receptors display subtype-selective coupling to Gs. Mol. Pharmacol. 1992, 41, 889–893. [Google Scholar] [PubMed]
- Konig, B.; Gratzel, M. Site of dopamine d1 receptor binding to Gs protein mapped with synthetic peptides. Biochim. Biophys. Acta 1994, 1223, 261–266. [Google Scholar] [CrossRef]


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roguski, A.; Gill, A.C. The Role of the Mammalian Prion Protein in the Control of Sleep. Pathogens 2017, 6, 58. https://doi.org/10.3390/pathogens6040058
Roguski A, Gill AC. The Role of the Mammalian Prion Protein in the Control of Sleep. Pathogens. 2017; 6(4):58. https://doi.org/10.3390/pathogens6040058
Chicago/Turabian StyleRoguski, Amber, and Andrew C. Gill. 2017. "The Role of the Mammalian Prion Protein in the Control of Sleep" Pathogens 6, no. 4: 58. https://doi.org/10.3390/pathogens6040058
APA StyleRoguski, A., & Gill, A. C. (2017). The Role of the Mammalian Prion Protein in the Control of Sleep. Pathogens, 6(4), 58. https://doi.org/10.3390/pathogens6040058