
Tillage Practices and Residue Management Manipulate Soil Bacterial and Fungal Communities and Networks in Maize Agroecosystems

Abstract
:1. Introduction
2. Materials and Methods
2.1. Field Experiment Design
2.2. Soil Sampling and Physiochemical Variables
2.3. Miseq Sequencing
2.4. Bioinformatics Analysis
2.5. Data Analysis
3. Results
3.1. Bacterial and Fungal Alpha-Diversity
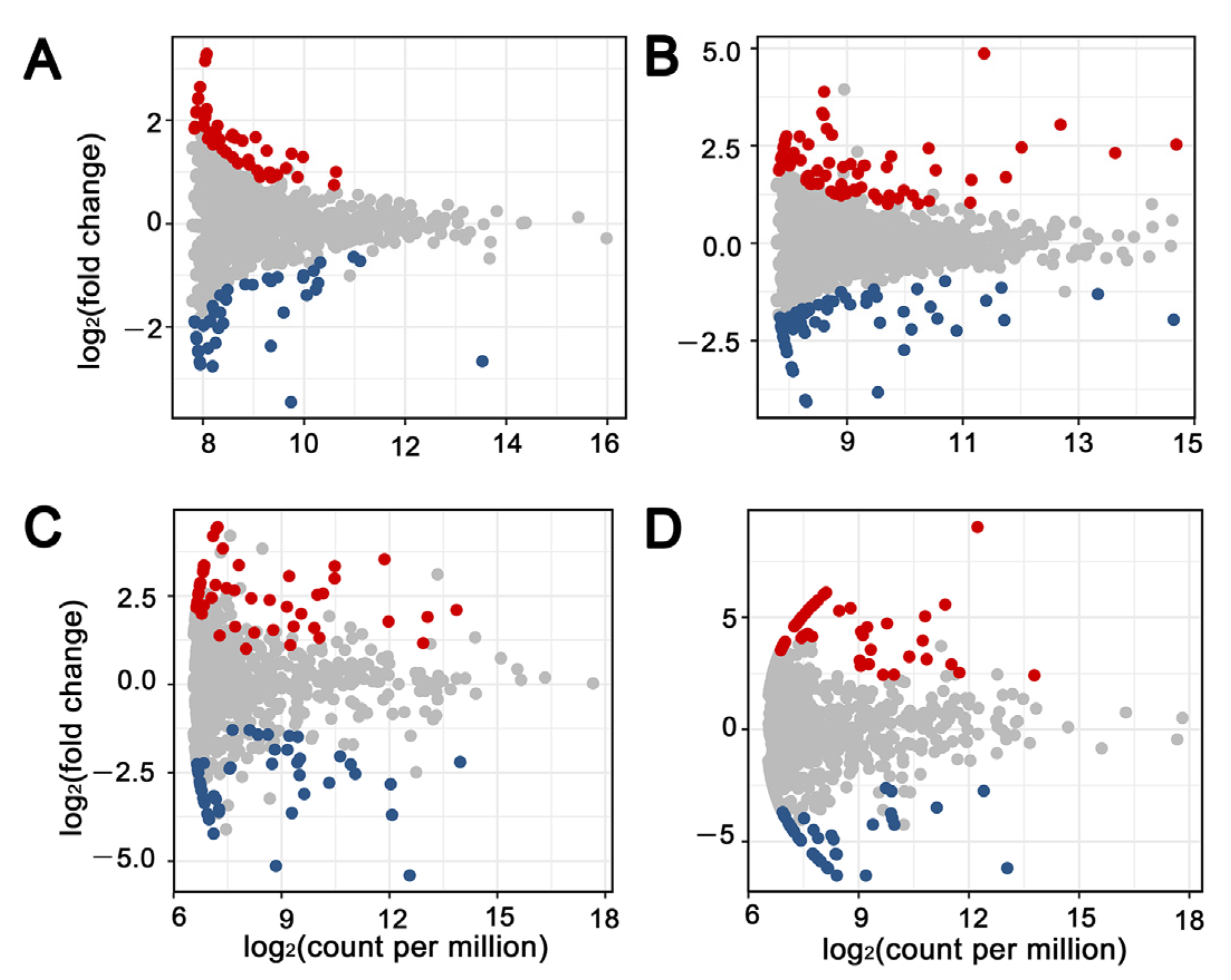
3.2. Bacterial and Fungal Community Composition
3.3. Co-Occurrence Networks
3.4. Predicted Bacterial and Fungal Functions
4. Discussion
4.1. Effects of Tillage Practices on the Soil Bacterial and Fungal Communities
4.2. Effects of Residue Management on Soil Bacterial and Fungal Communities
4.3. Bacterial and Fungal Co-Occurrence Networks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- FAO. Status of the World’s Soil Resources. 2015. Available online: http://www.fao.org/documents/card/en/c/c6814873-efc3-41db-b7d3-2081a10ede50 (accessed on 11 April 2022).
