Genomic Diversity and Hotspot Mutations in 30,983 SARS-CoV-2 Genomes: Moving Toward a Universal Vaccine for the “Confined Virus”?

 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Results
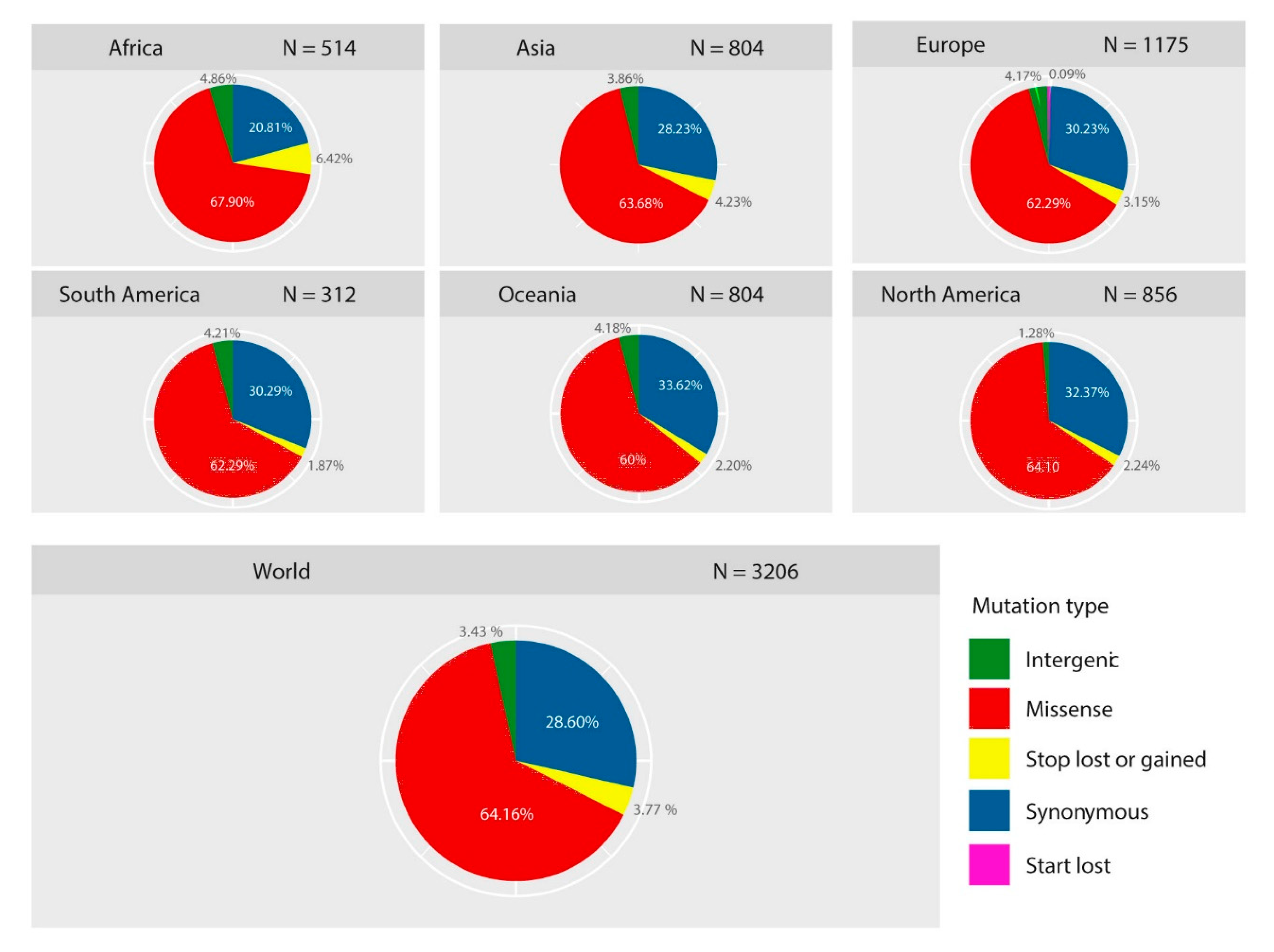
2.1. Diversity of Genetic Variants of SARS-CoV-2 in Different Geographic Areas
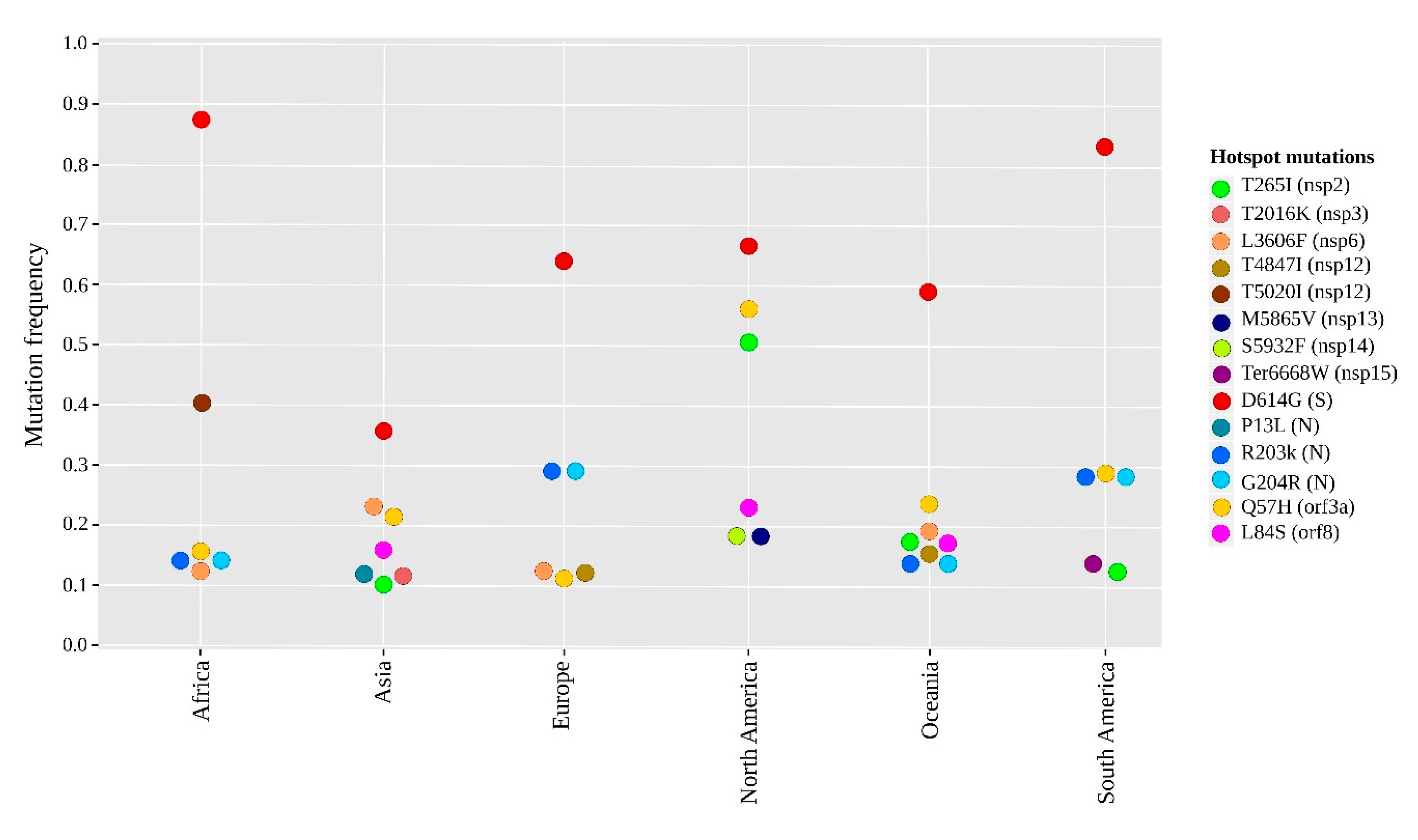
2.2. Geographical Distribution of the SARS-CoV-2 Hotspot Mutations
2.3. The Distribution of Hotspot Mutation Patterns of SARS-CoV-2 over Time
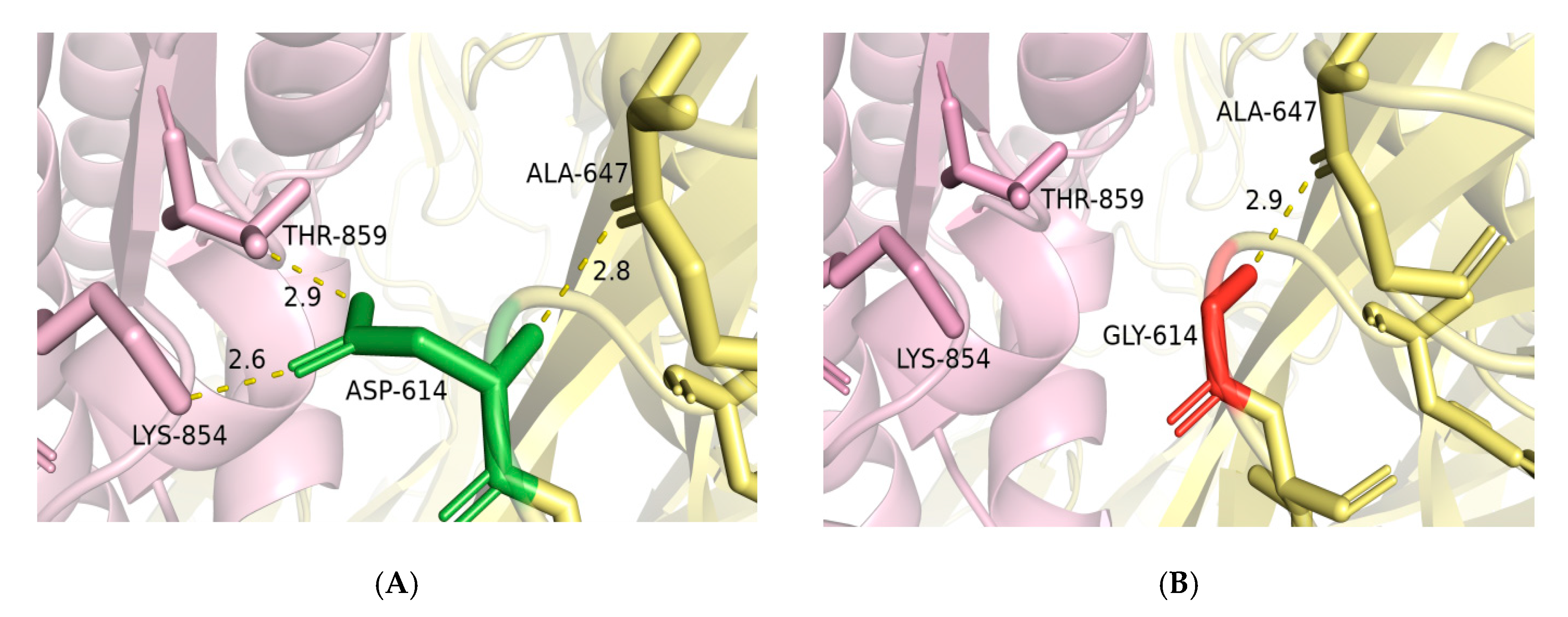
2.4. Mutagenesis of D614G and Impact of RBD Mutations on the Binding Ability of Spike to ACE2
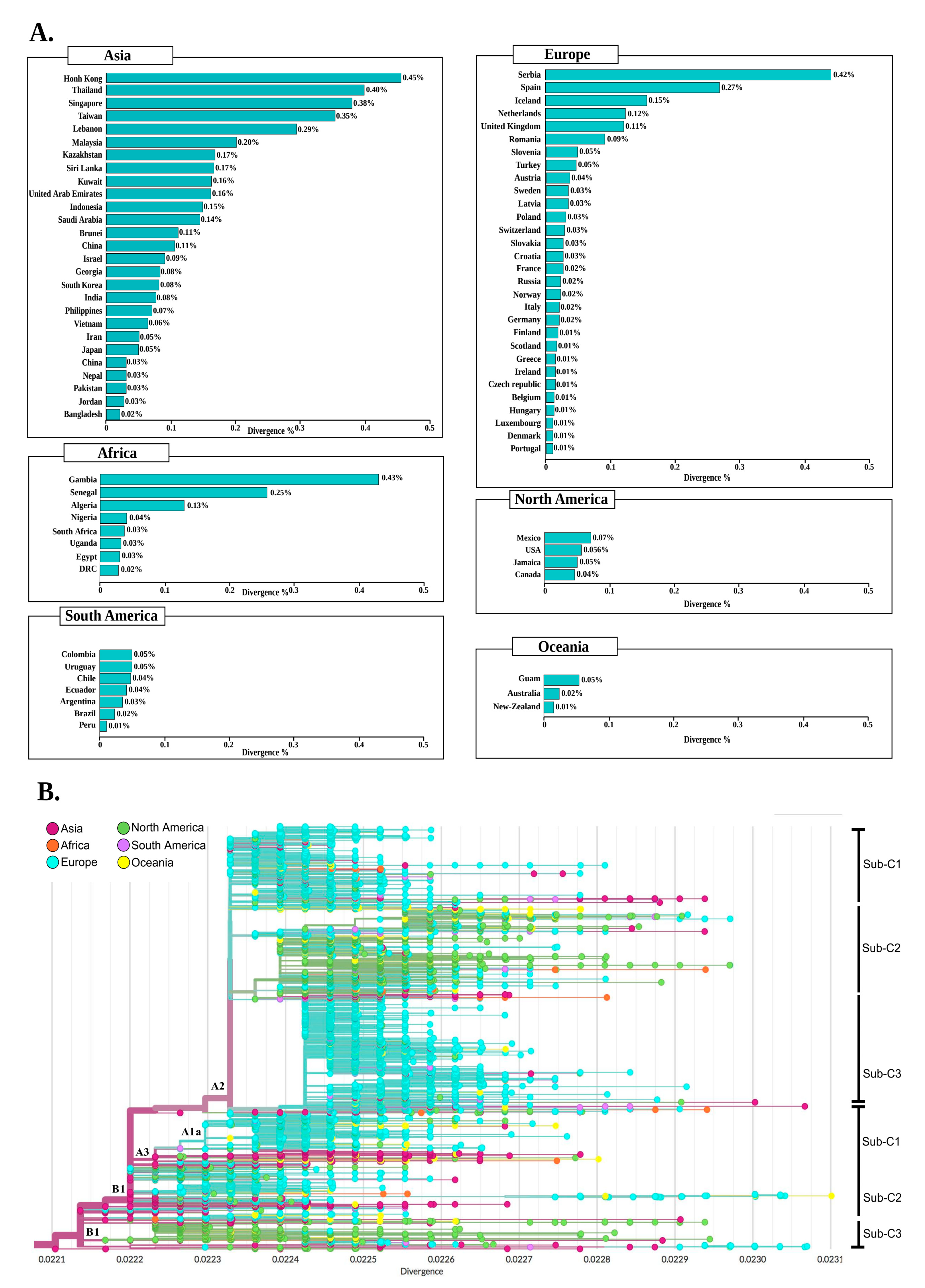
2.5. Clustering and Divergence of SARS-CoV-2 Genomes
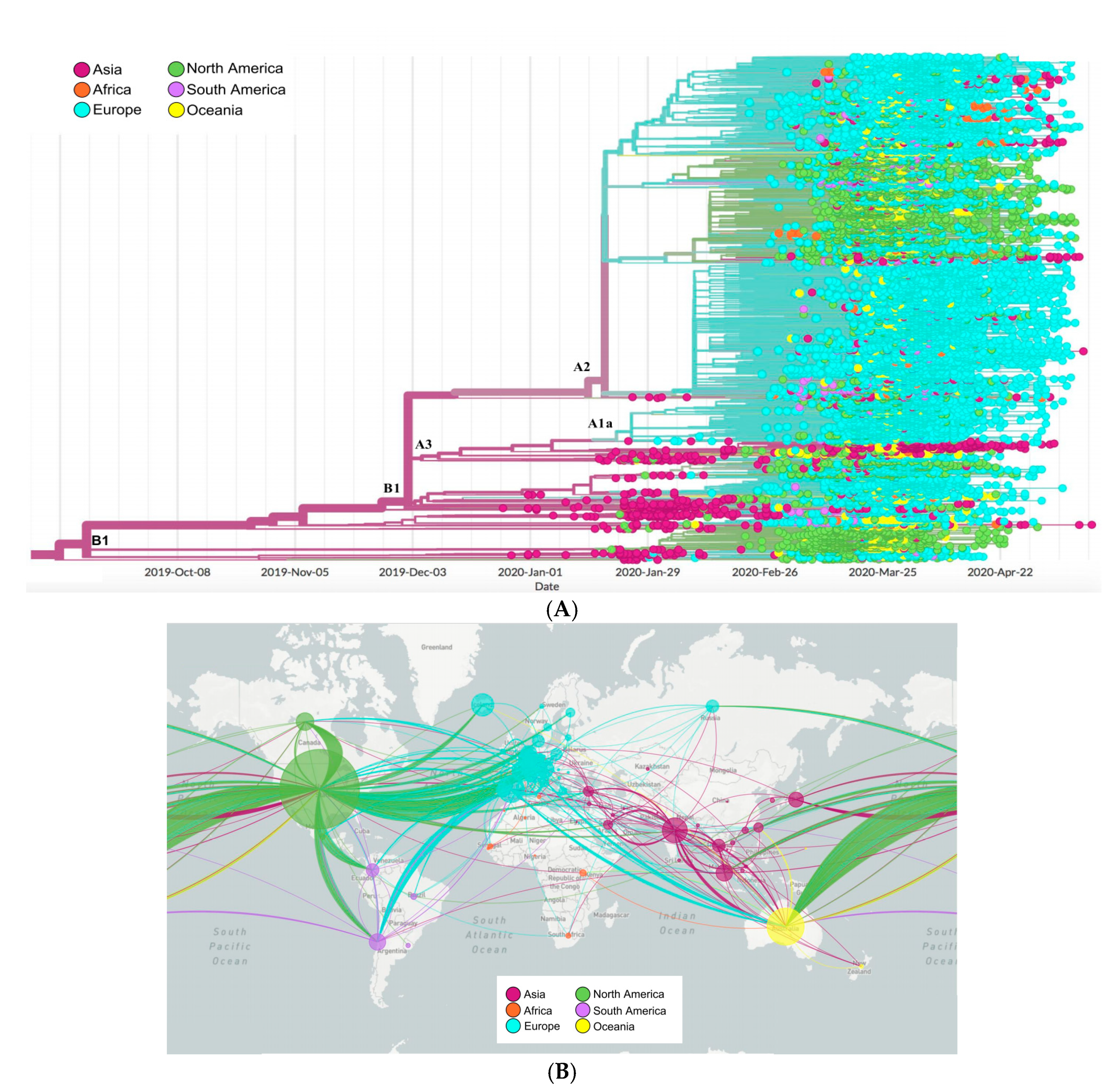
2.6. Phylogenetics and Spatio Dynamics of SARS-CoV-2
3. Discussion
4. Materials and Methods
4.1. Data Collection
4.2. Variant Calling Analysis
4.3. D614G Mutagenesis Analysis
4.4. RBD Mutations and Spike/ACE2 Binding Affinity
4.5. Clustering and Divergence Analysis
4.6. Phylogenetic and Spatio-Dynamic Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mackenzie, J.S.; Smith, D.W. COVID-19: A novel zoonotic disease caused by a coronavirus from China: What we know and what we don’t. Microbiol. Aust. 2020, 41, 45–50. [Google Scholar] [CrossRef]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Sohrabi, C.; Alsafi, Z.; O’Neill, N.; Khan, M.; Kerwan, A.; Al-Jabir, A.; Iosifidis, C.; Agha, R. World Health Organization declares global emergency: A review of the 2019 novel coronavirus (COVID-19). Int. J. Surg. 2020, 76, 71–76. [Google Scholar] [CrossRef]
- Cuevas, J.M.; Geller, R.; Garijo, R.; López-Aldeguer, J.; Sanjuán, R. Extremely High Mutation Rate of HIV-1 In Vivo. PLoS Biol. 2015, 13, e1002251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouse, B.T.; Sehrawat, S. Immunity and immunopathology to viruses: What decides the outcome? Nat. Rev. Immunol. 2010, 10, 514–526. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Peng, Y.; Huang, B.; Ding, X.; Wang, X.; Niu, P.; Meng, J.; Zhu, Z.; Zhang, Z.; Wang, J.; et al. Genome Composition and Divergence of the Novel Coronavirus (2019-nCoV) Originating in China. Cell Host Microbe 2020, 27, 325–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, Y.A. Properties of Coronavirus and SARS-CoV-2. Malays. J. Pathol. 2020, 42, 3–11. [Google Scholar] [PubMed]
