
Dietary Efficacy Evaluation by Applying a Prediction Model Using Clinical Fecal Microbiome Data of Colorectal Disease to a Controlled Animal Model from an Obesity Perspective
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects and Sample Collection
2.2. Experimental Design for High-Fat Diet Model in Rat
2.3. DNA Extraction and NGS Library Preparation for Sequencing of 16S rRNA Amplicon
2.4. Microbiome Profiling
2.5. Data Preprocessing through Taxonomic Accumulation
2.6. Risk Assessment Model
2.7. Statistical Analysis
3. Results
3.1. Clinical Characteristics of Subjects
3.2. Comparison of Alpha and Beta Diversity between Healthy Controls and IBD Patients
3.3. Differences in Gut Microbiome Abundance between Healthy Controls and IBD Patients
3.4. Comparison of Alpha and Beta Diversity between Healthy Controls and CRC Patients
3.5. Differences in Gut Microbiome Abundance between Healthy Controls and CRC Patients
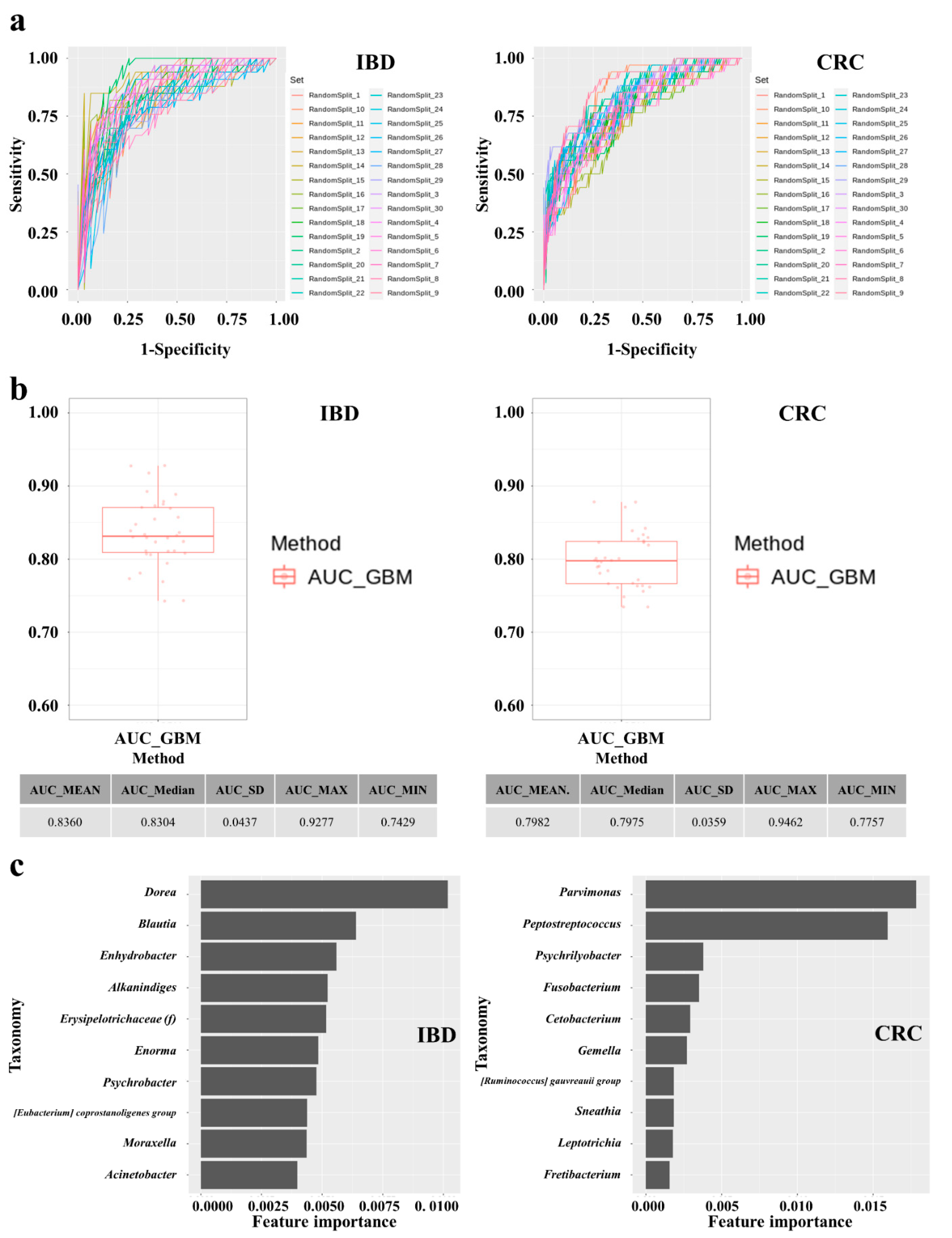
3.6. Establishment and Performances of IBD and CRC Risk Assessment Models
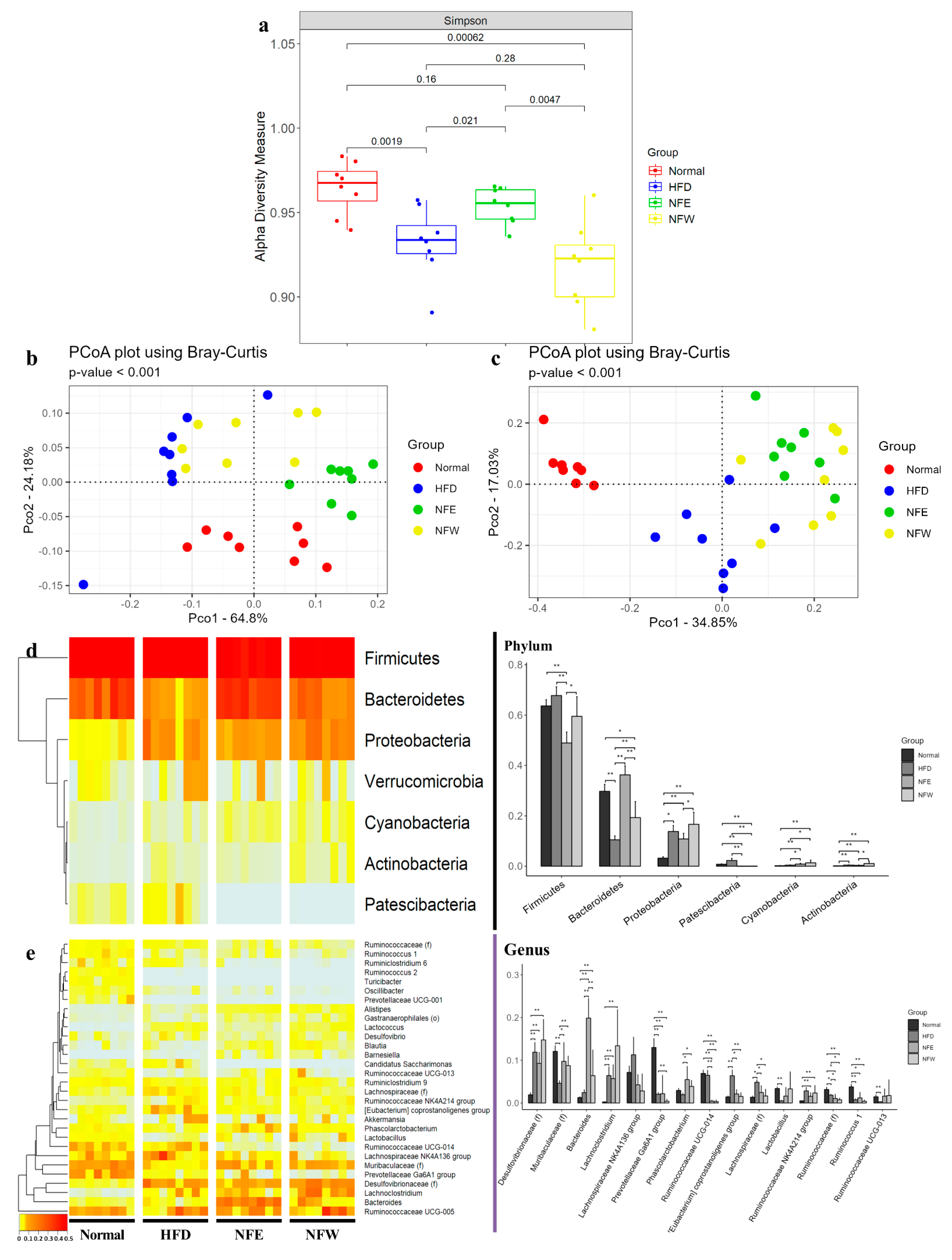
3.7. Body Weight Measure for 6 Weeks and Comparison of Alpha and Beta Diversity in HFD Induced Rat Model
3.8. Differences in Microbial Composition in HFD Induced Rat Model
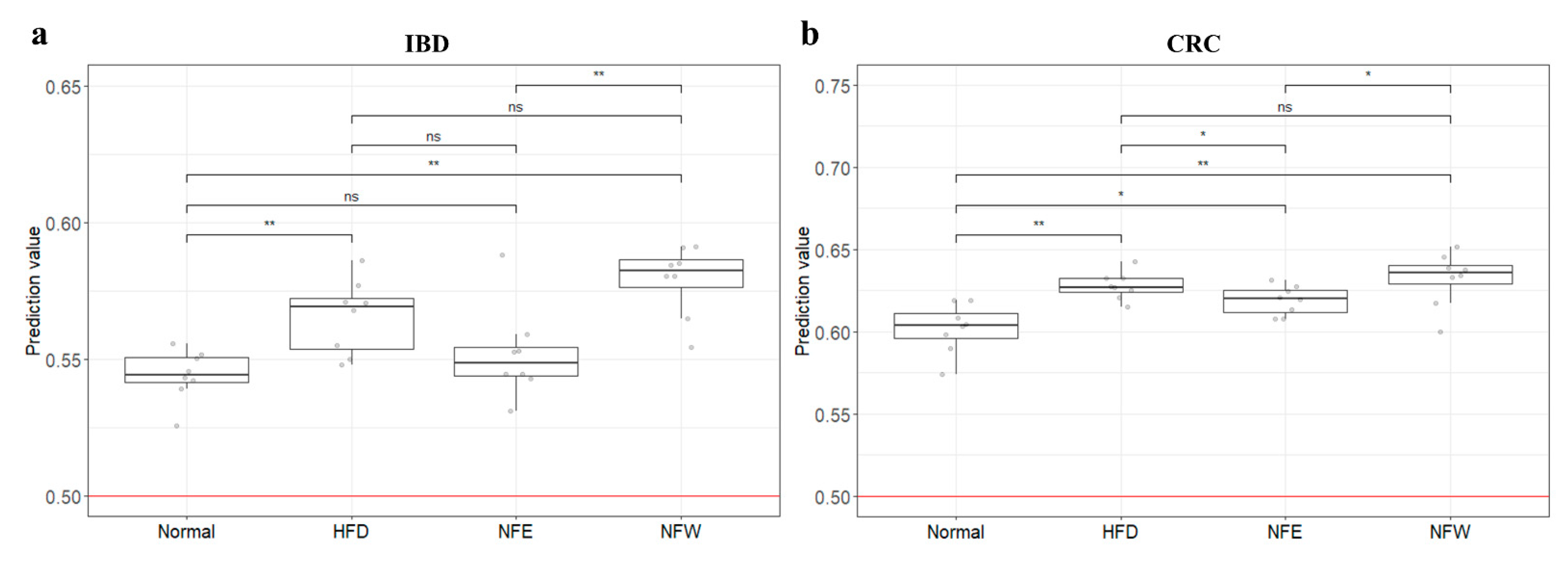
3.9. External Validation of Risk Assessment Models
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ng, M.; Fleming, T.; Robinson, M.; Thomson, B.; Graetz, N.; Margono, C.; Mullany, E.C.; Biryukov, S.; Abbafati, C.; Abera, S.F. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2014, 384, 766–781. [Google Scholar] [CrossRef]
- Yang, Z.-H.; Miyahara, H.; Takeo, J.; Katayama, M. Diet high in fat and sucrose induces rapid onset of obesity-related metabolic syndrome partly through rapid response of genes involved in lipogenesis, insulin signalling and inflammation in mice. Diabetol. Metab. Syndr. 2012, 4, 32. [Google Scholar] [CrossRef]
- Calle, E.E.; Kaaks, R. Overweight, obesity and cancer: Epidemiological evidence and proposed mechanisms. Nat. Rev. Cancer 2004, 4, 579–591. [Google Scholar] [CrossRef]