- Tilman, D. Global environmental impacts of agricultural expansion: The need for sustainable and efficient practices. Proc. Natl. Acad. Sci. USA 1999, 96, 5995–6000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartman, K.; van der Heijden, M.G.A.; Wittwer, R.A.; Banerjee, S.; Walser, J.-C.; Schlaeppi, K. Cropping practices manipulate abundance patterns of root and soil microbiome members paving the way to smart farming. Microbiome 2018, 6, 14. [Google Scholar] [CrossRef] [PubMed]
- Matson, P.A.; Parton, W.J.; Power, A.G.; Swift, M.J. Agricultural Intensification and Ecosystem Properties. Science 1997, 277, 504–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hobbs, P.R.; Sayre, K.; Gupta, R. The role of conservation agriculture in sustainable agriculture. Philos. Trans. R. Soc. 2008, 363, 543–555. [Google Scholar] [CrossRef]
- Sengupta, A.; Dick, W.A. Bacterial community diversity in soil under two tillage practices as determined by pyrosequencing. Microb. Ecol. 2015, 70, 853–859. [Google Scholar] [CrossRef]
- Basic, F.; Kisic, I.; Mesic, M.; Nestroy, O.; Butorac, A. Tillage and crop management effects on soil erosion in central Croatia. Soil Tillage Res. 2004, 78, 197–206. [Google Scholar] [CrossRef]
- Gu, S.; Guo, X.; Cai, Y.; Zhang, Z.; Wu, S.; Li, X.; Zhang, H.; Yang, W. Residue management alters microbial diversity and activity without affecting their community composition in black soil, Northeast China. Peer J. 2018, 6, e5754. [Google Scholar] [CrossRef] [Green Version]
- Sun, R.B.; Li, W.Y.; Dong, W.X.; Tian, Y.P.; Hu, C.S.; Liu, B.B. Tillage changes vertical distribution of soil bacterial and fungal communities. Front. Microbiol. 2018, 9, 699. [Google Scholar] [CrossRef]
- Lehtinen, T.; Schlatter, N.; Baumgarten, A.; Bechini, L.; Krüger, J.; Grignani, C.; Zavattaro, L.; Costamagna, C.; Spiegel, H. Effect of crop residue incorporation on soil organic carbon and greenhouse gas emissions in European agricultural soils. Soil Use Manag. 2014, 30, 524–538. [Google Scholar] [CrossRef]
- Malhi, S.; Nyborg, M.; Goddard, T.; Puurveen, D. Long-term tillage, straw and N rate effects on quantity and quality of organic C and N in a Gray Luvisol soil. Nutr. Cycl. Agroecosyst. 2011, 90, 21–22. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Liu, B.Y.; Liu, S.L.; Qi, J.Y.; Wang, X.; Pu, C.; Li, S.S.; Zhang, X.Z.; Yang, X.G.; Lal, R.; et al. Sustaining crop production in China’s cropland by crop residue retention: A meta-analysis. Land Degrad. Dev. 2019, 31, 694–709. [Google Scholar] [CrossRef]
- Hadas, A.; Kautsky, L.; Goek, M.; Erman Kara, E. Rates of decomposition of plant residues and available nitrogen in soil, related to residue composition through simulation of carbon and nitrogen turnover. Soil Biol. Biochem. 2004, 36, 255–266. [Google Scholar] [CrossRef]
- Ye, R.W.; Thomas, S.M. Microbial nitrogen cycles: Physiology, genomics and applications. Curr. Opin. Microbiol. 2001, 4, 307–312. [Google Scholar] [CrossRef]
- McKinley, V.L. Effects of Land Use and Restoration on Soil Microbial Communities. In Understanding Terrestrial Microbial Communities; Hurst, C.J., Ed.; Springer International Publishing: Cham, Switzerland, 2019; pp. 173–242. [Google Scholar]
- Guo, L.; Zheng, S.; Cao, C.; Li, C. Tillage practices and straw-returning methods affect topsoil bacterial community and organic C under a rice-wheat cropping system in central China. Sci. Rep. 2016, 6, 33155. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.R.; Blair, P.L.; Boyd, C.; Cody, B.; Hazel, A.; Hedrick, A.; Kathuria, H.; Khurana, P.; Kramer, B.; Muterspaw, K.; et al. Microbial community responses to soil tillage and crop rotation in a corn/soybean agroecosystem. Ecol. Evol. 2016, 6, 8075–8084. [Google Scholar] [CrossRef]
