The Hidden Cost of Modern Medical Interventions: How Medical Advances Have Shaped the Prevalence of Human Fungal Disease



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Corticosteroids
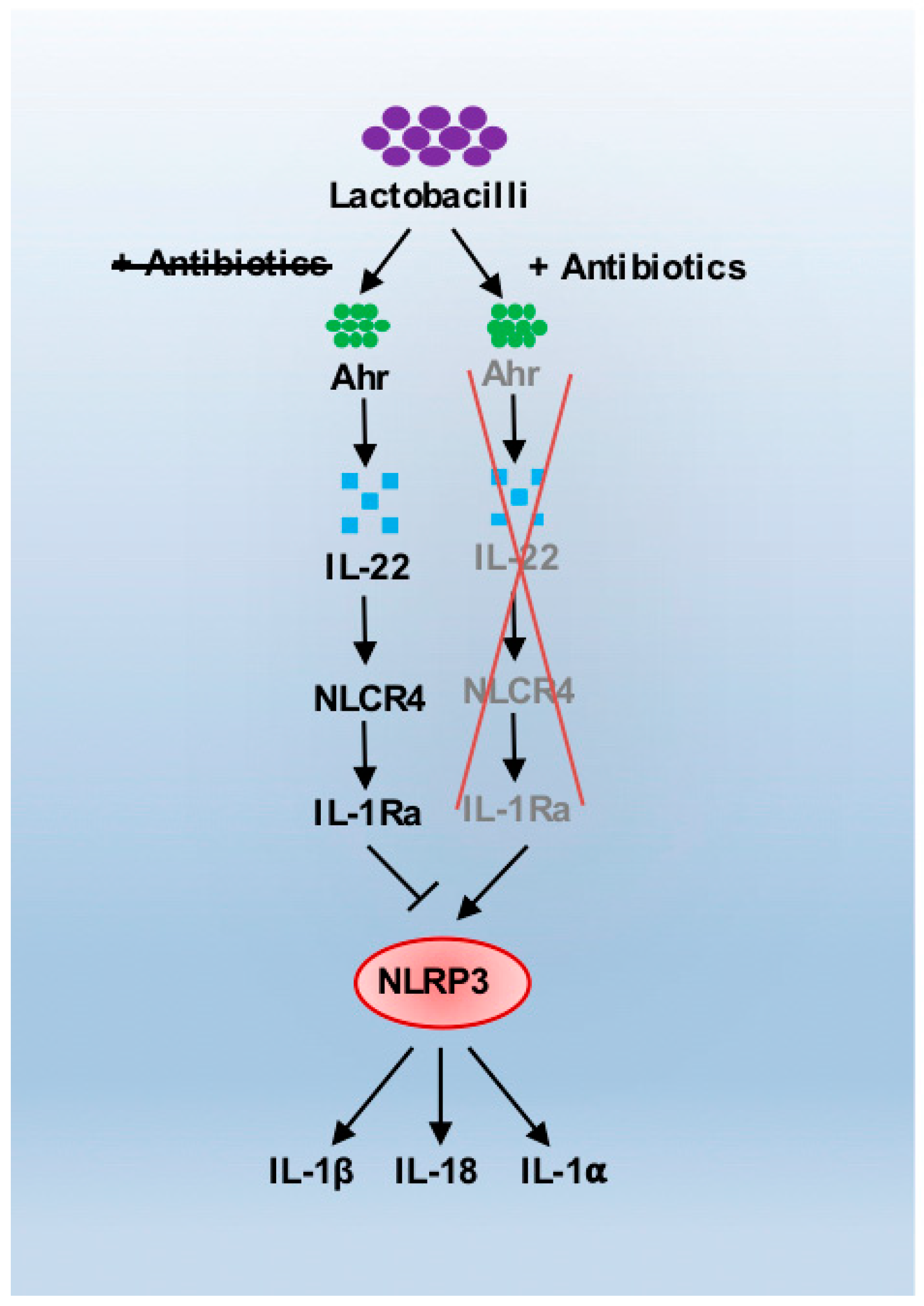
3. Antibiotics
4. Monoclonal Antibodies
5. Anti-Cancer Therapies
6. Conclusions
Funding
Conflicts of Interest
References
- Roser, M. Life Expectancy. Published online at OurWorldInData.org. Available online: https://ourworldindata.org/life-expectancy (accessed on 31 December 2018).
