Genomic Epidemiology of Streptococcus suis Sequence Type 7 Sporadic Infections in the Guangxi Zhuang Autonomous Region of China


Abstract
:1. Introduction
2. Results
2.1. MCG Analysis and Serotyping of S. suis ST7 Sporadic Strains
2.2. Phylogenetic Relationships among the S. suis ST7 Sporadic and Epidemic Strains
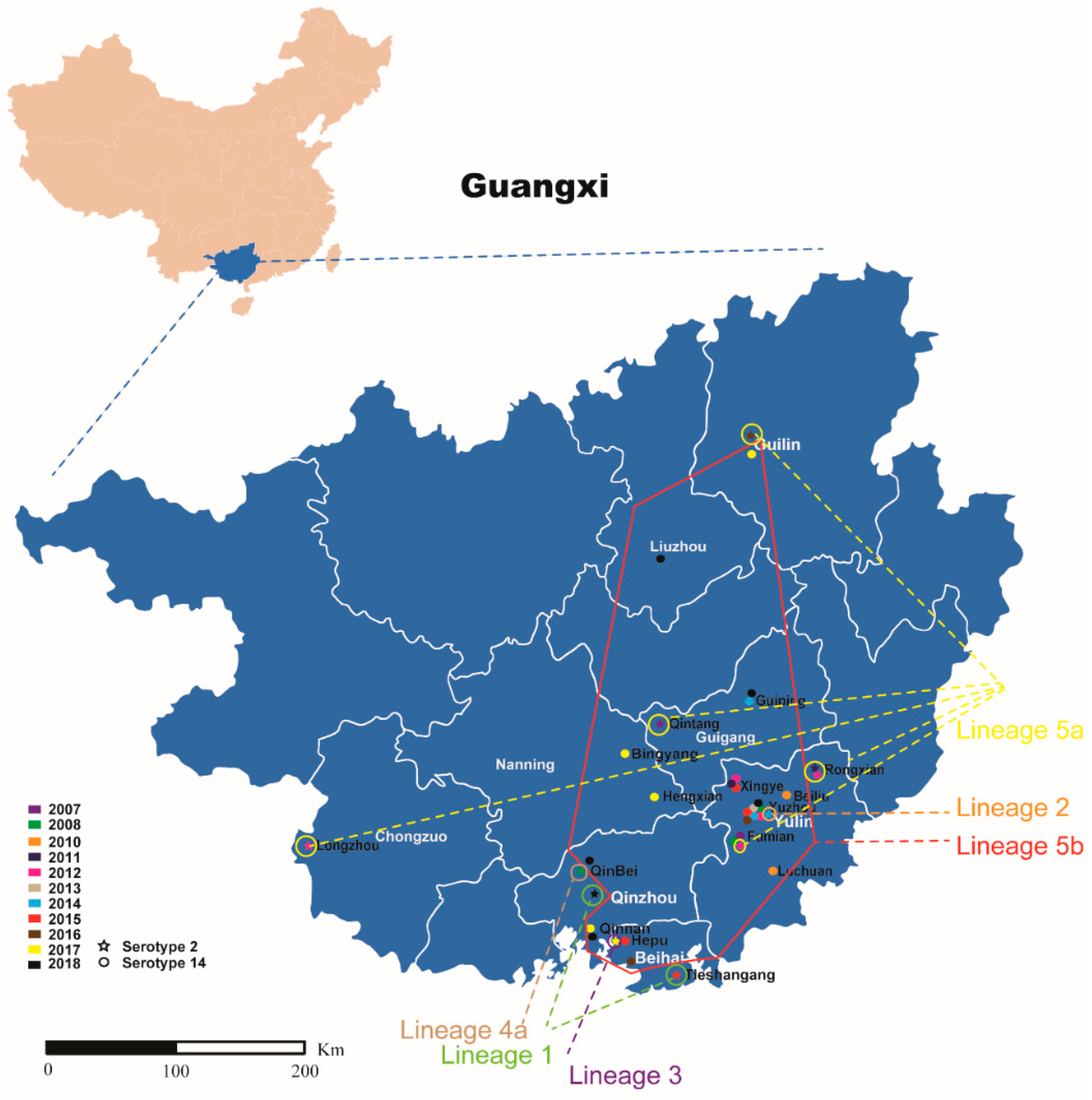
2.3. Geographic Distribution of S. suis ST7 Sporadic Strains
2.4. Virulence Genes of S. suis ST7 Sporadic Strains
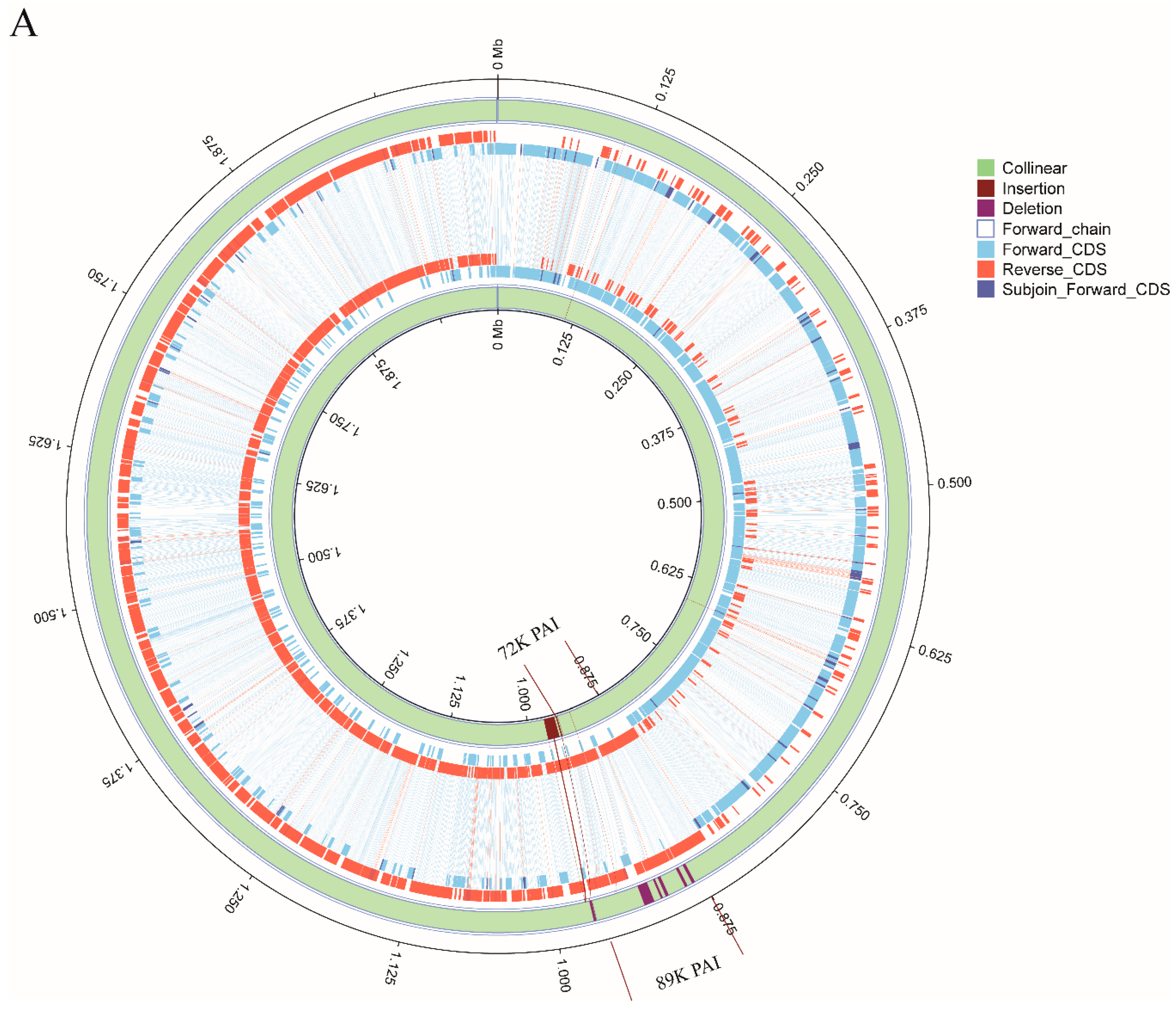
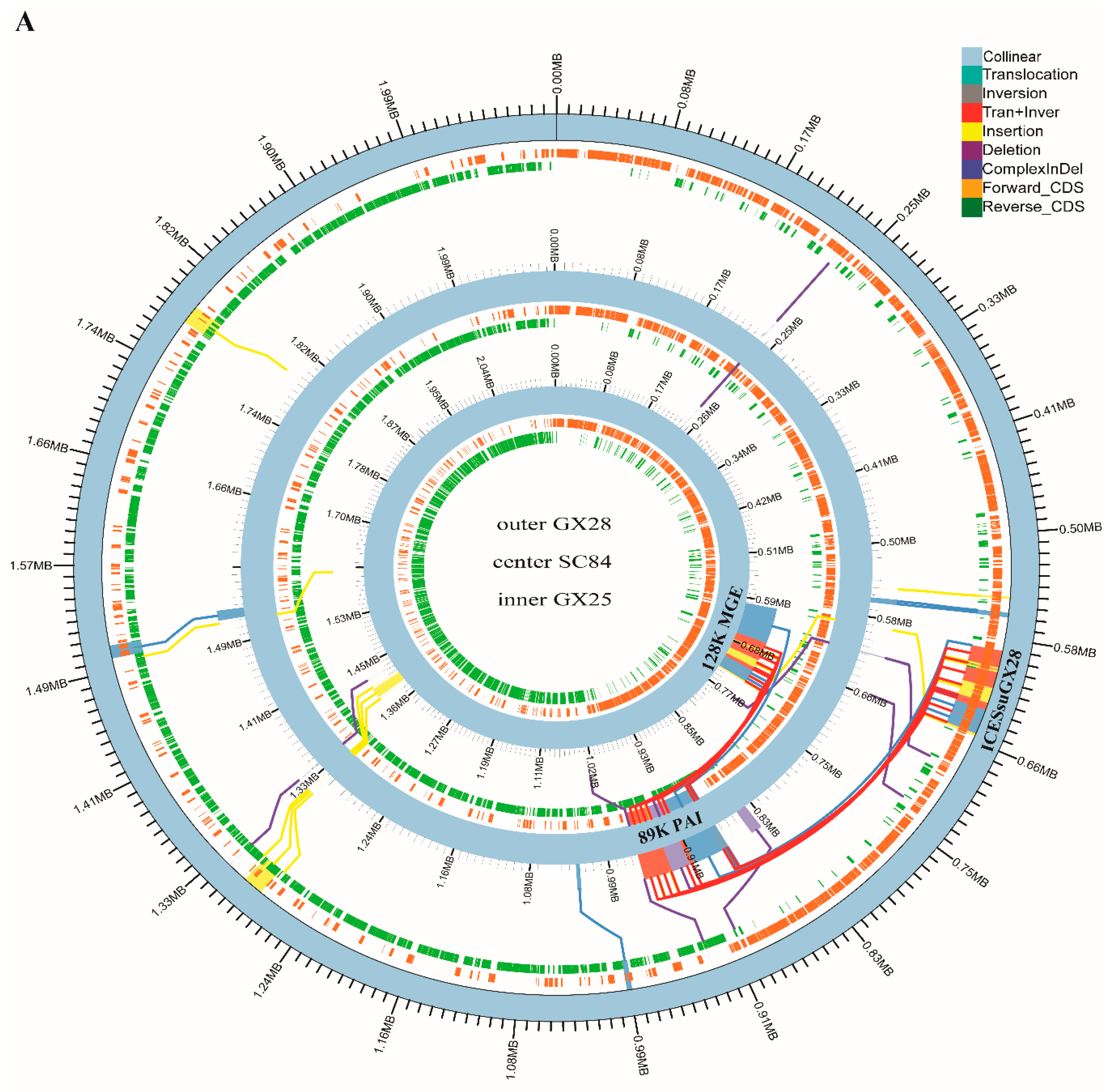
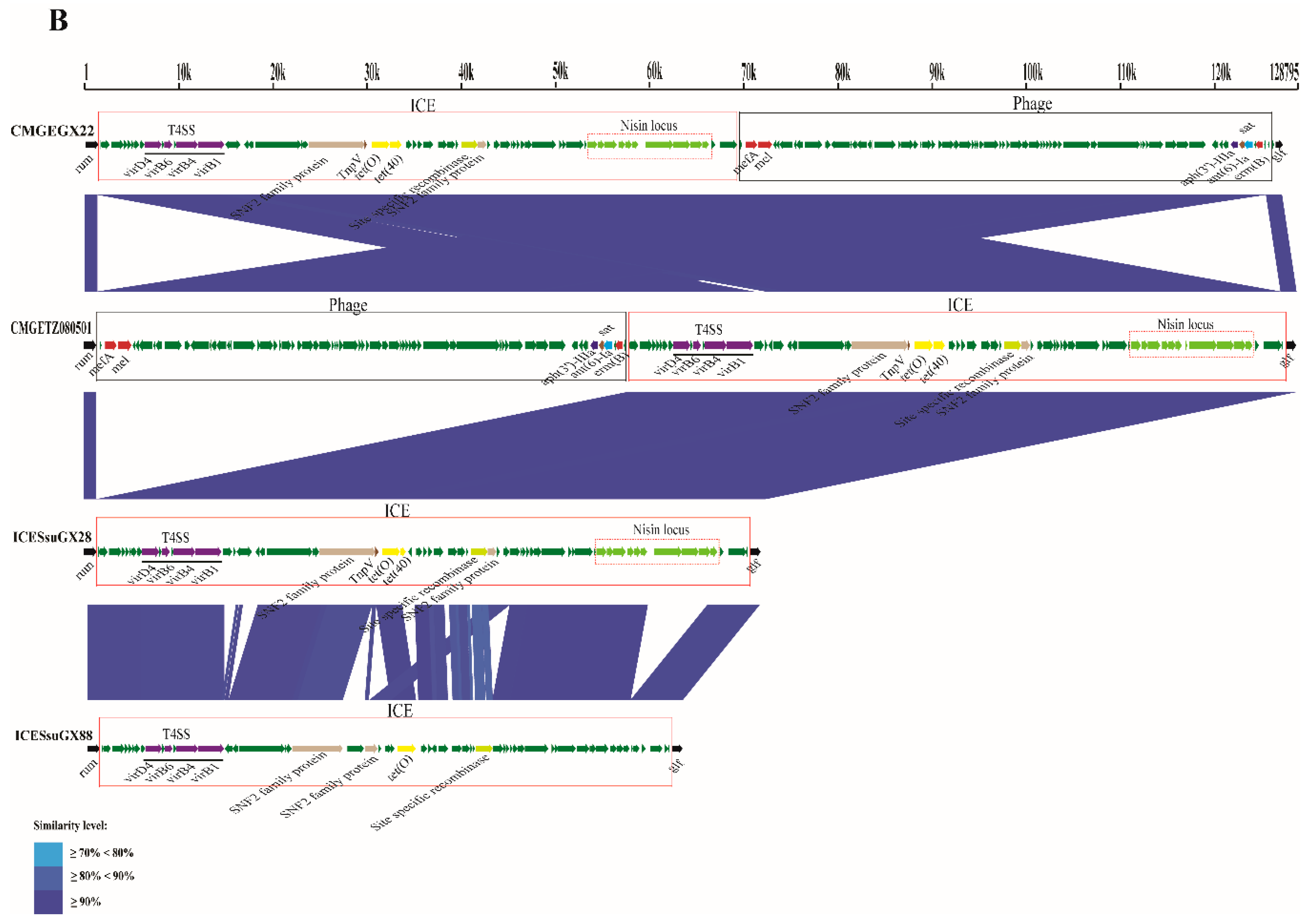
2.5. Whole Genome Synteny Analysis
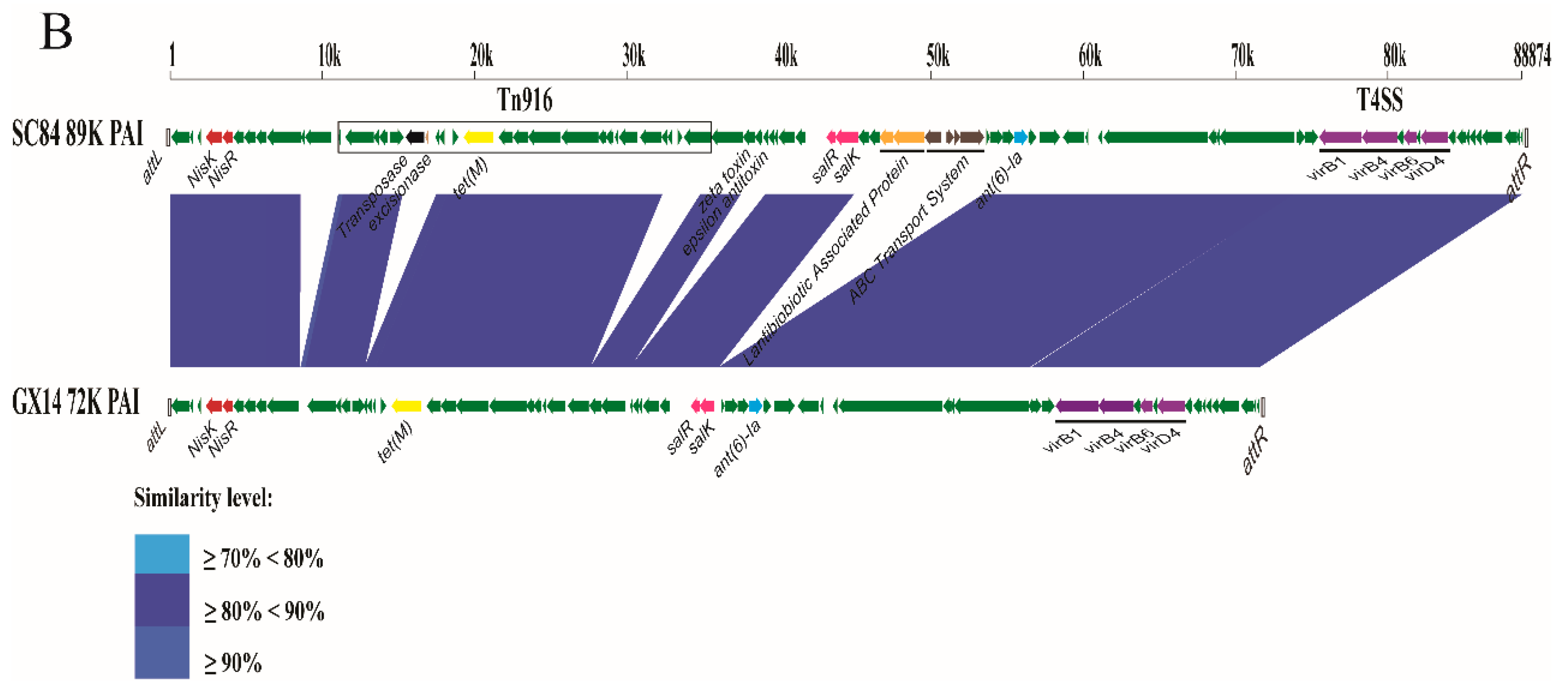
2.6. Identification of Lineage-related Genetic Characteristics
2.7. Antimicrobial Susceptibility Profiles
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains, Chromosomal DNA Sequencing and Bioinformatic Analysis
4.2. Phylogenetic Analysis
4.3. Whole Genome Synteny Analysis
4.4. Detection of Antibiotic Resistance Determinants and Antimicrobial Susceptibility Profiles
4.5. Nucleotide Sequence Accession Numbers
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Ethical Approval
References
- Huong, V.T.; Ha, N.; Huy, N.T.; Horby, P.; Nghia, H.D.; Thiem, V.D.; Zhu, X.; Hoa, N.T.; Hien, T.T.; Zamora, J.; et al. Epidemiology, clinical manifestations, and outcomes of Streptococcus suis infection in humans. Emerg. Infect. Dis. 2014, 20, 1105–1114. [Google Scholar] [CrossRef]
- Goyette-Desjardins, G.; Auger, J.P.; Xu, J.; Segura, M.; Gottschalk, M. Streptococcus suis, an important pig pathogen and emerging zoonotic agent—An update on the worldwide distribution based on serotyping and sequence typing. Emerg. Microbes Infect. 2014, 3, e45. [Google Scholar] [CrossRef] [PubMed]
- Wertheim, H.F.; Nguyen, H.N.; Taylor, W.; Lien, T.T.; Ngo, H.T.; Nguyen, T.Q.; Nguyen, B.N.; Nguyen, H.H.; Nguyen, H.M.; Nguyen, C.T.; et al. Streptococcus suis, an important cause of adult bacterial meningitis in northern Vietnam. PLoS ONE 2009, 4, e5973. [Google Scholar] [CrossRef] [PubMed]
- Mai, N.T.; Hoa, N.T.; Nga, T.V.; Linh le, D.; Chau, T.T.; Sinh, D.X.; Phu, N.H.; Chuong, L.V.; Diep, T.S.; Campbell, J.; et al. Streptococcus suis meningitis in adults in Vietnam. Clin. Infect. Dis. 2008, 46, 659–667. [Google Scholar] [PubMed]
- Yu, H.; Jing, H.; Chen, Z.; Zheng, H.; Zhu, X.; Wang, H.; Wang, S.; Liu, L.; Zu, R.; Luo, L.; et al. Human Streptococcus suis outbreak, Sichuan, China. Emerg. Infect. Dis. 2006, 12, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Zhu, X.; Jing, H.; Du, H.; Segura, M.; Zheng, H.; Kan, B.; Wang, L.; Bai, X.; Zhou, Y.; et al. Streptococcus suis sequence type 7 outbreak, Sichuan, China. Emerg. Infect. Dis. 2006, 12, 1203–1208. [Google Scholar] [CrossRef]