- Du, L.; He, Y.; Zhou, Y.; Liu, S.; Zheng, B.-J.; Jiang, S. The spike protein of SARS-CoV--a target for vaccine and therapeutic development. Nat. Rev. Microbiol. 2009, 7, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Prado, E.; Simbaña-Rivera, K.; Gómez-Barreno, L.; Rubio-Neira, M.; Guaman, L.P.; Kyriakidis, N.C.; Muslin, C.; Jaramillo, A.M.G.; Barba-Ostria, C.; Cevallos-Robalino, D. Clinical, molecular and epidemiological characterization of the SARS-CoV2 virus and the Coronavirus disease 2019 (COVID-19), a comprehensive literature review. Diagn. Microbiol. Infect. Dis. 2020, 98, 115094. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Chen, W.; Zhou, Y.-S.; Lian, J.-Q.; Zhang, Z.; Du, P.; Gong, L.; Zhang, Y.; Cui, H.-Y.; Geng, J.-J.; et al. SARS-CoV-2 invades host cells via a novel route: CD147-spike protein. BioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Grant, O.C.; Montgomery, D.; Ito, K.; Woods, R.J. Analysis of the SARS-CoV-2 spike protein glycan shield: Implications for immune recognition. BioRxiv Prepr. Serv. Biol. 2020. [Google Scholar] [CrossRef] [Green Version]
- Amanat, F.; Krammer, F. SARS-CoV-2 Vaccines: Status Report. Immunity 2020, 52, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zeng, H.; Gu, J.; Li, H.; Zheng, L.; Zou, Q. Progress and Prospects on Vaccine Development against SARS-CoV-2. Vaccines 2020, 8, 153. [Google Scholar] [CrossRef] [Green Version]
- Tu, Y.-F.; Chien, C.-S.; Yarmishyn, A.A.; Lin, Y.-Y.; Luo, Y.-H.; Lin, Y.-T.; Lai, W.-Y.; Yang, D.-M.; Chou, S.-J.; Yang, Y.-P.; et al. A Review of SARS-CoV-2 and the Ongoing Clinical Trials. Int. J. Mol. Sci. 2020, 21, 2657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rausch, J.W.; Capoferri, A.A.; Katusiime, M.G.; Patro, S.C.; Kearney, M.F. Low genetic diversity may be an Achilles heel of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 24614–24616. [Google Scholar] [CrossRef] [PubMed]