- Schmidt, R.; Mitchell, J.; Scow, K. Cover cropping and no-till increase diversity and symbiotroph:saprotroph ratios of soil fungal communities. Soil Biol. Biochem. 2019, 129, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Bu, R.; Ren, T.; Lei, M.; Liu, B.; Li, X.; Cong, R.; Zhang, Y.; Lu, J. Tillage and straw-returning practices effect on soil dissolved organic matter, aggregate fraction and bacteria community under rice-rice-rapeseed rotation system. Agric. Ecosyst. Environ. 2020, 287, 106681. [Google Scholar] [CrossRef]
- Zuber, S.M.; Villamil, M.B. Meta-analysis approach to assess effect of tillage on microbial biomass and enzyme activities. Soil Biol. Biochem. 2016, 97, 176–187. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, C.; Tu, C.; Hoyt, G.D.; DeForest, J.L.; Hu, S. Long-term no-tillage and organic input management enhanced the diversity and stability of soil microbial community. Sci. Total Environ. 2017, 609, 341–347. [Google Scholar] [CrossRef]
- Wang, H.; Li, X.; Li, X.; Wang, J.; Li, X.; Guo, Q.; Yu, Z.; Yang, T.; Zhang, H. Long-term no-tillage and different residue amounts alter soil microbial community composition and increase the risk of maize root rot in northeast China. Soil Tillage Res. 2020, 196, 104452. [Google Scholar] [CrossRef]
- Babujia, L.C.; Hungria, M.; Franchini, J.C.; Brookes, P.C. Microbial biomass and activity at various soil depths in a Brazilian oxisol after two decades of no-tillage and conventional tillage. Soil Biol. Biochem. 2010, 42, 2174–2181. [Google Scholar] [CrossRef]
- Pastorelli, R.; Vignozzi, N.; Landi, S.; Piccolo, R.; Orsini, R.; Seddaiu, G.; Roggero, P.P.; Pagliai, M. Consequences on macroporosity and bacterial diversity of adopting a no-tillage farming system in a clayish soil of Central Italy. Soil Biol. Biochem. 2013, 66, 78–93. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, M.; Tang, L.; Che, R.; Chen, H.; Blumfield, T.; Boyd, S.; Nouansyvong, M.; Xu, Z. Long-Term Harvest Residue Retention Could Decrease Soil Bacterial Diversities Probably Due to Favouring Oligotrophic Lineages. Microb. Ecol. 2018, 76, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Ye, G.; Lin, Y.; Liu, D.; Chen, Z.; Luo, J.; Bolan, N.; Fan, J.; Ding, W. Long-term application of manure over plant residues mitigates acidification, builds soil organic carbon and shifts prokaryotic diversity in acidic Ultisols. Appl. Soil Ecol. 2019, 133, 24–33. [Google Scholar] [CrossRef]
- Allison, S.D.; Martiny, J.B.H. Resistance, resilience, and redundancy in microbial communities. Proc. Natl. Acad. Sci. USA 2008, 105, 11512–11519. [Google Scholar] [CrossRef] [Green Version]
- Gao, C.; Montoya, L.; Xu, L.; Madera, M.; Hollingsworth, J.; Purdom, E.; Singan, V.; Vogel, J.; Hutmacher, R.B.; Dahlberg, J.A.; et al. Fungal community assembly in drought-stressed sorghum shows stochasticity, selection, and universal ecological dynamics. Nat. Commun. 2020, 11, 34–47. [Google Scholar] [CrossRef] [Green Version]
- Mora-Gómez, J.; Elosegi, A.; Duarte, S.; Cássio, F.; Pascoal, C.; Romaní, A.M. Differences in the sensitivity of fungi and bacteria to season and invertebrates affect leaf litter decomposition in a Mediterranean stream. FEMS Microbiol. Ecol. 2016, 92, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Wagg, C.; Schlaeppi, K.; Banerjee, S.; Kuramae, E.E.; van der Heijden, M.G.A. Fungal-bacterial diversity and microbiome complexity predict ecosystem functioning. Nat. Commun. 2019, 10, 4841. [Google Scholar] [CrossRef]