- Rodríguez-Hernández, H.; Simental-Mendía, L.E.; Rodríguez-Ramírez, G.; Reyes-Romero, M.A. Obesity and inflammation: Epidemiology, risk factors, and markers of inflammation. Int. J. Endocrinol. 2013, 2013, 678159. [Google Scholar] [CrossRef]
- Kershaw, E.E.; Flier, J.S. Adipose tissue as an endocrine organ. J. Clin. Endocrinol. Metab. 2004, 89, 2548–2556. [Google Scholar] [CrossRef]
- DiBaise, J.K.; Zhang, H.; Crowell, M.D.; Krajmalnik-Brown, R.; Decker, G.A.; Rittmann, B.E. Gut microbiota and its possible relationship with obesity. Mayo Clin. Proc. 2008, 83, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Itzkowitz, S.H.; Yio, X. Inflammation and cancer IV. Colorectal cancer in inflammatory bowel disease: The role of inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G7–G17. [Google Scholar] [CrossRef]
- Zhang, Y.-Z.; Li, Y.-Y. Inflammatory bowel disease: Pathogenesis. World J. Gastroenterol. 2014, 20, 91–99. [Google Scholar] [CrossRef]
- Glassner, K.L.; Abraham, B.P.; Quigley, E.M. The microbiome and inflammatory bowel disease. J. Allergy Clin. Immunol. 2020, 145, 16–27. [Google Scholar] [CrossRef]
- Kostic, A.D.; Xavier, R.J.; Gevers, D. The microbiome in inflammatory bowel disease: Current status and the future ahead. Gastroenterology 2014, 146, 1489–1499. [Google Scholar] [CrossRef] [Green Version]
- Saus, E.; Iraola-Guzmán, S.; Willis, J.R.; Brunet-Vega, A.; Gabaldón, T. Microbiome and colorectal cancer: Roles in carcinogenesis and clinical potential. Mol. Aspects Med. 2019, 69, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Guo, B.; Gao, R.; Zhu, Q.; Qin, H. Microbiota disbiosis is associated with colorectal cancer. Front. Microbiol. 2015, 6, 20. [Google Scholar] [CrossRef] [PubMed]
- Marchesi, J.R.; Dutilh, B.E.; Hall, N.; Peters, W.H.; Roelofs, R.; Boleij, A.; Tjalsma, H. Towards the human colorectal cancer microbiome. PLoS ONE 2011, 6, e20447. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Liu, F.; Ling, Z.; Tong, X.; Xiang, C. Human intestinal lumen and mucosa-associated microbiota in patients with colorectal cancer. PLoS ONE 2012, 7, e39743. [Google Scholar]
