

Uses of Multi-Objective Flux Analysis for Optimization of Microbial Production of Secondary Metabolites


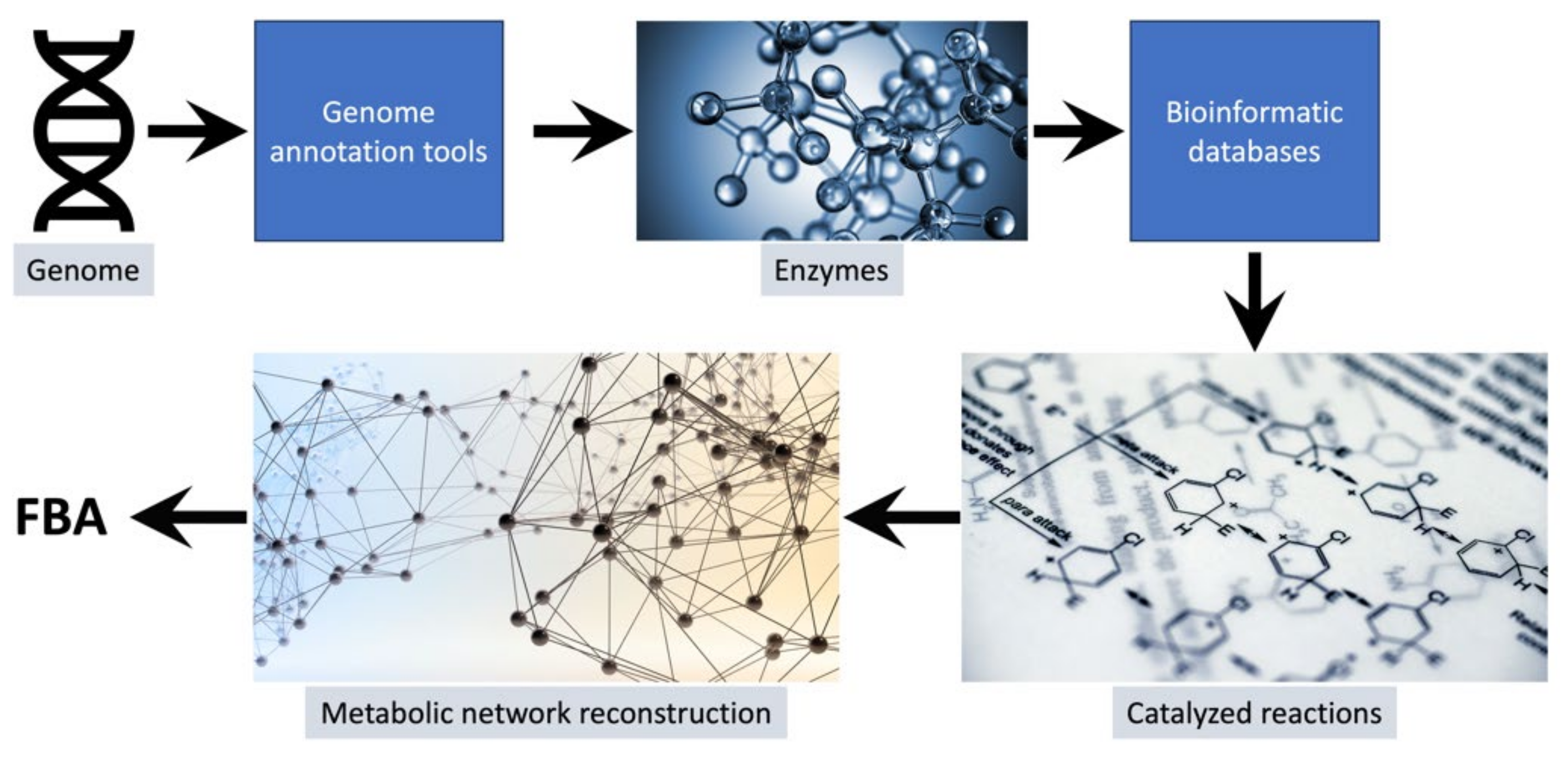{kind=link}
{kind=link}
{kind=link}
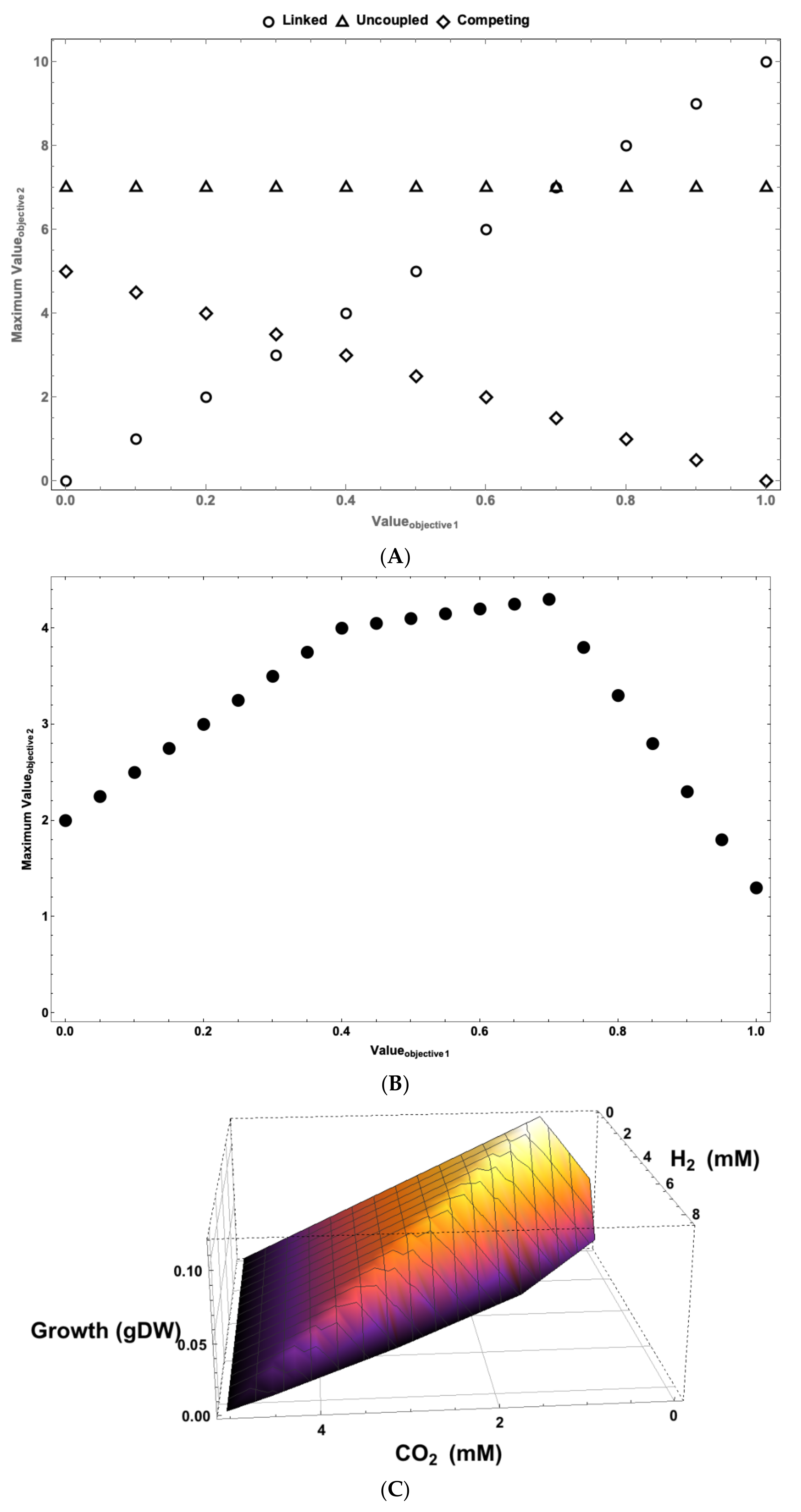{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Optimization of Microbial Production of Secondary Metabolites
3. System-Level Computational Models of Metabolism
3.1. Flux Balance Analysis
3.2. Multi-Objective Optimization
3.2.1. OptKnock
3.2.2. OptStrain
3.2.3. MultiMetEval
3.2.4. Multi-Objective Flux Analysis (MOFA)
3.3. System-Level Analysis of Microbial Communities
3.3.1. OptCom
3.3.2. Community and Systems-Level Interactive Optimization (CASINO)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shibl, A.A.; Isaac, A.; Ochsenkuhn, M.A.; Cárdenas, A.; Fei, C.; Behringer, G.; Arnoux, M.; Drou, N.; Santos, M.P.; Gunsalus, K.C.; et al. Diatom modulation of select bacteria through use of two unique secondary metabolites. Proc. Natl. Acad. Sci. USA 2020, 117, 27445–27555. [Google Scholar] [CrossRef] [PubMed]
- Demain, A.L. Valuable Secondary Metabolites from Fungi. In Biosynthesis and Molecular Genetics of Fungal Secondary Metabolites; Martín, J.-F., García-Estrada, C., Zeilinger, S., Eds.; Springer: New York, NY, USA, 2014; pp. 1–15. [Google Scholar]
- Smith, M.A.L. Large scale production of secondary metabolites. In Current Issues in Plant Molecular and Cellular Biology: Proceedings of the VIIIth International Congress on Plant Tissue and Cell Culture, Florence, Italy, 12–17 June, 1995; Terzi, M., Cella, R., Falavigna, A., Eds.; Springer: Berlin/Heidelberg, Germany, 1995; pp. 669–674. [Google Scholar]
- Murphy, D.J. Alkaloids. In Encyclopedia of Applied Plant Sciences, 2nd ed.; Thomas, B., Murray, B.G., Murphy, D.J., Eds.; Academic Press: Oxford, UK, 2017; pp. 118–124. [Google Scholar]
- Rokem, J.S.; Lantz, A.E.; Nielsen, J. Systems biology of antibiotic production by microorganisms. Nat. Prod. Rep. 2007, 24, 1262–1287. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, M.S.; Thodey, K.; Trenchard, I.; Smolke, C.D. Advancing secondary metabolite biosynthesis in yeast with synthetic biology tools. FEMS Yeast Res. 2012, 12, 144–170. [Google Scholar] [CrossRef] [PubMed]
- Beites, T.; Mendes, M.V. Chassis optimization as a cornerstone for the application of synthetic biology based strategies in microbial secondary metabolism. Front. Microbiol. 2015, 6, 906. [Google Scholar] [CrossRef]