- Bongomin, F.; Gago, S.; Oladele, R.O.; Denning, D.W. Global and Multi-National Prevalence of Fungal Diseases-Estimate Precision. J. Fungi 2017, 3, 57. [Google Scholar] [CrossRef]
- Rodrigues, M.L.; Albuquerque, P.C. Searching for a change: The need for increased support for public health and research on fungal diseases. PLoS Negl. Trop. Dis. 2018, 12, e0006479. [Google Scholar] [CrossRef] [PubMed]
- Lionakis, M.S.; Iliev, I.D.; Hohl, T.M. Immunity against fungi. JCI Insight 2017, 2, e93156. [Google Scholar] [CrossRef] [PubMed]
- Drummond, R.A.; Lionakis, M.S. Mechanistic insights into the role of C-type lectin receptor/CARD9 signaling in human antifungal immunity. Front. Cell Infect. Microbiol. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Ferwerda, B.; Ferwerda, G.; Plantinga, T.S.; Willment, J.A.; van Spriel, A.B.; Venselaar, H.; Elbers, C.C.; Johnson, M.D.; Cambi, A.; Huysamen, C.; et al. Human Dectin-1 Deficiency and Mucocutaneous Fungal Infections. N. Eng. J. Med. 2009, 361, 1760–1767. [Google Scholar] [CrossRef] [Green Version]
- Marr, K.A. Fungal infections in hematopoietic stem cell transplant recipients. Med. Mycol. 2008, 46, 293–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, G.D.; Denning, D.W.; Gow, N.A.R.; Levitz, S.M.; Netea, M.G.; White, T.C. Hidden Killers: Human Fungal Infections. Sci. Transl. Med. 2012, 4, 165rv113. [Google Scholar] [CrossRef] [PubMed]
- McKay, L.I.; Cidlowski, J.A. Molecular Control of Immune/Inflammatory Responses: Interactions Between Nuclear Factor-κB and Steroid Receptor-Signaling Pathways. Endocr. Rev. 1999, 20, 435–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lionakis, M.S.; Netea, M.G.; Holland, S.M. Mendelian genetics of human susceptibility to fungal infection. Cold Spring Harbor Perspect. Med. 2014, 4, a019638. [Google Scholar] [CrossRef] [PubMed]
- Richards, D.F.; Fernandez, M.; Caulfield, J.; Hawrylowicz, C.M. Glucocorticoids drive human CD8+ T cell differentiation towards a phenotype with high IL-10 and reduced IL-4, IL-5 and IL-13 production. Eur. J. Immunol. 2000, 30, 2344–2354. [Google Scholar] [CrossRef] [Green Version]
- Luther, C.; Adamopoulou, E.; Stoeckle, C.; Brucklacher-Waldert, V.; Rosenkranz, D.; Stoltze, L.; Lauer, S.; Poeschel, S.; Melms, A.; Tolosa, E. Prednisolone Treatment Induces Tolerogenic Dendritic Cells and a Regulatory Milieu in Myasthenia Gravis Patients. J. Immunol. 2009, 183, 841. [Google Scholar] [CrossRef] [PubMed]
- Hooper, K.M.; Barlow, P.G.; Stevens, C.; Henderson, P. Inflammatory Bowel Disease Drugs: A Focus on Autophagy. J. Crohns Colitis 2017, 11, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Williamson, P.R.; Jarvis, J.N.; Panackal, A.A.; Fisher, M.C.; Molloy, S.F.; Loyse, A.; Harrison, T.S. Cryptococcal meningitis: Epidemiology, immunology, diagnosis and therapy. Nat. Rev. Neurol. 2017, 13, 13–24. [Google Scholar] [CrossRef]
- Johansson, Å.C.; Ohlsson, S.; Pettersson, Å.; Bengtsson, A.A.; Selga, D.; Hansson, M.; Hellmark, T. Impaired phagocytosis and reactive oxygen species production in phagocytes is associated with systemic vasculitis. Arthritis Res. Ther. 2016, 18, 92. [Google Scholar] [CrossRef] [PubMed]
- Saffar, A.S.; Ashdown, H.; Gounni, A.S. The molecular mechanisms of glucocorticoids-mediated neutrophil survival. Curr. Drug Targets 2011, 12, 556–562. [Google Scholar] [CrossRef]