- Abidha, C.A.; Nyiro, J.; Kamau, E.; Abdullahi, O.; Nokes, D.J.; Agoti, C.N. Transmission and evolutionary dynamics of human coronavirus OC43 strains in coastal Kenya investigated by partial spike sequence analysis, 2015–2016. Virus Evol. 2020, 6. [Google Scholar] [CrossRef]
- Koyama, T.; Weeraratne, D. Emergence of Drift Variants That May Affect COVID-19 Vaccine Development and Antibody Treatment. Pathogens 2020, 9, 324. [Google Scholar] [CrossRef]
- Lai, A.; Bergna, A.; Caucci, S.; Clementi, N.; Vicenti, I.; Dragoni, F.; Cattelan, A.M.; Menzo, S.; Pan, A.; Callegaro, A.; et al. Molecular tracing of SARS-CoV-2 in Italy in the first three months of the epidemic. MedRxiv 2020. [Google Scholar] [CrossRef]
- Islam, M.R.; Hoque, M.N.; Rahman, M.S.; Alam, A.S.M.R.U.; Akther, M.; Puspo, J.A.; Akter, S.; Sultana, M.; Crandall, K.A.; Hossain, M.A. Genome-wide analysis of SARS-CoV-2 virus strains circulating worldwide implicates heterogeneity. Sci. Rep. 2020, 10, 14004. [Google Scholar] [CrossRef]
- Chattopadhyay, S.; Weissman, S.J.; Minin, V.N.; Russo, T.A.; Dykhuizen, D.E.; Sokurenko, E.V. High frequency of hotspot mutations in core genes of Escherichia coli due to short-term positive selection. Proc. Natl. Acad. Sci. USA 2009, 106, 12412–12417. [Google Scholar] [CrossRef] [Green Version]
- Alm, E.; Broberg, E.K.; Connor, T.; Hodcroft, E.B.; Komissarov, A.B.; Maurer-Stroh, S.; Melidou, A.; Neher, R.A.; O’Toole, Á.; Pereyaslov, D.; et al. Geographical and temporal distribution of SARS-CoV-2 clades in the WHO European Region, January to June 2020. Eurosurveillance 2020, 25, 2001410. [Google Scholar] [CrossRef] [PubMed]
- Laamarti, M.; Alouane, T.; Kartti, S.; Chemao-Elfihri, M.W.; Hakmi, M.; Essabbar, A.; Laamart, M.; Hlali, H.; Allam, L.; Hafidi, N.E.L.; et al. Large scale genomic analysis of 3067 SARS-CoV-2 genomes reveals a clonal geo-distribution and a rich genetic variations of hotspots mutations. BioRxiv 2020. [Google Scholar] [CrossRef]