- Deng, Y.; Jiang, Y.H.; Yang, Y.; He, Z.; Luo, F.; Zhou, J. Molecular ecological network analyses. BMC Bioinform. 2012, 13, 113. [Google Scholar] [CrossRef] [Green Version]
- Ling, N.; Zhu, C.; Xue, C.; Chen, H.; Duan, Y.H.; Peng, C.; Guo, S.W.; Shen, Q.R. Insight into how organic amendments can shape the soil microbiome in long-term field experiments as revealed by network analysis. Soil Biol. Biochem. 2016, 99, 137–149. [Google Scholar] [CrossRef]
- Yang, W.; Guan, Y.; Zhai, C.; Wang, T.; Shi, D.; Chen, J.; Sun, W.; Gu, S. Response of fungal communities and co-occurrence network patterns to compost amendment in black soil of Northeast China. Front. Microbiol. 2019, 10, 1562. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Yang, Z.; Guan, Y.; Zhai, C.; Shi, D.; Chen, J.; Wang, T.; Gu, S. Dose-dependent effect of compost amendment on soil bacterial community composition and co-occurrence network patterns in soybean agroecosystem. Arch. Agron. Soil Sci. 2019, 66, 1027–1041. [Google Scholar] [CrossRef]
- Yu, Y.; Wu, M.; Petropoulos, E.; Zhang, J.; Nie, J.; Liao, Y.; Li, Z.; Lin, X.; Feng, Y. Responses of paddy soil bacterial community assembly to different long-term fertilizations in southeast China. Sci. Total Environ. 2019, 656, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Walder, F.; Büchi, L.; Meyer, M.; Held, A.Y.; Gattinger, A.; Keller, T.; Charles, R.; van der Heijden, M.G.A. Agricultural intensification reduces microbial network complexity and the abundance of keystone taxa in roots. ISME J. 2019, 13, 1722–1736. [Google Scholar] [CrossRef] [Green Version]
- Siyu, G.; Shuai, W.; Yupeng, G.; Cheng, Z.; Zehui, Z.; Ayodeji, B.; Xingjun, G.; Wei, Y. Arbuscular mycorrhizal fungal community was affected by tillage practices rather than residue management in black soil of northeast China. Soil Tillage Res. 2020, 198, 104552. [Google Scholar]
- Baker, G.C.; Smith, J.J.; Cowan, D.A. Review and re-analysis of domain-specific 16S primers. J. Microbiol. Methods 2003, 55, 541–555. [Google Scholar] [CrossRef] [Green Version]
- Gardes, M.; Bruns, T.D. ITS primers with enhanced specificity for basidiomycetes-application to the identification of mycorrhizae and rusts. Mol. Ecol. 1993, 2, 113–118. [Google Scholar] [CrossRef]
- Ihrmark, K.; Bodeker, I.T.M.; Cruz-Martinez, K.; Friberg, H.; Kubartova, A.; Schenck, J.; Strid, Y.; Stenlid, J.; Brandstrom-Durling, M.; Clemmensen, K.E.; et al. New primers to amplify the fungal ITS2 region—Evaluation by 454-sequencing of artificial and natural communities. FEMS Microbiol. Ecol. 2012, 82, 666–677. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [Green Version]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012, 6, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, R.H.; Tedersoo, L.; Ryberg, M.; Kristiansson, E.; Hartmann, M.; Unterseher, M.; Porter, T.M.; Bengtsson-Palme, J.; Walker, D.M.; de Sousa, F.; et al. A comprehensive, automatically updated fungal its sequence dataset for reference-based chimera control in environmental sequencing efforts. Microbes Environ. 2015, 30, 145–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Wagner, H. Vegan: Community Ecology Package Version 2.0-6. 2013. Available online: http://CRAN.R-project.org/package=vegan (accessed on 11 February 2013).