- Timsit, J.-F.; Bassetti, M.; Cremer, O.; Daikos, G.; de Waele, J.; Kallil, A.; Kipnis, E.; Kollef, M.; Laupland, K.; Paiva, J.-A.; et al. Rationalizing antimicrobial therapy in the ICU: A narrative review. Intensiv. Care Med. 2019, 45, 172–189. [Google Scholar] [CrossRef]
- Liu, M.; Huang, S.; Guo, L.; Li, H.; Wang, F.; Zhang, Q.I.; Song, G. Clinical features and risk factors for blood stream infections of Candida in neonates. Exp. Ther. Med. 2015, 10, 1139–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Ami, R.; Olshtain-Pops, K.; Krieger, M.; Oren, I.; Bishara, J.; Dan, M.; Wiener-Well, Y.; Weinberger, M.; Zimhony, O.; Chowers, M.; et al. Antibiotic Exposure as a Risk Factor for Fluconazole-Resistant Candida Bloodstream Infection. Antimicrob. Agents Chemother. 2012, 56, 2518–2523. [Google Scholar] [CrossRef] [Green Version]
- Allonsius, C.N.; van den Broek, M.F.L.; De Boeck, I.; Kiekens, S.; Oerlemans, E.F.M.; Kiekens, F.; Foubert, K.; Vandenheuvel, D.; Cos, P.; Delputte, P.; et al. Interplay between Lactobacillus rhamnosus GG and Candida and the involvement of exopolysaccharides. Microb. Biotech. 2017, 10, 1753–1763. [Google Scholar] [CrossRef] [Green Version]
- Zelante, T.; Iannitti, R.G.; Cunha, C.; De Luca, A.; Giovannini, G.; Pieraccini, G.; Zecchi, R.; D’Angelo, C.; Massi-Benedetti, C.; Fallarino, F.; et al. Tryptophan Catabolites from Microbiota Engage Aryl Hydrocarbon Receptor and Balance Mucosal Reactivity via Interleukin-22. Immunity 2013, 39, 372–385. [Google Scholar] [CrossRef] [Green Version]
- Borghi, M.; De Luca, A.; Puccetti, M.; Jaeger, M.; Mencacci, A.; Oikonomou, V.; Pariano, M.; Garlanda, C.; Moretti, S.; Bartoli, A.; et al. Pathogenic NLRP3 Inflammasome Activity during Candida Infection Is Negatively Regulated by IL-22 via Activation of NLRC4 and IL-1Ra. Cell Host Microbe 2015, 18, 198–209. [Google Scholar] [CrossRef] [Green Version]
- Peters, B.M.; Palmer, G.E.; Nash, A.K.; Lilly, E.A.; Fidel, P.L., Jr.; Noverr, M.C. Fungal morphogenetic pathways are required for the hallmark inflammatory response during Candida albicans vaginitis. Infect Immun. 2014, 82, 532–543. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Carvalho, A.; Cunha, C.; Iannitti, R.G.; Pitzurra, L.; Giovannini, G.; Mencacci, A.; Bartolommei, L.; Moretti, S.; Massi-Benedetti, C.; et al. IL-22 and IDO1 Affect Immunity and Tolerance to Murine and Human Vaginal Candidiasis. PLoS Pathog. 2013, 9, e1003486. [Google Scholar] [CrossRef]
- Francino, M.P. Antibiotics and the Human Gut Microbiome: Dysbioses and Accumulation of Resistances. Front. Microbiol. 2016, 6, 1543. [Google Scholar] [CrossRef]
- Fan, D.; Coughlin, L.A.; Neubauer, M.M.; Kim, J.; Kim, M.S.; Zhan, X.; Simms-Waldrip, T.R.; Xie, Y.; Hooper, L.V.; Koh, A.Y. Activation of HIF-1[alpha] and LL-37 by commensal bacteria inhibits Candida albicans colonization. Nat. Med. 2015, 21, 808. [Google Scholar] [CrossRef]
- Lionakis, M.S. New insights into innate immune control of systemic candidiasis. Med. Mycol. 2014, 52, 555–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odds, F.C.; Davidson, A.D.; Jacobsen, M.D.; Tavanti, A.; Whyte, J.A.; Kibbler, C.C.; Ellis, D.H.; Maiden, M.C.J.; Shaw, D.J.; Gow, N.A.R. Candida albicans strain maintenance, replacement, and microvariation demonstrated by multilocus sequence typing. J. Clin. Microbiol. 2006, 44, 3647–3658. [Google Scholar] [CrossRef]