- Ye, C.; Zheng, H.; Zhang, J.; Jing, H.; Wang, L.; Xiong, Y.; Wang, W.; Zhou, Z.; Sun, Q.; Luo, X.; et al. Clinical, experimental, and genomic differences between intermediately pathogenic, highly pathogenic, and epidemic Streptococcus suis. J. Infect. Dis. 2009, 199, 97–107. [Google Scholar] [CrossRef]
- Chen, C.; Zhang, W.; Zheng, H.; Lan, R.; Wang, H.; Du, P.; Bai, X.; Ji, S.; Meng, Q.; Jin, D.; et al. Minimum core genome sequence typing of bacterial pathogens: A unified approach for clinical and public health microbiology. J. Clin. Microbiol. 2013, 51, 2582–2591. [Google Scholar] [CrossRef]
- Athey, T.B.; Teatero, S.; Takamatsu, D.; Wasserscheid, J.; Dewar, K.; Gottschalk, M.; Fittipaldi, N. Population Structure and Antimicrobial Resistance Profiles of Streptococcus suis Serotype 2 Sequence Type 25 Strains. PLoS ONE 2016, 11, e0150908. [Google Scholar] [CrossRef]
- Athey, T.B.; Auger, J.P.; Teatero, S.; Dumesnil, A.; Takamatsu, D.; Wasserscheid, J.; Dewar, K.; Gottschalk, M.; Fittipaldi, N. Complex Population Structure and Virulence Differences among Serotype 2 Streptococcus suis Strains Belonging to Sequence Type 28. PLoS ONE 2015, 10, e0137760. [Google Scholar] [CrossRef]
- Zheng, X.; Zheng, H.; Lan, R.; Ye, C.; Wang, Y.; Zhang, J.; Jing, H.; Chen, C.; Segura, M.; Gottschalk, M.; et al. Identification of genes and genomic islands correlated with high pathogenicity in Streptococcus suis using whole genome tiling microarrays. PLoS ONE 2011, 6, e17987. [Google Scholar]
- Du, P.; Zheng, H.; Zhou, J.; Lan, R.; Ye, C.; Jing, H.; Jin, D.; Cui, Z.; Bai, X.; Liang, J.; et al. Detection of Multiple Parallel Transmission Outbreak of Streptococcus suis Human Infection by Use of Genome Epidemiology, China, 2005. Emerg. Infect. Dis. 2017, 23, 204. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Wang, M.; Hao, H.; Yang, R.; Xie, J.; Su, J.; Lin, M.; Cui, Y.; Jiang, Y. Genomic epidemiological investigation of a Streptococcus suis outbreak in Guangxi, China, 2016. Infect. Genet. Evol. 2019, 68, 249–252. [Google Scholar] [CrossRef]