- Stefanelli, P.; Faggioni, G.; Lo Presti, A.; Fiore, S.; Marchi, A.; Benedetti, E.; Fabiani, C.; Anselmo, A.; Ciammaruconi, A.; Fortunato, A.; et al. Whole genome and phylogenetic analysis of two SARS-CoV-2 strains isolated in Italy in January and February 2020: Additional clues on multiple introductions and further circulation in Europe. Eurosurveillance 2020, 25, 2000305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Worobey, M.; Pekar, J.; Larsen, B.B.; Nelson, M.I.; Hill, V.; Joy, J.B.; Rambaut, A.; Suchard, M.A.; Wertheim, J.O.; Lemey, P. The emergence of SARS-CoV-2 in Europe and North America. Science 2020, eabc8169. [Google Scholar] [CrossRef]
- Sheikh, J.A.; Singh, J.; Singh, H.; Jamal, S.; Khubaib, M.; Kohli, S.; Dobrindt, U.; Rahman, S.A.; Ehtesham, N.Z.; Hasnain, S.E. Emerging genetic diversity among clinical isolates of SARS-CoV-2: Lessons for today. Infect. Genet. Evol. 2020, 84, 104330. [Google Scholar] [CrossRef]
- Katsidzira, L.; Gwaunza, L.; Hakim, J.G. The Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Epidemic in Zimbabwe: Quo Vadis? Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Massinga Loembé, M.; Tshangela, A.; Salyer, S.J.; Varma, J.K.; Ouma, A.E.O.; Nkengasong, J.N. COVID-19 in Africa: The spread and response. Nat. Med. 2020, 26, 999–1003. [Google Scholar] [CrossRef]
- Poterico, J.A.; Mestanza, O. Genetic variants and source of introduction of SARS-CoV-2 in South America. J. Med. Virol. 2020, 92, 2139–2145. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Foley, B.; Giorgi, E.E.; Bhattacharya, T.; Parker, M.D.; et al. Spike mutation pipeline reveals the emergence of a more transmissible form of SARS-CoV-2. BioRxiv 2020. [Google Scholar] [CrossRef]
- Hu, J.; He, C.-L.; Gao, Q.-Z.; Zhang, G.-J.; Cao, X.-X.; Long, Q.-X.; Deng, H.-J.; Huang, L.-Y.; Chen, J.; Wang, K.; et al. The D614G mutation of SARS-CoV-2 spike protein enhances viral infectivity and decreases neutralization sensitivity to individual convalescent sera. BioRxiv 2020. [Google Scholar] [CrossRef]
- Thanh Le, T.; Andreadakis, Z.; Kumar, A.; Gómez Román, R.; Tollefsen, S.; Saville, M.; Mayhew, S. The COVID-19 vaccine development landscape. Nat. Rev. Drug Discov. 2020, 19, 305–306. [Google Scholar] [CrossRef] [PubMed]