- Berkow, E.L.; Lockhart, S.R. Fluconazole resistance in Candida species: A current perspective. Infect. Drug Resist. 2017, 10, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Katsuno, G.; Takahashi, H.K.; Iwagaki, H.; Sugita, S.; Mori, S.; Saito, S.; Yoshino, T.; Nishibori, M.; Tanaka, N. The Effect of Ciprofloxacin on CD14 and Toll-like Receptor-4 Expression on Human Monocytes. Shock 2006, 25, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Jensen, J.-U.S.M.D.P.; Hein, L.M.D.; Lundgren, B.M.D.D.; Bestle, M.H.M.D.P.; Mohr, T.M.D.P.; Andersen, M.H.M.D.; Loken, J.M.D.; Tousi, H.M.D.; Soe-Jensen, P.M.D.; Lauritsen, A.O.M.D.; et al. Invasive Candida Infections and the Harm from Antibacterial Drugs in Critically Ill Patients: Data from a Randomized, Controlled Trial to Determine the Role of Ciprofloxacin, Piperacillin-Tazobactam, Meropenem, and Cefuroxime. Crit. Care Med. 2015, 43, 594–602. [Google Scholar] [CrossRef]
- Yang, J.H.; Bhargava, P.; McCloskey, D.; Mao, N.; Palsson, B.O.; Collins, J.J. Antibiotic-Induced Changes to the Host Metabolic Environment Inhibit Drug Efficacy and Alter Immune Function. Cell Host Microbe 2017, 22, 757–765.e753. [Google Scholar] [CrossRef] [PubMed]
- Bae, H.-B.; Zmijewski, J.W.; Deshane, J.S.; Tadie, J.-M.; Chaplin, D.D.; Takashima, S.; Abraham, E. AMP-activated protein kinase enhances the phagocytic ability of macrophages and neutrophils. FASEB J. 2011, 25, 4358–4368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, L.A.J.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalghatgi, S.; Spina, C.S.; Costello, J.C.; Liesa, M.; Morones-Ramirez, J.R.; Slomovic, S.; Molina, A.; Shirihai, O.S.; Collins, J.J. Bactericidal Antibiotics Induce Mitochondrial Dysfunction and Oxidative Damage in Mammalian Cells. Sci. Trans. Med. 2013, 5, 192ra185. [Google Scholar] [CrossRef] [PubMed]
- Scott, N.A.; Andrusaite, A.; Andersen, P.; Lawson, M.; Alcon-Giner, C.; Leclaire, C.; Caim, S.; Le Gall, G.; Shaw, T.; Connolly, J.P.R.; et al. Antibiotics induce sustained dysregulation of intestinal T cell immunity by perturbing macrophage homeostasis. Sci. Trans. Med. 2018, 10, eaao4755. [Google Scholar] [CrossRef] [PubMed]
- Maslowski, K.M.; Vieira, A.T.; Ng, A.; Kranich, J.; Sierro, F.; Yu, D.; Schilter, H.C.; Rolph, M.S.; Mackay, F.; Artis, D.; et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 2009, 461, 1282–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrêa-Oliveira, R.; Fachi, J.L.; Vieira, A.; Sato, F.T.; Vinolo, M.A.R. Regulation of immune cell function by short-chain fatty acids. Clin. Trans. Immunol. 2016, 5, e73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanov, I.I.; Frutos, R.d.L.; Manel, N.; Yoshinaga, K.; Rifkin, D.B.; Sartor, R.B.; Finlay, B.B.; Littman, D.R. Specific Microbiota Direct the Differentiation of IL-17-Producing T-Helper Cells in the Mucosa of the Small Intestine. Cell Host Microbe 2008, 4, 337–349. [Google Scholar] [CrossRef]