- Arbuckle, J.L. IBM SPSS Amos 20 User’s Guide; IBM Corporation: New York, NY, USA, 2011. [Google Scholar]
- Hooper, D.; Coughlan, J.; Mullen, M.R. Structural equation modeling: Guidelines for determining model fit. Electron. J. Bus. Res. Methods 2008, 6, 53–60. [Google Scholar]
- Goslee, S.C.; Urban, D.L. The ecodist package for dissimilarity-based analysis of ecological data. J. Stat. Softw. 2007, 22, 1–19. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and disperation for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550–570. [Google Scholar] [CrossRef] [Green Version]
- Revelle, W. Psych: Procedures for Psychological, Psychometric, and Personality Research. R Package Version 1.5.1. 2015. Available online: http://CRAN.R-project.org/package=psych (accessed on 1 January 2015).
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Widder, S.; Besemer, K.; Singer, G.A.; Ceola, S.; Bertuzzo, E.; Quince, C. Fluvial network organization imprints on microbial co-occurrence networks. Proc. Natl. Acad. Sci. USA 2014, 111, 12799–12804. [Google Scholar] [CrossRef] [Green Version]
- Csardi, G.; Nepusz, T. The igraph software package for complex network research. InterJ. Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.V.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Nguyen, N.H.; Song, Z.W.; Bates, S.T.; Branco, S.; Tedersoo, L.; Menke, J.; Schilling, J.S.; Kennedy, P.G. FUNGuild: An open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 2016, 20, 241–248. [Google Scholar] [CrossRef]
- Tedersoo, L.; Bahram, M.; Põlme, S.; Kõljalg, U.; Yorou, N.S.; Wijesundera, R.; Ruiz, L.V.; Vasco-Palacios, A.M.; Thu, P.Q.; Suija, A.; et al. Fungal biogeography. Global diversity and geography of soil fungi. Science 2014, 346, 1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Carvalhais, L.C.; Rincon-Florez, V.; Crawford, M.; Dang, Y.P.; Dennis, P.G.; Schenk, P.M. One-time strategic tillage does not cause major impacts on soil microbial properties in a no-till Calcisol. Soil Tillage Res. 2016, 158, 91–99. [Google Scholar] [CrossRef]
- Tyler, H.L. Bacterial community composition under long-term reduced tillage and no till management. J. Appl. Microbiol. 2019, 126, 1797–1807. [Google Scholar] [CrossRef]
- Rousk, J.; Baath, E.; Brookes, P.C.; Lauber, C.L.; Lozupone, C.; Caporaso, J.G.; Knight, R.; Fierer, N. Soil bacterial and fungal communities across a pH gradient in an arable soil. ISME J. 2010, 4, 1340–1351. [Google Scholar] [CrossRef]
- Rahman, M.H.; Okubo, A.; Sugiyama, S.; Mayland, H.F. Physical, chemical and microbiological properties of an Andisol as related to land use and tillage practice. Soil Tillage Res. 2008, 101, 10–19. [Google Scholar] [CrossRef]
- Essel, E.; Xie, J.; Deng, C.; Peng, Z.; Wang, J.; Shen, J.; Xie, J.; Coulter, J.A.; Li, L. Bacterial and fungal diversity in rhizosphere and bulk soil under different long-term tillage and cereal/legume rotation. Soil Tillage Res. 2019, 194, 104302. [Google Scholar] [CrossRef]
- Khan, A.R. Influence of tillage on soil aeration. Soil Tillage Res. 1986, 8, 359. [Google Scholar] [CrossRef]