- Isabel, S.; Graña-Miraglia, L.; Gutierrez, J.M.; Bundalovic-Torma, C.; Groves, H.E.; Isabel, M.R.; Eshaghi, A.; Patel, S.N.; Gubbay, J.B.; Poutanen, T.; et al. Evolutionary and structural analyses of SARS-CoV-2 D614G spike protein mutation now documented worldwide. Sci. Rep. 2020, 10, 14031. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Gu, W.; Federman, S.; du Plessis, L.; Pybus, O.G.; Faria, N.R.; Wang, C.; Yu, G.; Bushnell, B.; Pan, C.-Y.; et al. Genomic surveillance reveals multiple introductions of SARS-CoV-2 into Northern California. Science 2020, 369, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Schulkins, A.; Chen, C.-N.; Deshayes, K.; Kenney, J.S. The SARS-CoV-2 Spike Protein D614G Mutation Shows Increasing Dominance and May Confer a Structural Advantage to the Furin Cleavage Domain. Preprints 2020. [Google Scholar] [CrossRef]
- Xiong, X.; Qu, K.; Ciazynska, K.A.; Hosmillo, M.; Carter, A.P.; Ebrahimi, S.; Ke, Z.; Scheres, S.H.W.; Bergamaschi, L.; Grice, G.L.; et al. A thermostable, closed SARS-CoV-2 spike protein trimer. Nat. Struct. Mol. Biol. 2020. [Google Scholar] [CrossRef]
- Noy-Porat, T.; Makdasi, E.; Alcalay, R.; Mechaly, A.; Levy, Y.; Bercovich-Kinori, A.; Zauberman, A.; Tamir, H.; Yahalom-Ronen, Y.; Israeli, M.a.; et al. A panel of human neutralizing mAbs targeting SARS-CoV-2 spike at multiple epitopes. Nat. Commun. 2020, 11, 4303. [Google Scholar] [CrossRef]
- Grubaugh, N.D.; Hanage, W.P.; Rasmussen, A.L. Making Sense of Mutation: What D614G Means for the COVID-19 Pandemic Remains Unclear. Cell 2020, 182, 794–795. [Google Scholar] [CrossRef]
- Tai, W.; He, L.; Zhang, X.; Pu, J.; Voronin, D.; Jiang, S.; Zhou, Y.; Du, L. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: Implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunol. 2020, 17, 613–620. [Google Scholar] [CrossRef] [Green Version]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [Green Version]
- Wong, A.H.M.; Tomlinson, A.C.A.; Zhou, D.; Satkunarajah, M.; Chen, K.; Sharon, C.; Desforges, M.; Talbot, P.J.; Rini, J.M. Receptor-binding loops in alphacoronavirus adaptation and evolution. Nat. Commun. 2017, 8, 1735. [Google Scholar] [CrossRef]