- Chen, C.; Tang, J.; Dong, W.; Wang, C.; Feng, Y.; Wang, J.; Zheng, F.; Pan, X.; Liu, D.; Li, M.; et al. A glimpse of streptococcal toxic shock syndrome from comparative genomics of S. suis 2 Chinese isolates. PLoS ONE 2007, 2, e315. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Ma, J.; Shang, K.; Hu, X.; Liang, Y.; Li, D.; Wu, Z.; Dai, L.; Chen, L.; Wang, L. Evolution and Diversity of the Antimicrobial Resistance Associated Mobilome in Streptococcus suis: A Probable Mobile Genetic Elements Reservoir for Other Streptococci. Front. Cell. Infect. Microbiol. 2016, 6, 118. [Google Scholar] [CrossRef] [PubMed]
- Fittipaldi, N.; Fuller, T.E.; Teel, J.F.; Wilson, T.L.; Wolfram, T.J.; Lowery, D.E.; Gottschalk, M. Serotype distribution and production of muramidase-released protein, extracellular factor and suilysin by field strains of Streptococcus suis isolated in the United States. Vet. Microbiol. 2009, 139, 310–317. [Google Scholar] [CrossRef]
- Fokas, S.; Fokas, S.; Tsironi, M.; Kalkani, M.; Dionysopouloy, M. Prevalence of inducible clindamycin resistance in macrolide-resistant Staphylococcus spp. Clin. Microbiol. Infect. 2005, 11, 337–340. [Google Scholar] [CrossRef]
- Syrogiannopoulos, G.A.; Grivea, I.N.; Ednie, L.M.; Bozdogan, B.; Katopodis, G.D.; Beratis, N.G.; Davies, T.A.; Appelbaum, P.C. Antimicrobial susceptibility and macrolide resistance inducibility of Streptococcus pneumoniae carrying erm(A), erm(B), or mef(A). Antimicrob. Agents Chemother. 2003, 47, 2699–2702. [Google Scholar] [CrossRef]
- Sparo, M.; Delpech, G.; Garcia Allende, N. Impact on Public Health of the Spread of High-Level Resistance to Gentamicin and Vancomycin in Enterococci. Front. Microbiol. 2018, 9, 3073. [Google Scholar] [CrossRef] [Green Version]
- Gurung, M.; Tamang, M.D.; Moon, D.C.; Kim, S.R.; Jeong, J.H.; Jang, G.C.; Jung, S.C.; Park, Y.H.; Lim, S.K. Molecular Basis of Resistance to Selected Antimicrobial Agents in the Emerging Zoonotic Pathogen Streptococcus suis. J. Clin. Microbiol. 2015, 53, 2332–2336. [Google Scholar] [CrossRef]
- Ramirez, M.S.; Tolmasky, M.E. Aminoglycoside modifying enzymes. Drug Resist. Updates 2010, 13, 151–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, H.; Lan, R.; Zheng, X.; Cui, Z.; Liu, Z.; Bai, X.; Ji, S.; Gottschalk, M.; Xu, J. Comparative genomic hybridization identifies virulence differences in Streptococcus suis. PLoS ONE 2014, 9, e87866. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Song, Y.; Wei, Z.; He, H.; Zhang, A.; Jin, M. Antimicrobial susceptibility, tetracycline and erythromycin resistance genes, and multilocus sequence typing of Streptococcus suis isolates from diseased pigs in China. J. Vet. Med Sci. 2013, 75, 583–587. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Dong, X.; Li, Z.; Zou, G.; Lin, L.; Wang, X.; Chen, H.; Gasser, R.B.; Li, J. Predominance of Streptococcus suis ST1 and ST7 in human cases in China, and detection of a novel sequence type, ST658. Virulence 2017, 8, 1031–1035. [Google Scholar] [CrossRef] [PubMed]