- Rahmat, E.; Kang, Y. Yeast metabolic engineering for the production of pharmaceutically important secondary metabolites. Appl. Microbiol. Biotechnol. 2020, 104, 4659–4674. [Google Scholar] [CrossRef]
- Ruprecht, C.; Bönisch, F.; Ilmberger, N.; Heyer, T.V.; Haupt, E.T.K.; Streit, W.R.; Rabausch, U. High level production of flavonoid rhamnosides by metagenome-derived glycosyltransferase C in Escherichia coli utilizing dextrins of starch as a single carbon source. Metab. Eng. 2019, 55, 212–219. [Google Scholar] [CrossRef]
- Shrestha, A.; Pandey, R.P.; Dhakal, D.; Parajuli, P.; Sohng, J.K. Biosynthesis of flavone C-glucosides in engineered Escherichia coli. Appl. Microbiol. Biotechnol. 2018, 102, 1251–1267. [Google Scholar] [CrossRef]
- Baedeker, M.; Schulz, G.E. Overexpression of a designed 2.2 kb gene of eukaryotic phenylalanine ammonia-lyase in Escherichia coli. FEBS Lett. 1999, 457, 57–60. [Google Scholar] [CrossRef]
- Oßwald, C.; Zipf, G.; Schmidt, G.; Maier, J.; Bernauer, H.S.; Müller, R.; Wenzel, S.C. Modular construction of a functional artificial epothilone polyketide pathway. ACS Synth. Biol. 2014, 3, 759–772. [Google Scholar] [CrossRef]
- Yoshimura, T.; Shibata, N.; Hamano, Y.; Yamanaka, K. Heterologous production of hyaluronic acid in an ε-poly-L-lysine producer, Streptomyces albulus. Appl. Environ. Microbiol. 2015, 81, 3631–3640. [Google Scholar] [CrossRef]
- Teusink, B.; Bakker, B.M.; Westerhoff, H.V. Control of frequency and amplitudes is shared by all enzymes in three models for yeast glycolytic oscillations. Biochim. Biophys. Acta BBA Bioenerg. 1996, 1275, 204–212. [Google Scholar] [CrossRef]
- Kacser, H.; Burns, J.A. (Eds.) Rate control of biological processes. Symp. Soc. Exp. Biol. 1973, 27, 65. [Google Scholar]
- Groen, A.K.; Wanders, R.J.; Westerhoff, H.V.; van der Meer, R.; Tager, J.M. Quantification of the contribution of various steps to the control of mitochondrial respiration. J. Biol. Chem. 1982, 257, 2754–2757. [Google Scholar] [CrossRef] [PubMed]
- Veenstra, T.D. Omics in Systems Biology: Current Progress and Future Outlook. Proteomics 2021, 21, 2000235. [Google Scholar] [CrossRef]
- Blencowe, M.; Arneson, D.; Ding, J.; Chen, Y.-W.; Saleem, Z.; Yang, X. Network modeling of single-cell omics data: Challenges, opportunities, and progresses. Emerg. Top. Life Sci. 2019, 3, 379–398. [Google Scholar]