- Fahey, J.R.; Lyons, B.L.; Olekszak, H.L.; Mourino, A.J.; Ratiu, J.J.; Racine, J.J.; Chapman, H.D.; Serreze, D.V.; Baker, D.L.; Hendrix, N.K. Antibiotic-associated Manipulation of the Gut Microbiota and Phenotypic Restoration in NOD Mice. Comp. Med. 2017, 67, 335–343. [Google Scholar]
- Puel, A.; Daffinger, R.; Natividad, A.; Chrabieh, M.; Barcenas-Morales, G.; Picard, C.; Cobat, A.; Ouachace-Chardin, M.; Toulon, A.; Bustamante, J.; et al. Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J. Exp. Med. 2010, 207, 291–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conti, H.R.; Shen, F.; Nayyar, N.; Stocum, E.; Sun, J.N.; Lindemann, M.J.; Ho, A.W.; Hai, J.H.; Yu, J.J.; Jung, J.W.; et al. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J. Exp. Med. 2009, 206, 299–311. [Google Scholar] [CrossRef] [Green Version]
- Deshmukh, H.S.; Liu, Y.; Menkiti, O.R.; Mei, J.; Dai, N.; O’Leary, C.E.; Oliver, P.M.; Kolls, J.K.; Weiser, J.N.; Worthen, G.S. The microbiota regulates neutrophil homeostasis and host resistance to Escherichia coli K1 sepsis in neonatal mice. Nat. Med. 2014, 20, 528–534. [Google Scholar] [CrossRef] [PubMed]
- Erny, D.; Hrabe de Angelis, A.L.; Jaitin, D.; Wieghofer, P.; Staszewski, O.; David, E.; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; Buch, T.; et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 2015, 18, 965. [Google Scholar] [CrossRef] [PubMed]
- Puig, L. Brodalumab: The first anti-IL-17 receptor agent for psoriasis. Drugs Today 2017, 53, 283–297. [Google Scholar] [CrossRef]
- Saunte, D.M.; Mrowietz, U.; Puig, L.; Zachariae, C. Candida infections in patients with psoriasis and psoriatic arthritis treated with interleukin-17 inhibitors and their practical management. Br. J. Dermatol. 2017, 177, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Stamatiades, G.A.; Ioannou, P.; Petrikkos, G.; Tsioutis, C. Fungal infections in patients with inflammatory bowel disease: A systematic review. Mycoses 2018, 61, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, D.C.; Yeaman, M.R.; Filler, S.G. Tumor Necrosis Factor Inhibition and Invasive Fungal Infections. Clin. Infect. Dis. 2005, 41, S208–S212. [Google Scholar]
- Ghez, D.; Calleja, A.; Protin, C.; Baron, M.; Ledoux, M.-P.; Damaj, G.; Dupont, M.; Dreyfus, B.; Ferrant, E.; Herbaux, C.; et al. Early-onset invasive aspergillosis and other fungal infections in patients treated with ibrutinib. Blood 2018. [Google Scholar] [CrossRef]
- Baron, M.; Zini, J.M.; Challan Belval, T.; Vignon, M.; Denis, B.; Alanio, A.; Malphettes, M. Fungal infections in patients treated with ibrutinib: Two unusual cases of invasive aspergillosis and cryptococcal meningoencephalitis. Leuk. Lymphoma 2017, 58, 2981–2982. [Google Scholar] [CrossRef]
- Bercusson, A.; Colley, T.; Shah, A.; Warris, A.; Armstrong-James, D. Ibrutinib blocks Btk-dependent NF-ĸB and NFAT responses in human macrophages during Aspergillus fumigatus phagocytosis. Blood 2018. [Google Scholar] [CrossRef]
- Singh, S.; Sadanandam, A.; Nannuru, K.C.; Varney, M.L.; Mayer-Ezell, R.; Bond, R.; Singh, R.K. Small-Molecule Antagonists for CXCR2 and CXCR1 Inhibit Human Melanoma Growth by Decreasing Tumor Cell Proliferation, Survival, and Angiogenesis. Clin. Cancer Res. 2009, 15, 2380. [Google Scholar] [CrossRef] [PubMed]