- Kim, K.-A.; Jung, I.-H.; Park, S.-H.; Ahn, Y.-T.; Huh, C.-S.; Kim, D.-H. Comparative analysis of the gut microbiota in people with different levels of ginsenoside Rb1 degradation to compound K. PLoS ONE 2013, 8, e62409. [Google Scholar]
- Iwai, S.; Fei, M.; Huang, D.; Fong, S.; Subramanian, A.; Grieco, K.; Lynch, S.V.; Huang, L. Oral and airway microbiota in HIV-infected pneumonia patients. J. Clin. Microbiol. 2012, 50, 2995–3002. [Google Scholar] [CrossRef]
- Hsiao, E.Y.; McBride, S.W.; Hsien, S.; Sharon, G.; Hyde, E.R.; McCue, T.; Codelli, J.A.; Chow, J.; Reisman, S.E.; Petrosino, J.F. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 2013, 155, 1451–1463. [Google Scholar] [CrossRef]
- Lloyd-Price, J.; Arze, C.; Ananthakrishnan, A.N.; Schirmer, M.; Avila-Pacheco, J.; Poon, T.W.; Andrews, E.; Ajami, N.J.; Bonham, K.S.; Brislawn, C.J. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 2019, 569, 655–662. [Google Scholar] [CrossRef]
- Jian, S.; Ban, J.; Ren, H.; Yan, H. Low genetic variation detected within the widespread mangrove species Nypa fruticans (Palmae) from Southeast Asia. Aquat. Bot. 2010, 92, 23–27. [Google Scholar] [CrossRef]
- Sugai, K.; Watanabe, S.; Kuishi, T.; Imura, S.; Ishigaki, K.; Yokota, M.; Yanagawa, S.; Suyama, Y. Extremely low genetic diversity of the northern limit populations of Nypa fruticans (Arecaceae) on Iriomote Island, Japan. Conserv. Genet. 2016, 17, 221–228. [Google Scholar] [CrossRef]
- Yusoff, N.A.; Lim, V.; Al-Hindi, B.; Abdul Razak, K.N.; Widyawati, T.; Anggraini, D.R.; Ahmad, M.; Asmawi, M.Z. Nypa fruticans Wurmb. Vinegar’s aqueous extract stimulates insulin secretion and exerts hepatoprotective effect on STZ-induced diabetic rats. Nutrients 2017, 9, 925. [Google Scholar] [CrossRef]
- Yusoff, N.A.; Ahmad, M.; Al-Hindi, B.; Widyawati, T.; Yam, M.F.; Mahmud, R.; Abdul Razak, K.N.; Asmawi, M.Z. Aqueous extract of Nypa fruticans Wurmb. vinegar alleviates postprandial hyperglycemia in normoglycemic rats. Nutrients 2015, 7, 7012–7026. [Google Scholar] [CrossRef] [Green Version]