- Kerdsin, A.; Oishi, K.; Sripakdee, S.; Boonkerd, N.; Polwichai, P.; Nakamura, S.; Uchida, R.; Sawanpanyalert, P.; Dejsirilert, S. Clonal dissemination of human isolates of Streptococcus suis serotype 14 in Thailand. J. Med. Microbiol. 2009, 58, 1508–1513. [Google Scholar] [CrossRef]
- Okura, M.; Takamatsu, D.; Maruyama, F.; Nozawa, T.; Nakagawa, I.; Osaki, M.; Sekizaki, T.; Gottschalk, M.; Kumagai, Y.; Hamada, S. Genetic analysis of capsular polysaccharide synthesis gene clusters from all serotypes of Streptococcus suis: Potential mechanisms for generation of capsular variation. Appl. Environ. Microbiol. 2013, 79, 2796–2806. [Google Scholar] [CrossRef]
- King, S.J.; Leigh, J.A.; Heath, P.J.; Luque, I.; Tarradas, C.; Dowson, C.G.; Whatmore, A.M. Development of a multilocus sequence typing scheme for the pig pathogen Streptococcus suis: Identification of virulent clones and potential capsular serotype exchange. J. Clin. Microbiol. 2002, 40, 3671–3680. [Google Scholar] [CrossRef]
- Xu, J.; Fu, S.; Liu, M.; Xu, Q.; Bei, W.; Chen, H.; Tan, C. The two-component system NisK/NisR contributes to the virulence of Streptococcus suis serotype 2. Microbiol. Res. 2014, 169, 541–546. [Google Scholar] [CrossRef]
- Zhao, Y.; Liu, G.; Li, S.; Wang, M.; Song, J.; Wang, J.; Tang, J.; Li, M.; Hu, F. Role of a type IV-like secretion system of Streptococcus suis 2 in the development of streptococcal toxic shock syndrome. J. Infect. Dis. 2011, 204, 274–281. [Google Scholar] [CrossRef]
- Li, M.; Wang, C.; Feng, Y.; Pan, X.; Cheng, G.; Wang, J.; Ge, J.; Zheng, F.; Cao, M.; Dong, Y.; et al. SalK/SalR, a two-component signal transduction system, is essential for full virulence of highly invasive Streptococcus suis serotype 2. PLoS ONE 2008, 3, e2080. [Google Scholar] [CrossRef]
- Wang, J.; Gao, Y.; Teng, K.; Zhang, J.; Sun, S.; Zhong, J. Restoration of bioactive lantibiotic suicin from a remnant lan locus of pathogenic Streptococcus suis serotype 2. Appl. Environ. Microbiol. 2014, 80, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Ji, S.; Lan, R.; Liu, Z.; Bai, X.; Zhang, W.; Gottschalk, M.; Xu, J. Population analysis of Streptococcus suis isolates from slaughtered swine by use of minimum core genome sequence typing. J. Clin. Microbiol. 2014, 52, 3568–3572. [Google Scholar] [CrossRef] [PubMed]