- Six, J.; Elliott, E.T.; Paustian, K. Soil macroaggregate turnover and microaggregate formation: A mechanism for C sequestration under no-tillage agriculture. Soil Biol. Biochem. 2000, 32, 2099–2103. [Google Scholar] [CrossRef]
- Piazza, G.; Ercoli, L.; Nuti, M.; Pellegrino, E. Interaction between conservation tillage and nitrogen fertilization shapes prokaryotic and fungal diversity at different soil depths: Evidence from a 23-year field experiment in the Mediterranean area. Front. Microbiol. 2019, 10, 2047. [Google Scholar] [CrossRef] [Green Version]
- Lacerda, G.V.; Noronha, M.F.; Cabral, L.; Delforno, T.P.; de Sousa, S.T.P.; Fernandes, P.I.; Melo, T.S.; Oliveira, V.M. Land use and seasonal effects on the soil microbiome of a Brazilian dry forest. Front. Microbiol. 2019, 10, 648–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filippidou, S.; Jaussi, M.; Junier, T.; Wunderlin, T.; Jeanneret, N.; Palmieri, F.; Palmieri, I.; Roussel-Delif, L.; Vieth-Hillebrand, A.; Vetter, A.; et al. Anoxybacillus geothermalis sp. nov., a facultatively anaerobic, endospore-forming bacterium isolated from mineral deposits in a geothermal station. Int. J. Syst. Evol. Microbiol. 2016, 66, 2944–2951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-kahem Al-balawi, T.H.; Wood, A.L.; Solis, A.; Cooper, T.; Barabote, R.D. Anoxybacillus sp. strain UARK-01, a new thermophilic soil bacterium with hyperthermostable alkaline laccase activity. Curr. Microbiol. 2017, 74, 762–771. [Google Scholar] [CrossRef] [PubMed]
- Degrune, F.; Theodorakopoulos, N.; Dufrene, M.; Colinet, G.; Bodson, B.; Hiel, M.P.; Taminiau, B.; Nezer, C.; Daube, G.; Vandenbol, M. No favorable effect of reduced tillage on microbial community diversity in a silty loam soil (Belgium). Agric. Ecosyst. Environ. 2016, 224, 12–21. [Google Scholar] [CrossRef]
- Lienhard, P.; Tivet, F.; Chabanne, A.; Dequiedt, S.; Lelièvre, M.; Sayphoummie, S.; Leudphanane, B.; Chemidlin Prévost-Bouré, N.; Séguy, L.; Maron, P.A.; et al. No-till and cover crops shift soil microbial abundance and diversity in Laos tropical grasslands. Agron. Sustain. Dev. 2013, 33, 375–384. [Google Scholar] [CrossRef] [Green Version]
- Gruninger, R.J.; Puniya, A.K.; Callaghan, T.M.; Edwards, J.E.; Youssef, N.; Dagar, S.S.; Fliegerova, K.; Griffith, G.W.; Forster, R.; Tsang, A.; et al. Anaerobic fungi (phylum Neocallimastigomycota): Advances in understanding their taxonomy, life cycle, ecology, role and biotechnological potential. FEMS Microbiol. Ecol. 2014, 90, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Govaerts, B.; Mezzalama, M.; Sayre, K.D.; Crossa, J.; Nicol, J.M.; Deckers, J. Long-term consequences of tillage, residue management, and crop rotation on maize/wheat root rot and nematode populations in subtropical highlands. Appl. Soil Ecol. 2006, 32, 305–315. [Google Scholar] [CrossRef]
- Young, I.M.; Ritz, K. Tillage, habitat space and function of soil microbes. Soil Tillage Res. 2000, 53, 201–213. [Google Scholar] [CrossRef]
- Schroeder, K.L.; Paulitz, T.C. Root diseases of wheat and barley during the transition from conventional tillage to direct seeding. Plant Dis. 2006, 90, 1247–1253. [Google Scholar] [CrossRef] [Green Version]