- Cho, J.S.; Gu, C.; Han, T.H.; Ryu, J.Y.; Lee, S.Y. Reconstruction of context-specific genome-scale metabolic models using multiomics data to study metabolic rewiring. Curr. Opin. Syst. Biol. 2019, 15, 1–11. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef]
- Seaver, S.M.D.; Liu, F.; Zhang, Q.; Jeffryes, J.; Faria, J.P.; Edirisinghe, J.N.; Mundy, M.; Chia, N.; Noor, E.; Beber, M.E.; et al. The ModelSEED Biochemistry Database for the integration of metabolic annotations and the reconstruction, comparison and analysis of metabolic models for plants, fungi and microbes. Nucleic Acids Res. 2021, 49, D575–D588. [Google Scholar] [CrossRef] [PubMed]
- Caspi, R.; Foerster, H.; Fulcher, C.A.; Kaipa, P.; Krummenacker, M.; Latendresse, M.; Paley, S.; Rhee, S.Y.; Shearer, A.G.; Tisser, C.; et al. The MetaCyc Database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 2008, 36, D623–D631. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Marcišauskas, S.; Sánchez, B.J.; Domenzain, I.; Hermansson, D.; Agren, R.; Nielsen, J.; Kerkhoven, E.J. RAVEN 2.0: A versatile toolbox for metabolic network reconstruction and a case study on Streptomyces coelicolor. PLoS Comput. Biol. 2018, 14, e1006541. [Google Scholar] [CrossRef]
- Latendresse, M.; Ong, W.K.; Karp, P.D. Metabolic modeling with MetaFlux. In Microbial Systems Biology: Methods and Protocols; Humana Press: New York, NY, USA, 2022; pp. 259–289. [Google Scholar]
- Karlsen, E.; Schulz, C.; Almaas, E. Automated generation of genome-scale metabolic draft reconstructions based on KEGG. BMC Bioinform. 2018, 19, 467. [Google Scholar] [CrossRef]
- Machado, D.; Andrejev, S.; Tramontano, M.; Patil, K.R. Fast automated reconstruction of genome-scale metabolic models for microbial species and communities. Nucleic Acids Res. 2018, 46, 7542–7553. [Google Scholar] [CrossRef] [PubMed]
- Dias, O.; Rocha, M.; Ferreira, E.C.; Rocha, I. Reconstructing genome-scale metabolic models with merlin. Nucleic Acids Res. 2015, 43, 3899–3910. [Google Scholar] [CrossRef]
- Swainston, N.; Smallbone, K.; Mendes, P.; Kell, D.B.; Paton, N.W. The SuBliMinaL Toolbox: Automating steps in the reconstruction of metabolic networks. J. Integr. Bioinform. 2011, 8, 187–203. [Google Scholar] [CrossRef]
- Arkin, A.P.; Cottingham, R.W.; Henry, C.S.; Harris, N.L.; Stevens, R.L.; Maslov, S.; Dehal, P.; Ware, D.; Perez, F.; Canon, S.; et al. KBase: The United States department of energy systems biology knowledgebase. Nat. Biotechnol. 2018, 36, 566. [Google Scholar] [CrossRef]