- Hoa, N.T.; Chieu, T.T.; Nghia, H.D.; Mai, N.T.; Anh, P.H.; Wolbers, M.; Baker, S.; Campbell, J.I.; Chau, N.V.; Hien, T.T.; et al. The antimicrobial resistance patterns and associated determinants in Streptococcus suis isolated from humans in southern Vietnam, 1997–2008. BMC Infect. Dis. 2011, 11, 6. [Google Scholar] [CrossRef] [PubMed]
- Nurjadi, D.; Olalekan, A.O.; Layer, F.; Shittu, A.O.; Alabi, A.; Ghebremedhin, B.; Schaumburg, F.; Hofmann-Eifler, J.; Van Genderen, P.J.; Caumes, E.; et al. Emergence of trimethoprim resistance gene dfrG in Staphylococcus aureus causing human infection and colonization in sub-Saharan Africa and its import to Europe. J. Antimicrob. Chemother. 2014, 69, 2361–2368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, P.; Yang, M.; Zhang, A.; Wu, J.; Chen, B.; Hua, Y.; Yu, J.; Xiao, J.; Jin, M. Complete genome sequence of Streptococcus suis serotype 14 strain JS14. J. Bacteriol. 2011, 193, 2375–2376. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Wang, W.; Tang, M.; Shao, J.; Dai, C.; Zhang, W.; Fan, H.; Yao, H.; Zong, J.; Chen, D.; et al. Comparative genomic analysis shows that Streptococcus suis meningitis isolate SC070731 contains a unique 105K genomic island. Gene 2014, 535, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Yang, X.; Qin, J.; Lu, N.; Cheng, G.; Wu, N.; Pan, Y.; Li, J.; Zhu, L.; Wang, X.; et al. Metagenome-wide analysis of antibiotic resistance genes in a large cohort of human gut microbiota. Nat. Commun. 2013, 4, 2151. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.; Zhang, Q.; Song, Y.; Zhang, Z.; Zhang, A.; Xiao, J.; Jin, M. Characterization of Spectinomycin Resistance in Streptococcus suis Leads to Two Novel Insights into Drug Resistance Formation and Dissemination Mechanism. Antimicrob. Agents Chemother. 2016, 60, 6390–6392. [Google Scholar] [CrossRef]
- Marie, J.; Morvan, H.; Berthelot-Herault, F.; Sanders, P.; Kempf, I.; Gautier-Bouchardon, A.V.; Jouy, E.; Kobisch, M. Antimicrobial susceptibility of Streptococcus suis isolated from swine in France and from humans in different countries between 1996 and 2000. J. Antimicrob. Chemother. 2002, 50, 201–209. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lineage | Strain Name | Accession No. | City | Year | Symptom | PAI/GI | PEN G (BP: > 4 μg/mL) | CEF (BP: > 4 μg/mL) | TET (BP: > 8 μg/mL) | ERY (BP: > 1 μg/mL) | AZI (BP: >1 μg/mL) | CLI (BP: > 1 μg/mL) | STR (BP: > 250 μg/mL) | KAN (BP: > 250 μg/mL) | SPE (BP: > 256 μg/mL) | GEN (BP: > 250 μg/mL) | TRI (BP *: > 4 μg/mL) | Antibiotic Resistance Genes |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Lineage 1 | GX49 | SRR8523142 | Beihai | 2015 | Sepsis | ICESsuGX88 | 0.064 | 0.047 | 16 | 0.19 | 0.38 | 0.5 | 2 | 6 | 32 | 6 | 1* | tet(O) |