- Rockx, B.; Donaldson, E.; Frieman, M.; Sheahan, T.; Corti, D.; Lanzavecchia, A.; Baric, R.S. Escape from human monoclonal antibody neutralization affects in vitro and in vivo fitness of severe acute respiratory syndrome coronavirus. J. Infect. Dis. 2010, 201, 946–955. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, R.; Wang, M.; Wei, G.-W. Mutations Strengthened SARS-CoV-2 Infectivity. J. Mol. Biol. 2020, 432, 5212–5226. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Giorgi, E.E.; Marichannegowda, M.H.; Foley, B.; Xiao, C.; Kong, X.-P.; Chen, Y.; Gnanakaran, S.; Korber, B.; Gao, F. Emergence of SARS-CoV-2 through recombination and strong purifying selection. Sci. Adv. 2020, 6, eabb9153. [Google Scholar] [CrossRef]
- Ou, J.; Zhou, Z.; Dai, R.; Zhang, J.; Lan, W.; Zhao, S.; Wu, J.; Seto, D.; Cui, L.; Zhang, G.; et al. Emergence of RBD mutations in circulating SARS-CoV-2 strains enhancing the structural stability and human ACE2 receptor affinity of the spike protein. BioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Uludağ, H.; Parent, K.; Aliabadi, H.M.; Haddadi, A. Prospects for RNAi Therapy of COVID-19. Front. Bioeng. Biotechnol. 2020, 8. [Google Scholar] [CrossRef]
- Liu, C.; Zhou, Q.; Li, Y.; Garner, L.V.; Watkins, S.P.; Carter, L.J.; Smoot, J.; Gregg, A.C.; Daniels, A.D.; Jervey, S.; et al. Research and Development on Therapeutic Agents and Vaccines for COVID-19 and Related Human Coronavirus Diseases. ACS Cent. Sci. 2020, 6, 315–331. [Google Scholar] [CrossRef]
- Ghosh, S.; Firdous, S.M.; Nath, A. siRNA could be a potential therapy for COVID-19. EXCLI J. 2020, 19, 528–531. [Google Scholar] [CrossRef]
- Shi, Y.; Yang, D.H.; Xiong, J.; Jia, J.; Huang, B.; Jin, Y.X. Inhibition of genes expression of SARS coronavirus by synthetic small interfering RNAs. Cell Res. 2005, 15, 193–200. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Zhang, Y.; Fu, L.; Yu, C.; Li, X.; Li, Y.; Zhang, X.; Rong, Z.; Wang, Y.; Ning, H.; et al. siRNA targeting the leader sequence of SARS-CoV inhibits virus replication. Gene Ther. 2005, 12, 751–761. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.J.; Huang, H.W.; Liu, C.Y.; Hong, C.F.; Chan, Y.L. Inhibition of SARS-CoV replication by siRNA. Antivir. Res. 2005, 65, 45–48. [Google Scholar] [CrossRef]