- Thiele, I.; Palsson, B.O. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc. 2010, 5, 93–121. [Google Scholar] [CrossRef] [PubMed]
- Navid, A. Curating COBRA models of microbial metabolism. In Microbial Systems Biology: Methods and Protocols; Springer: Berlin/Heidelberg, Germany, 2022; pp. 321–338. [Google Scholar]
- Griesemer, M.; Kimbrel, J.A.; Zhou, C.E.; Navid, A.; D’haeseleer, P. Combining multiple functional annotation tools increases coverage of metabolic annotation. BMC Genom. 2018, 19, 948. [Google Scholar] [CrossRef] [PubMed]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32 (Suppl. S1), D277. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; Van Wezel, G.P.; Medema, M.H.; Weber, T. antiSMASH 6.0: Improving cluster detection and comparison capabilities. Nucleic Acids Res. 2021, 49, W29–W35. [Google Scholar] [CrossRef]
- Medema, M.H.; Blin, K.; Cimermancic, P.; De Jager, V.; Zakrzewski, P.; Fischbach, M.A.; Weber, T.; Takano, E.; Breitling, R. antiSMASH: Rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011, 39 (Suppl. S2), W339–W346. [Google Scholar] [CrossRef] [PubMed]
- Skinnider, M.A.; Dejong, C.A.; Rees, P.N.; Johnston, C.W.; Li, H.; Webster, A.L.H.; Wyatt, M.A.; Magarvey, N.A. Genomes to natural products prediction informatics for secondary metabolomes (PRISM). Nucleic Acids Res. 2015, 43, 9645–9662. [Google Scholar] [CrossRef] [PubMed]
- Skinnider, M.A.; Merwin, N.J.; Johnston, C.W.; Magarvey, N.A. PRISM 3: Expanded prediction of natural product chemical structures from microbial genomes. Nucleic Acids Res. 2017, 45, W49–W54. [Google Scholar] [CrossRef]
- Schuetz, R.; Kuepfer, L.; Sauer, U. Systematic evaluation of objective functions for predicting intracellular fluxes in Escherichia coli. Mol. Syst. Biol. 2007, 3, 119. [Google Scholar] [CrossRef]
- Feist, A.M.; Palsson, B.O. The biomass objective function. Curr. Opin. Microbiol. 2010, 13, 344–349. [Google Scholar] [CrossRef]
- Burgard, A.P.; Maranas, C.D. Optimization-based framework for inferring and testing hypothesized metabolic objective functions. Biotechnol. Bioeng. 2003, 82, 670–677. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Stettner, A.; Reznik, E.; Segrè, D.; Paschalidis, I.C. (Eds.) Learning cellular objectives from fluxes by inverse optimization. In Proceedings of the 2015 IEEE 54th Annual Conference on Decision and Control (CDC), Osaka, Japan, 15 December 2015; IEEE: Piscataway, NJ, USA. [Google Scholar]