| GX88 | SRR8523131 | Qinzhou | 2018 | Meningitis | ICESsuGX88 | 0.064 | 0.064 | 24 | 48 | >256 | >256 | 2 | 4 | 32 | 4 | 0.75 * | tet(O) | |
| Lineage 2 | GX47 | SRR8523138 | Yulin | 2014 | Sepsis | ICESsuGX88 | 0.064 | 0.064 | 24 | >256 | >256 | >256 | 4 | 3 | 32 | 6 | 3 * | erm(B), tet(O) |
| Lineage 3 | GX79 | SRR8523125 | Beihai | 2017 | Meningitis | / | 0.047 | 0.047 | 24 | >256 | >256 | >256 | >1024 | >256 | 16 | 12 | 0.5 * | aph(3’)-IIIa, erm(B), tet(O), sat4, ant(6)-Ia |
| Lineage 4a | GX14 | SRR8523149 | Qinzhou | 2008 | STSLS | 72K | 0.094 | 0.047 | 24 | 0.19 | 0.38 | 0.38 | >1024 | 8 | 24 | 8 | 0.75 * | ant(6)-Ia, tet(M) |
| Lineage 5a | GX18 | SRR8523148 | Yulin | 2011 | Sepsis | ICESsuGX81 | 0.094 | 0.125 | 24 | 0.75 | 0.75 | 0.75 | 3 | 6 | 24 | 8 | 0.5 * | tet(O), tet(40) |
| GX21 | SRR8523152 | Yulin | 2012 | Sepsis | CMGETZ080501 | 0.032 | 0.064 | 24 | >256 | >256 | >256 | 512 | >256 | 16 | 8 | 0.75 * | aph(3’)-IIIa, erm(B), tet(O), tet(40), sat4, mefA, mel,ant(6)-Ia | |
| GX22 | SRR8523135 | Chongzuo | 2012 | Sepsis | CMGEGX22 | 0.047 | 0.064 | 32 | >256 | >256 | >256 | >1024 | >256 | 24 | 4 | 0.25 * | aph(3’)-IIIa, erm(B), tet(O), tet(40), sat4, mefA, mel,ant(6)-Ia | |
| GX98 | SRR8523127 | Guilin | 2016 | / | CMGEGX22 | 0.047 | 0.047 | 24 | >256 | >256 | >256 | >1024 | >256 | 16 | 6 | 0.38 * | aph(3’)-IIIa, erm(B), tet(O), tet(40), sat4, mefA, mel,ant(6)-Ia | |
| GX9 | SRR8523146 | Guigang | 2007 | Meningitis | CMGETZ080501 | 1.5 | 2 | 16 | >256 | >256 | >256 | >1024 | >256 | 24 | 3 | >32 * | aph(3’)-IIIa, erm(B), tet(O), tet(40), sat4, mefA, mel | |
| GX25 | SRR8523133 | Yulin | 2012 | Sepsis | CMGETZ080501 | 0.047 | 0.064 | 16 | >256 | >256 | >256 | >1024 | >256 | 24 | 6 | 3 * | aph(3’)-IIIa, erm(B), tet(O), tet(40), sat4, mefA, mel,ant(6)-Ia | |
| Lineage 5b | GX24 | SRR8523136 | Yulin | 2012 | Meningitis | ICESsuGX81 | 0.064 | 0.094 | 16 | >256 | >256 | >256 | 2 | 3 | 24 | 6 | 0.75 * | erm(B), tet(O), tet(40) |
| GX37 | SRR8523140 | Guigang | 2014 | Sepsis | ICESsuGX81 | 0.047 | 0.094 | 24 | 0.75 | 0.75 | 0.5 | 6 | 4 | 48 | 8 | 0.5 * | tet(O), tet(40) | |
| GX89 | SRR8523132 | Guigang | 2018 | Meningitis | ICESsuGX81 | 0.064 | 0.064 | 24 | 0.5 | 0.5 | 0.75 | 2 | 3 | 24 | 2 | 0.38 * | tet(O), tet(40) | |