- Hodgson, J. The pandemic pipeline. Nat. Biotechnol. 2020, 38, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Feng, P.; Liu, K.; Wu, M.; Lin, H. Computational Identification of Small Interfering RNA Targets in SARS-CoV-2. Virol. Sin. 2020, 35, 359–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data—From vision to reality. Eurosurveillance 2017, 22, 30494. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Kiyotani, K.; Toyoshima, Y.; Nemoto, K.; Nakamura, Y. Bioinformatic prediction of potential T cell epitopes for SARS-Cov-2. J. Hum. Genet. 2020, 65, 569–575. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Weng, G.; Wang, E.; Wang, Z.; Liu, H.; Zhu, F.; Li, D.; Hou, T. HawkDock: A web server to predict and analyze the protein-protein complex based on computational docking and MM/GBSA. Nucleic Acids Res. 2019, 47, W322–W330. [Google Scholar] [CrossRef]
- Levandowsky, M.; Winter, D. Distance between Sets. Nature 1971, 234, 34–35. [Google Scholar] [CrossRef]
- Ukkonen, E. Approximate string-matching with q-grams and maximal matches. Theor. Comput. Sci. 1992, 92, 191–211. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Sagulenko, P.; Puller, V.; Neher, R.A. TreeTime: Maximum-likelihood phylodynamic analysis. Virus Evol. 2018, 4. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutations | ΔGBind (kcal/mol) | ΔΔG 1 (kcal/mol) | Effect on Spike/ACE2 |
|---|---|---|---|
| V367F | −62.47 | 3.53 | Potentially decreased binding affinity |
| S477N | −62.69 | 3.31 | |
| R408I | −62.8 | 3.2 | |
| V483A | −63.85 | 2.15 | |
| A522S | −64.03 | 1.97 | |
| G339D | −64.08 | 1.92 | |
| N354D | −64.39 | 1.61 | |
| K356N | −64.81 | 1.19 | |
| H519Q | −64.84 | 1.16 | |
| Wild Type | −66 | 0 | Wild-type MMGBSA value |
| N440K | −67.88 | −1.88 | Potentially increased binding affinity |
| N450K | −67.88 | −1.88 | |
| D364Y | −68.24 | −2.24 | |
| S477R | −69.86 | −3.86 |
| Cluster | Sub-Cluster | Countries | Jaccard Distance | Geographic Areas |
|---|---|---|---|---|
| Cluster 1 | SC-1 | Brunei, Guam | 0.22 | Asia, Oceania |
| Cluster 2 | SC-2 | Kazakhstan, Georgia | 0.22 | Asia |
| Cluster 2 | SC-3 | Nigeria, Serbia, Croatia, Ireland, Peru | 0.26 | Africa, Europe, South America |
| Cluster 2 | SC-4 | Vietnam, Jordan | 0.27 | Asia |
| Cluster 2 | SC-5 | Sri Lanka, Kuwait | 0.30 | Asia |