- Tamunaidu, P.; Saka, S. Chemical characterization of various parts of nipa palm (Nypa fruticans). Ind. Crops. Prod. 2011, 34, 1423–1428. [Google Scholar] [CrossRef]
- Lee, Y.-H.; Kim, W.-K.; Jung, H.-A.; Oh, W.-K. Analysis of nutritional components and antioxidant activity of nipa palm (Nypa fruticans Wurmb) Flower stalk. Korean J. Food & Nutr. 2017, 30, 1080–1086. [Google Scholar]
- Yusoff, N.A.; Yam, M.F.; Beh, H.K.; Razak, K.N.A.; Widyawati, T.; Mahmud, R.; Ahmad, M.; Asmawi, M.Z. Antidiabetic and antioxidant activities of Nypa fruticans Wurmb. vinegar sample from Malaysia. Asian Pac. J. Trop. Med. 2015, 8, 595–605. [Google Scholar] [CrossRef]
- Martin, F.; Boris, N.; Kengne, S.; Chia, T.; Guy, T.; Gabin, A.; Ngondil, J.; Innocent, G. Antioxidant and postprandial glucose-lowering potential of the hydroethanolic extract of Nypa fruticans seed mesocarp. Biol. Med. (Aligarh) 2017, 9, 2. [Google Scholar] [CrossRef]
- Sukairi, A.H.; Sabri, W.M.A.W.; Yusop, S.A.T.W.; Asaruddin, M.R. Phytochemical screening, antidiabetic and antioxidant properties of Nypa frutican sap. Mater. Today: Proc. 2019, 19, 1738–1744. [Google Scholar]
- Reza, H.; Haq, W.M.; Das, A.K.; Rahman, S.; Jahan, R.; Rahmatullah, M. Anti-hyperglycemic and antinociceptive activity of methanol leaf and stem extract of Nypa fruticans Wurmb. Pak. J. Pharm. 2011, 24, 485–488. [Google Scholar]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Kwon, S.; Lee, B.; Yoon, S. CASPER: Context-aware scheme for paired-end reads from high-throughput amplicon sequencing. In Proceedings of the BMC Bioinform; 2014; pp. 1–11. [Google Scholar]
- Bokulich, N.A.; Subramanian, S.; Faith, J.J.; Gevers, D.; Gordon, J.I.; Knight, R.; Mills, D.A.; Caporaso, J.G. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 2013, 10, 57–59. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ. 2016, 4, e2584. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic. Acids. Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Duchesnay, E. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Manichanh, C.; Borruel, N.; Casellas, F.; Guarner, F. The gut microbiota in IBD. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 599–608. [Google Scholar] [CrossRef]