- Karr, J.R.; Sanghvi, J.C.; Macklin, D.N.; Gutschow, M.V.; Jacobs, J.M.; Bolival, B.; Assad-Garcia, N.; Glass, J.I.; Covert, M.W. A whole-cell computational model predicts phenotype from genotype. Cell 2012, 150, 389–401. [Google Scholar] [CrossRef]
- Karr, J.R.; Takahashi, K.; Funahashi, A. The principles of whole-cell modeling. Curr. Opin. Microbiol. 2015, 27, 18–24. [Google Scholar] [CrossRef]
- Purcell, O.; Jain, B.; Karr, J.R.; Covert, M.W.; Lu, T.K. Towards a whole-cell modeling approach for synthetic biology. Chaos 2013, 23, 025112. [Google Scholar] [CrossRef] [PubMed]
- Birch, E.W.; Udell, M.; Covert, M.W. Incorporation of flexible objectives and time-linked simulation with flux balance analysis. J. Theor. Biol. 2014, 345, 12–21. [Google Scholar] [CrossRef]
- Almaas, E.; Kovacs, B.; Vicsek, T.; Oltvai, Z.N.; Barabasi, A.L. Global organization of metabolic fluxes in the bacterium Escherichia coli. Nature 2004, 427, 839–843. [Google Scholar] [CrossRef]
- Almaas, E.; Oltvai, Z.N.; Barabasi, A.L. The activity reaction core and plasticity of metabolic networks. PLoS Comput. Biol. 2005, 1, e68. [Google Scholar] [CrossRef] [PubMed]
- Schuetz, R.; Zamboni, N.; Zampieri, M.; Heinemann, M.; Sauer, U. Multidimensional optimality of microbial metabolism. Science 2012, 336, 601–604. [Google Scholar] [CrossRef] [PubMed]
- Navid, A.; Almaas, E. (Eds.) Genome-scale reconstruction of the metabolic network in Yersinia pestis CO922007. Mol. Biosyst. 2009, 5, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Chaudhury, S.; Abdulhameed, M.D.M.; Singh, N.; Tawa, G.J.; D’haeseleer, P.M.; Zemla, A.T.; Navid, A.; Zhou, C.E.; Franklin, M.C.; Cheung, J.; et al. Rapid countermeasure discovery against Francisella tularensis based on a metabolic network reconstruction. PLoS ONE 2013, 8, e63369. [Google Scholar] [CrossRef] [PubMed]
- Navid, A.; Almaas, E. Genome-level transcription data of Yersinia pestis analyzed with a New metabolic constraint-based approach. BMC Syst. Biol. 2012, 6, 150. [Google Scholar] [CrossRef]
- Presta, L.; Bosi, E.; Mansouri, L.; Dijkshoorn, L.; Fani, R.; Fondi, M. Constraint-based modeling identifies new putative targets to fight colistin-resistant A. baumannii infections. Sci. Rep. 2017, 7, 3706. [Google Scholar] [CrossRef]