| GX91 | SRR8523117 | Liuzhou | 2018 | / | ICESsuGX81 | 0.047 | 0.064 | 16 | >256 | >256 | >256 | >1024 | 12 | >1024 | 4 | 0.38 * | spw, erm(B), tet(O), ant(6)-Ia, tet(40) | |
| GX50 | SRR8523121 | Beihai | 2015 | Sepsis | ICESsuGX81 | 0.064 | 0.38 | 12 | 0.125 | 0.25 | 0.5 | 2 | 4 | 24 | 6 | 0.75 * | tet(O), tet(40) | |
| GX80 | SRR8523124 | Qinzhou | 2017 | Meningitis | ICESsuGX81 | 0.047 | 0.125 | 12 | >256 | >256 | >256 | 2 | 4 | 24 | 4 | 0.75 * | tet(O), tet(40) | |
| GX48 | SRR8523141 | Yulin | 2015 | Sepsis | ICESsuGX81 | 0.064 | 0.064 | 16 | 0.5 | 0.75 | 0.75 | 6 | 8 | 24 | 8 | 1 * | tet(O), tet(40) | |
| GX27 | SRR8523134 | Yulin | 2012 | Sepsis | ICESsuGX81 | 0.064 | 0.125 | 16 | 0.38 | 0.38 | 0.75 | 2 | 2 | 48 | 4 | 0.5 * | tet(O), tet(40) | |
| GX70 | SRR8523118 | Beihai | 2016 | Sepsis | ICESsuGX81 | 0.047 | 0.19 | 16 | 0.38 | 0.5 | 0.75 | 2 | 2 | 32 | 4 | 0.19 * | tet(O), tet(40) | |
| GX85 | SRR8523115 | Qinzhou | 2018 | Meningitis | ICESsuGX81 | 0.064 | 0.064 | 16 | 0.75 | 0.75 | 0.75 | 2 | 3 | 48 | 4 | 1 * | tet(O), tet(40) | |
| GX81 | SRR8523123 | Nanning | 2017 | Sepsis | ICESsuGX81 | 0.094 | 0.094 | 12 | 48 | >256 | >256 | 8 | 12 | 24 | 6 | 2 * | tet(O), tet(40) | |
| GX87 | SRR8523130 | Qinzhou | 2018 | Sepsis | ICESsuGX81 | 0.047 | 0.094 | 24 | >256 | >256 | >256 | >1024 | >256 | 32 | >256 | 0.25 * | AAC(6′)-Ie, APH(2″)-Ia, erm(B), tet(O), ant(6)-Ia, tet(40) | |
| GX16 | SRR8523150 | Yulin | 2010 | Sepsis | ICESsuGX81 | 0.094 | 0.064 | 16 | 0.75 | 0.75 | 0.75 | 2 | 2 | 32 | 4 | 0.38 * | tet(O), tet(40) | |
| GX39 | SRR8523137 | Yulin | 2014 | / | ICESsuGX81 | 0.064 | 0.064 | 16 | >256 | >256 | >256 | >1024 | >256 | 16 | >256 | >32 * | dfrG, erm(B), tet(O), ant(6)-Ia, tet(40), AAC(6′)-Ie-APH(2″)-Ia | |
| GX8 | SRR8523145 | Yulin | 2007 | Sepsis | ICESsuGX81 | 0.094 | 0.094 | 12 | >256 | >256 | >256 | 3 | 8 | 24 | 6 | 0.25 * | erm(B), tet(O), tet(40) | |
| GX11 | SRR8523143 | Yulin | 2008 | Sepsis | ICESsuGX81 | 0.094 | 0.094 | 24 | 0.75 | 0.75 | 0.5 | 3 | 8 | 32 | 6 | 1 * | tet(O), tet(40) | |
| GX13 | SRR8523144 | Yulin | 2008 | Meningitis | ICESsuGX81 | 0.064 | 0.094 | 24 | >256 | >256 | >256 | 4 | 6 | 32 | 4 | 0.25 * | erm(B), tet(O), tet(40) | |
| GX17 | SRR8523147 | Yulin | 2010 | Sepsis | ICESsuGX81 | 0.064 | 0.094 | 16 | 0.5 | 0.5 | 0.75 | 8 | 6 | 32 | 3 | 0.094 * | tet(O), tet(40) | |
| GX83 | SRR8523122 | Nanning | 2017 | Sepsis | ICESsuGX81 | 0.094 | 0.094 | 12 | 0.75 | 0.75 | 0.38 | 4 | 6 | 24 | 8 | 3 * | tet(O), tet(40) | |
| GX19 | SRR8523151 | Yulin | 2011 | Meningitis | ICESsuGX81 | 0.047 | 0.094 | 16 | 0.75 | 0.75 | 0.5 | 12 | 8 | 32 | 3 | 0.125 * | tet(O), tet(40) | |