- Franzosa, E.A.; Sirota-Madi, A.; Avila-Pacheco, J.; Fornelos, N.; Haiser, H.J.; Reinker, S.; Vatanen, T.; Hall, A.B.; Mallick, H.; McIver, L.J. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol. 2019, 4, 293–305. [Google Scholar] [CrossRef]
- Nemoto, H.; Kataoka, K.; Ishikawa, H.; Ikata, K.; Arimochi, H.; Iwasaki, T.; Ohnishi, Y.; Kuwahara, T.; Yasutomo, K. Reduced diversity and imbalance of fecal microbiota in patients with ulcerative colitis. Dig. Dis. Sci. 2012, 57, 2955–2964. [Google Scholar] [CrossRef]
- Vrakas, S.; Mountzouris, K.C.; Michalopoulos, G.; Karamanolis, G.; Papatheodoridis, G.; Tzathas, C.; Gazouli, M. Intestinal bacteria composition and translocation of bacteria in inflammatory bowel disease. PLoS ONE 2017, 12, e0170034. [Google Scholar] [CrossRef]
- Jandhyala, S.M.; Talukdar, R.; Subramanyam, C.; Vuyyuru, H.; Sasikala, M.; Reddy, D.N. Role of the normal gut microbiota. World J. Gastroenterol. 2015, 21, 8787. [Google Scholar] [CrossRef]
- Ferreira, C.M.; Vieira, A.T.; Vinolo, M.A.R.; Oliveira, F.A.; Curi, R.; Martins, F.d.S. The central role of the gut microbiota in chronic inflammatory diseases. J. Immunol. Res. 2014, 2014, 689492. [Google Scholar] [CrossRef]
- Cotillard, A.; Kennedy, S.P.; Kong, L.C.; Prifti, E.; Pons, N.; Le Chatelier, E.; Almeida, M.; Quinquis, B.; Levenez, F.; Galleron, N. Dietary intervention impact on gut microbial gene richness. Nature 2013, 500, 585–588. [Google Scholar] [CrossRef] [PubMed]
- Hansen, L.; Roager, H.M.; Søndertoft, N.B.; Gøbel, R.J.; Kristensen, M.; Vallès-Colomer, M.; Vieira-Silva, S.; Ibrügger, S.; Lind, M.V.; Mærkedahl, R.B. A low-gluten diet induces changes in the intestinal microbiome of healthy Danish adults. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- van der Merwe, M. Gut microbiome changes induced by a diet rich in fruits and vegetables. Int. J. Food Sci. Nutr. 2021, 72, 665–669. [Google Scholar] [CrossRef]
- Lukens, J.R.; Gurung, P.; Vogel, P.; Johnson, G.R.; Carter, R.A.; McGoldrick, D.J.; Bandi, S.R.; Calabrese, C.R.; Walle, L.V.; Lamkanfi, M. Dietary modulation of the microbiome affects autoinflammatory disease. Nature 2014, 516, 246–249. [Google Scholar] [CrossRef]