| Cluster 2 | SC-6 | Greece, Portugal | 0.32 | Europe |
| Cluster 2 | SC-7 | Singapore, Thailand | 0.35 | Asia |
| Cluster 2 | SC-8 | Finland, Poland | 0.35 | Europe |
| Cluster 2 | SC-9 | Slovenia, Jamaica | 0.35 | Europe, North America |
| Cluster 2 | SC-10 | Denmark, Iceland | 0.36 | Europe |
| Cluster 2 | SC-11 | Germany, Russia | 0.36 | Europe |
| Cluster 1 | SC-12 | Hungary, Latvia | 0.41 | Europe |
| Cluster 1 | SC-13 | Chile, Brazil | 0.43 | South America |
| Cluster 1 | SC-14 | Iran, Pakistan | 0.43 | Asia |
| Cluster 2 | SC-15 | Netherland, Belgium, Austria | 0.49 | Europe |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alouane, T.; Laamarti, M.; Essabbar, A.; Hakmi, M.; Bouricha, E.M.; Chemao-Elfihri, M.W.; Kartti, S.; Boumajdi, N.; Bendani, H.; Laamarti, R.; et al. Genomic Diversity and Hotspot Mutations in 30,983 SARS-CoV-2 Genomes: Moving Toward a Universal Vaccine for the “Confined Virus”? Pathogens 2020, 9, 829. https://doi.org/10.3390/pathogens9100829
Alouane T, Laamarti M, Essabbar A, Hakmi M, Bouricha EM, Chemao-Elfihri MW, Kartti S, Boumajdi N, Bendani H, Laamarti R, et al. Genomic Diversity and Hotspot Mutations in 30,983 SARS-CoV-2 Genomes: Moving Toward a Universal Vaccine for the “Confined Virus”? Pathogens. 2020; 9(10):829. https://doi.org/10.3390/pathogens9100829
Chicago/Turabian StyleAlouane, Tarek, Meriem Laamarti, Abdelomunim Essabbar, Mohammed Hakmi, El Mehdi Bouricha, M. W. Chemao-Elfihri, Souad Kartti, Nasma Boumajdi, Houda Bendani, Rokia Laamarti, and et al. 2020. "Genomic Diversity and Hotspot Mutations in 30,983 SARS-CoV-2 Genomes: Moving Toward a Universal Vaccine for the “Confined Virus”?" Pathogens 9, no. 10: 829. https://doi.org/10.3390/pathogens9100829
APA StyleAlouane, T., Laamarti, M., Essabbar, A., Hakmi, M., Bouricha, E. M., Chemao-Elfihri, M. W., Kartti, S., Boumajdi, N., Bendani, H., Laamarti, R., Ghrifi, F., Allam, L., Aanniz, T., Ouadghiri, M., El Hafidi, N., El Jaoudi, R., Benrahma, H., Attar, J. E., Mentag, R., ... Ibrahimi, A. (2020). Genomic Diversity and Hotspot Mutations in 30,983 SARS-CoV-2 Genomes: Moving Toward a Universal Vaccine for the “Confined Virus”? Pathogens, 9(10), 829. https://doi.org/10.3390/pathogens9100829