- Swamydas, M.; Gao, J.-L.; Break, T.J.; Johnson, M.D.; Jaeger, M.; Rodriguez, C.A.; Lim, J.K.; Green, N.M.; Collar, A.L.; Fischer, B.G.; et al. CXCR1-mediated neutrophil degranulation and fungal killing promote Candida clearance and host survival. Sci. Trans. Med. 2016, 8, ra310–ra322. [Google Scholar] [CrossRef] [PubMed]
- Drummond, R.A.; Collar, A.L.; Swamydas, M.; Rodriguez, C.A.; Lim, J.K.; Mendez, L.M.; Fink, D.L.; Hsu, A.P.; Zhai, B.; Karauzum, H.; et al. CARD9-dependent neutrophil recruitment protects against fungal invasion of the central nervous system. PLoS Pathog. 2015, 11, e1005293. [Google Scholar] [CrossRef] [PubMed]
- Krisenko, M.O.; Geahlen, R.L. Calling in SYK: SYK’s dual role as a tumor promoter and tumor suppressor in cancer. Biochim. Biophys. Acta 2015, 1853, 254–263. [Google Scholar] [CrossRef]
- Friedberg, J.W.; Sharman, J.; Sweetenham, J.; Johnston, P.B.; Vose, J.M.; Lacasce, A.; Schaefer-Cutillo, J.; De Vos, S.; Sinha, R.; Leonard, J.P.; et al. Inhibition of Syk with fostamatinib disodium has significant clinical activity in non-Hodgkin lymphoma and chronic lymphocytic leukemia. Blood 2010, 115, 2578–2585. [Google Scholar] [CrossRef] [PubMed]
- Whitney, P.G.; Bär, E.; Osorio, F.; Rogers, N.C.; Schraml, B.U.; Deddouche, S.; LeibundGut-Landmann, S.; Reis e Sousa, C. Syk Signaling in Dendritic Cells Orchestrates Innate Resistance to Systemic Fungal Infection. PLoS Pathog. 2014, 10, e1004276. [Google Scholar] [CrossRef] [PubMed]
- Said-Sadier, N.; Padilla, E.; Langsley, G.; Ojcius, D.M. Aspergillus fumigatus Stimulates the NLRP3 Inflammasome through a Pathway Requiring ROS Production and the Syk Tyrosine Kinase. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Brossart, P.; Kotthoff, P. Dexamethasone Promotes Fungal Infection By Inhibition of APC Activation with Beta-Glucans Via STAT-3 and NF-κb. Blood 2016, 128, 3710. [Google Scholar]
- Deepe, G.S., Jr.; McGuinness, M. Interleukin-1 and host control of pulmonary histoplasmosis. J. Infect. Dis. 2006, 194, 855–864. [Google Scholar] [CrossRef]
- Daver, N.; Kontoyiannis, D.P. Checkpoint inhibitors and aspergillosis in AML: The double hit hypothesis. Lancet Oncol. 2017, 18, 1571–1573. [Google Scholar] [CrossRef]
- Grimaldi, D.; Pradier, O.; Hotchkiss, R.S.; Vincent, J.-L. Nivolumab plus interferon-beta in the treatment of intractable mucormycosis. Lancet Infect Dis. 2017, 17, 18. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clark, C.; Drummond, R.A. The Hidden Cost of Modern Medical Interventions: How Medical Advances Have Shaped the Prevalence of Human Fungal Disease. Pathogens 2019, 8, 45. https://doi.org/10.3390/pathogens8020045
Clark C, Drummond RA. The Hidden Cost of Modern Medical Interventions: How Medical Advances Have Shaped the Prevalence of Human Fungal Disease. Pathogens. 2019; 8(2):45. https://doi.org/10.3390/pathogens8020045
Chicago/Turabian StyleClark, Callum, and Rebecca A. Drummond. 2019. "The Hidden Cost of Modern Medical Interventions: How Medical Advances Have Shaped the Prevalence of Human Fungal Disease" Pathogens 8, no. 2: 45. https://doi.org/10.3390/pathogens8020045
APA StyleClark, C., & Drummond, R. A. (2019). The Hidden Cost of Modern Medical Interventions: How Medical Advances Have Shaped the Prevalence of Human Fungal Disease. Pathogens, 8(2), 45. https://doi.org/10.3390/pathogens8020045