- Wang, D.D.; Nguyen, L.H.; Li, Y.; Yan, Y.; Ma, W.; Rinott, E.; Ivey, K.L.; Shai, I.; Willett, W.C.; Hu, F.B. The gut microbiome modulates the protective association between a Mediterranean diet and cardiometabolic disease risk. Nat. Med. 2021, 27, 333–343. [Google Scholar] [CrossRef]
- Cammarota, G.; Ianiro, G.; Ahern, A.; Carbone, C.; Temko, A.; Claesson, M.J.; Gasbarrini, A.; Tortora, G. Gut microbiome, big data and machine learning to promote precision medicine for cancer. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 635–648. [Google Scholar] [CrossRef]
- Prifti, E.; Chevaleyre, Y.; Hanczar, B.; Belda, E.; Danchin, A.; Clément, K.; Zucker, J.-D. Interpretable and accurate prediction models for metagenomics data. GigaScience 2020, 9, giaa010. [Google Scholar] [CrossRef]
- Kaul, A.; Mandal, S.; Davidov, O.; Peddada, S.D. Analysis of microbiome data in the presence of excess zeros. Front. Microbiol. 2017, 8, 2114. [Google Scholar] [CrossRef]
- Badal, V.D.; Wright, D.; Katsis, Y.; Kim, H.-C.; Swafford, A.D.; Knight, R.; Hsu, C.-N. Challenges in the construction of knowledge bases for human microbiome-disease associations. Microbiome 2019, 7, 1–15. [Google Scholar] [CrossRef]
- Shang, Q.; Shan, X.; Cai, C.; Hao, J.; Li, G.; Yu, G. Dietary fucoidan modulates the gut microbiota in mice by increasing the abundance of Lactobacillus and Ruminococcaceae. Food Funct. 2016, 7, 3224–3232. [Google Scholar] [CrossRef]
- Catana, C.S.; Magdas, C.; Tabaran, F.A.; Craciun, E.C.; Deak, G.; Magdas, V.A.; Cozma, V.; Gherman, C.M.; Berindan-Neagoe, I.; Dumitrascu, D.L. Comparison of two models of inflammatory bowel disease in rats. Adv. Clin. Exp. Med. 2018, 27, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Chi, M.M.; Scull, B.P.; Rigby, R.; Schwerbrock, N.M.; Magness, S.; Jobin, C.; Lund, P.K. High-fat diet: Bacteria interactions promote intestinal inflammation which precedes and correlates with obesity and insulin resistance in mouse. PLoS ONE 2010, 5, e12191. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wei, X.; Sun, Y.; Du, J.; Li, X.; Xun, Z.; Li, Y.C. High-fat diet promotes experimental colitis by inducing oxidative stress in the colon. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 317, G453–G462. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease Type | Age and Sex | Control | Case | p-Value |
|---|---|---|---|---|
| CRC | Age (mean) Sex (M:F) | 62.75 292 (106:186) | 63.5 111(61:50) | 0.462 0.001 |
| IBD | Age (mean) Sex (M:F) | 39.99 103 (30:73) | 38.27 109(82:27) | 0.313 <0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seo, H.; Kwon, C.-O.; Park, J.-H.; Kang, C.-S.; Shin, T.-S.; Yang, E.-Y.; Jung, J.W.; Moon, B.-S.; Kim, Y.-K. Dietary Efficacy Evaluation by Applying a Prediction Model Using Clinical Fecal Microbiome Data of Colorectal Disease to a Controlled Animal Model from an Obesity Perspective. Microorganisms 2022, 10, 1833. https://doi.org/10.3390/microorganisms10091833
Seo H, Kwon C-O, Park J-H, Kang C-S, Shin T-S, Yang E-Y, Jung JW, Moon B-S, Kim Y-K. Dietary Efficacy Evaluation by Applying a Prediction Model Using Clinical Fecal Microbiome Data of Colorectal Disease to a Controlled Animal Model from an Obesity Perspective. Microorganisms. 2022; 10(9):1833. https://doi.org/10.3390/microorganisms10091833
Chicago/Turabian StyleSeo, Hochan, Cheol-O Kwon, Joo-Hyun Park, Chil-Sung Kang, Tae-Seop Shin, Eun-Young Yang, Jin Woo Jung, Byoung-Seok Moon, and Yoon-Keun Kim. 2022. "Dietary Efficacy Evaluation by Applying a Prediction Model Using Clinical Fecal Microbiome Data of Colorectal Disease to a Controlled Animal Model from an Obesity Perspective" Microorganisms 10, no. 9: 1833. https://doi.org/10.3390/microorganisms10091833
APA StyleSeo, H., Kwon, C. -O., Park, J. -H., Kang, C. -S., Shin, T. -S., Yang, E. -Y., Jung, J. W., Moon, B. -S., & Kim, Y. -K. (2022). Dietary Efficacy Evaluation by Applying a Prediction Model Using Clinical Fecal Microbiome Data of Colorectal Disease to a Controlled Animal Model from an Obesity Perspective. Microorganisms, 10(9), 1833. https://doi.org/10.3390/microorganisms10091833