- Navarro-Noya, Y.E.; Gómez-Acata, S.; Montoya-Ciriaco, N.; Rojas-Valdez, A.; Suárez-Arriaga, M.C.; Valenzuela-Encinas, C.; Jiménez-Bueno, N.; Verhulst, N.; Govaerts, B.; Dendooven, L. Relative impacts of tillage, residue management and crop-rotation on soil bacterial communities in a semi-arid agroecosystem. Soil Biol. Biochem. 2013, 65, 86–95. [Google Scholar] [CrossRef]
- Chen, H.; Liang, Q.; Gong, Y.; Kuzyakov, Y.; Fan, M.; Plante, A.F. Reduced tillage and increased residue retention increase enzyme activity and carbon and nitrogen concentrations in soil particle size fractions in a long-term field experiment on Loess Plateau in China. Soil Tillage Res. 2019, 194, 104296. [Google Scholar] [CrossRef]
- Peng, C.; Lai, S.; Luo, X.; Lu, J.; Huang, Q.; Chen, W. Effects of long term rice straw application on the microbial communities of rapeseed rhizosphere in a paddyupland rotation system. Sci. Total Environ. 2016, 557–558, 231–239. [Google Scholar] [CrossRef] [PubMed]
- De la Cruz-Barrón, M.; Cruz-Mendoza, A.; Navarro-Noya, Y.E.; Ruiz-Valdiviezo, V.M.; Ortíz-Gutiérrez, D.; Ramírez-Villanueva, D.A.; Luna-Guido, M.; Thierfelder, C.; Wall, P.C.; Verhulst, N.; et al. The bacterial community structure and dynamics of carbon and nitrogen when maize (Zea Mays, L.) and its neutral detergent fibre were added to soil from zimbabwe with contrasting management practices. Microb. Ecol. 2017, 73, 135–152. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, A.L.; Sheaffer, C.C.; Wyse, D.L.; Staley, C.; Gould, T.J.; Sadowsky, M.J. Structure of bacterial communities in soil following cover crop and organic fertilizer incorporation. Appl. Microbiol. Biotechnol. 2016, 100, 9331–9341. [Google Scholar] [CrossRef]
- Tláskal, V.; Voříšková, J.; Baldrian, P. Bacterial succession on decomposing leaf litter exhibits a specific occurrence pattern of cellulolytic taxa and potential decomposers of fungal mycelia. FEMS Microbiol. Ecol. 2016, 92, fiw177. [Google Scholar] [CrossRef]
- Ha, Z.T.; Li, T.; Li, Y.Z.; Zhao, D.Q.; Han, J.; Liu, Y.; Liao, Y.C. Relationship between the microbial community and catabolic diversity in response to conservation tillage. Soil Tillage Res. 2020, 196, 104431. [Google Scholar]
- Marschner, P.; Umar, S.; Baumann, K. The microbial community composition changes rapidly in the early stages of decomposition of wheat residue. Soil Biol. Biochem. 2011, 43, 445–451. [Google Scholar] [CrossRef]
- Hernández-Restrepo, M.; Groenewald, J.Z.; Elliott, M.L.; Canning, G.; McMillan, V.E.; Crous, P.W. Take-all or nothing. Stud. Mycol. 2016, 83, 19–48. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, R.L. First report of Exserohilum pedicellatum on Zea mays in Australia. Plant Pathol. 2003, 52, 404. [Google Scholar] [CrossRef]
- Geml, J.; Davis, D.D.; Geiser, D.M. Systematics of the genus Sphaerobolus based on molecular and morphological data, with the description of Sphaerobolus ingoldii sp nov. Mycologia 2005, 97, 680–694. [Google Scholar] [CrossRef]
- Gleason, F.H.; Marano, A.V.; Digby, A.L.; Al-Shugairan, N.; Lilje, O.; Steciow, M.M.; Barrera, M.D.; Inaba, S.; Nakagiri, A. Patterns of utilization of different carbon sources by Chytridiomycota. Hydrobiologia 2011, 659, 55–64. [Google Scholar] [CrossRef]