| GX86 | SRR8523129 | Yulin | 2018 | Sepsis | ICESsuGX81 | 0.064 | 0.064 | 16 | >256 | >256 | >256 | >1024 | 8 | >1024 | 3 | 0.25 * | spw, erm(B), tet(O), ant(6)-Ia, tet(40) | |
| GX28 | SRR8523139 | Yulin | 2013 | Sepsis | ICESsuGX81 | 0.047 | 0.064 | 16 | >256 | >256 | >256 | >1024 | 8 | >1024 | 4 | 0.75 * | spw, erm(B), tet(O), ant(6)-Ia, tet(40) | |
| GX95 | SRR8523128 | / | 2013 | / | ICESsuGX81 | 0.047 | 0.064 | 24 | >256 | >256 | >256 | >1024 | 6 | >1024 | 2 | 0.19 * | spw, erm(B), tet(O), ant(6)-Ia, tet(40) | |
| GX51 | SRR8523120 | Yulin | 2015 | Sepsis | ICESsuGX81 | 0.047 | 0.047 | 16 | >256 | >256 | >256 | >1024 | 2 | >1024 | 8 | 0.75 * | spw, erm(B), tet(O), ant(6)-Ia, tet(40) | |
| GX64 | SRR8523119 | Yulin | 2016 | / | ICESsuGX81 | 0.094 | 0.064 | 16 | >256 | >256 | >256 | >1024 | 12 | >1024 | 6 | 0.75 * | spw, erm(B), tet(O), ant(6)-Ia, tet(40) | |
| GX84 | SRR8523116 | Guilin | 2017 | / | ICESsuGX81 | 0.064 | 0.064 | 16 | >256 | >256 | >256 | >1024 | 8 | >1024 | 3 | 0.5 * | spw, erm(B), tet(O), ant(6)-Ia, tet(40) | |
| GX97 | SRR8523126 | / | 2018 | / | ICESsuGX81 | 0.047 | 0.064 | 16 | >256 | >256 | >256 | >1024 | 4 | >1024 | 1.5 | 0.38 * | spw, erm(B), tet(O), ant(6)-Ia, tet(40) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, M.; Du, P.; Wang, J.; Lan, R.; Huang, J.; Luo, M.; Jiang, Y.; Zeng, J.; Quan, Y.; Shi, Z.; et al. Genomic Epidemiology of Streptococcus suis Sequence Type 7 Sporadic Infections in the Guangxi Zhuang Autonomous Region of China. Pathogens 2019, 8, 187. https://doi.org/10.3390/pathogens8040187
Wang M, Du P, Wang J, Lan R, Huang J, Luo M, Jiang Y, Zeng J, Quan Y, Shi Z, et al. Genomic Epidemiology of Streptococcus suis Sequence Type 7 Sporadic Infections in the Guangxi Zhuang Autonomous Region of China. Pathogens. 2019; 8(4):187. https://doi.org/10.3390/pathogens8040187
Chicago/Turabian StyleWang, Mingliu, Pengcheng Du, Jianping Wang, Ruiting Lan, Jun Huang, Ming Luo, Yan Jiang, Jun Zeng, Yi Quan, Zhaohui Shi, and et al. 2019. "Genomic Epidemiology of Streptococcus suis Sequence Type 7 Sporadic Infections in the Guangxi Zhuang Autonomous Region of China" Pathogens 8, no. 4: 187. https://doi.org/10.3390/pathogens8040187
APA StyleWang, M., Du, P., Wang, J., Lan, R., Huang, J., Luo, M., Jiang, Y., Zeng, J., Quan, Y., Shi, Z., & Zheng, H. (2019). Genomic Epidemiology of Streptococcus suis Sequence Type 7 Sporadic Infections in the Guangxi Zhuang Autonomous Region of China. Pathogens, 8(4), 187. https://doi.org/10.3390/pathogens8040187