- de Vries, F.T.; Thébault, E.; Liiri, M.; Birkhofer, K.; Tsiafouli, M.A.; Bjørnlund, L.; Bracht Jørgensen, H.; Brady, M.V.; Christensen, S.; de Ruiter, P.C.; et al. Soil food web properties explain ecosystem services across European land use systems. Proc. Natl. Acad. Sci. USA 2013, 110, 14296–14301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Orenes, F.; Morugan-Coronado, A.; Zornoza, R.; Cerda, A.; Scow, K. Changes in soil microbial community structure influenced by agricultural management practices in a mediterranean agro-ecosystem. PLoS ONE 2013, 8, e80522. [Google Scholar] [CrossRef] [PubMed]
- Olff, H.; Alonso, D.; Berg, M.P.; Eriksson, B.K.; Loreau, M.; Piersma, T.; Rooney, N. Parallel ecological networks in ecosystems. Philos. Trans. R. Soc. B-Biol. Sci. 2009, 364, 1755–1779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinberger, Y.; Lavee, H.; Barness, G. Soil carbohydrates along a topoclimatic gradient in a Judean desert ecosystem. Land Degrad. Dev. 1999, 10, 523–530. [Google Scholar] [CrossRef]
- Zhou, J.; Deng, Y.; Luo, F.; He, Z.; Yang, Y. Phylogenetic molecular ecological network of soil microbial communities in response to elevated CO2. mBio 2011, 2, e00122-11. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Soil Variables | Bacteria | Fungi | ||||||
|---|---|---|---|---|---|---|---|---|
| 2017 | 2018 | 2017 | 2018 | |||||
| r | p | r | p | r | p | r | p | |
| AP | 0.11 | 0.15 | −0.05 | 0.63 | 0.00 | 0.47 | −0.03 | 0.57 |
| AK | 0.05 | 0.32 | 0.17 | 0.13 | 0.04 | 0.38 | −0.06 | 0.66 |
| NO3−-N | 0.04 | 0.29 | 0.08 | 0.33 | −0.02 | 0.46 | 0.14 | 0.18 |
| NH4+-N | −0.02 | 0.48 | −0.14 | 0.68 | −0.22 | 0.91 | 0.24 | 0.11 |
| SOM | 0.03 | 0.38 | 0.16 | 0.19 | 0.15 | 0.14 | 0.26 | 0.06 |
| pH | 0.60 | 0.001 | 0.17 | 0.18 | 0.08 | 0.28 | 0.44 | 0.003 |
| SM | 0.04 | 0.33 | 0.03 | 0.38 | −0.02 | 0.50 | 0.11 | 0.23 |
| SC | −0.03 | 0.56 | 0.37 | 0.02 | −0.04 | 0.50 | −0.14 | 0.84 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guan, Y.; Xu, B.; Zhang, X.; Yang, W. Tillage Practices and Residue Management Manipulate Soil Bacterial and Fungal Communities and Networks in Maize Agroecosystems. Microorganisms 2022, 10, 1056. https://doi.org/10.3390/microorganisms10051056
Guan Y, Xu B, Zhang X, Yang W. Tillage Practices and Residue Management Manipulate Soil Bacterial and Fungal Communities and Networks in Maize Agroecosystems. Microorganisms. 2022; 10(5):1056. https://doi.org/10.3390/microorganisms10051056
Chicago/Turabian StyleGuan, Yupeng, Bei Xu, Ximei Zhang, and Wei Yang. 2022. "Tillage Practices and Residue Management Manipulate Soil Bacterial and Fungal Communities and Networks in Maize Agroecosystems" Microorganisms 10, no. 5: 1056. https://doi.org/10.3390/microorganisms10051056
APA StyleGuan, Y., Xu, B., Zhang, X., & Yang, W. (2022). Tillage Practices and Residue Management Manipulate Soil Bacterial and Fungal Communities and Networks in Maize Agroecosystems. Microorganisms, 10(5), 1056. https://doi.org/10.3390/microorganisms10051056