- Folger, O.; Jerby, L.; Frezza, C.; Gottlieb, E.; Ruppin, E.; Shlomi, T. Predicting selective drug targets in cancer through metabolic networks. Mol. Syst. Biol. 2011, 7, 501. [Google Scholar] [CrossRef]
- Zielinski, D.C.; Jamshidi, N.; Corbett, A.J.; Bordbar, A.; Thomas, A.; Palsson, B.O. Systems biology analysis of drivers underlying hallmarks of cancer cell metabolism. Sci. Rep. 2017, 7, 41241. [Google Scholar] [CrossRef]
- Shlomi, T.; Benyamini, T.; Gottlieb, E.; Sharan, R.; Ruppin, E. Genome-scale metabolic modeling elucidates the role of proliferative adaptation in causing the Warburg effect. PLoS Comput. Biol. 2011, 7, e1002018. [Google Scholar] [CrossRef] [PubMed]
- Asgari, Y.; Zabihinpour, Z.; Salehzadeh-Yazdi, A.; Schreiber, F.; Masoudi-Nejad, A. Alterations in cancer cell metabolism: The Warburg effect and metabolic adaptation. Genomics 2015, 105, 275–281. [Google Scholar] [CrossRef]
- Fong, S.S.; Palsson, B.O. Metabolic gene-deletion strains of Escherichia coli evolve to computationally predicted growth phenotypes. Nat. Genet. 2004, 36, 1056–1058. [Google Scholar] [CrossRef]
- Segre, D.; Vitkup, D.; Church, G.M. Analysis of optimality in natural and perturbed metabolic networks. Proc. Natl. Acad. Sci. USA 2002, 99, 15112–15117. [Google Scholar] [CrossRef]
- Shlomi, T.; Berkman, O.; Ruppin, E. Regulatory on/off minimization of metabolic flux changes after genetic perturbations. Proc. Natl. Acad. Sci. USA 2005, 102, 7695–7700. [Google Scholar] [CrossRef]
- Herrgard, M.J.; Lee, B.S.; Portnoy, V.; Palsson, B.O. Integrated analysis of regulatory and metabolic networks reveals novel regulatory mechanisms in Saccharomyces cerevisiae. Genome Res. 2006, 16, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Wen, J.; Wang, G.; Yu, G.; Jia, X.; Chen, Y. In silico aided metabolic engineering of Streptomyces roseosporus for daptomycin yield improvement. Appl. Microbiol. Biotechnol. 2012, 94, 637–649. [Google Scholar] [CrossRef]
- Medema, M.H.; Trefzer, A.; Kovalchuk, A.; van den Berg, M.; Müller, U.; Heijne, W.; Wu, L.; Alam, M.T.; Ronning, C.M.; Nierman, W.C.; et al. The Sequence of a 1.8-Mb bacterial linear plasmid reveals a rich evolutionary reservoir of secondary metabolic pathways. Genome Biol. Evol. 2010, 2, 212–224. [Google Scholar] [CrossRef]
- Kiviharju, K.; Moilanen, U.; Leisola, M.; Eerikäinen, T. A chemostat study of Streptomyces peucetius var. caesius N47. Appl. Microbiol. Biotechnol. 2007, 73, 1267–1274. [Google Scholar] [CrossRef] [PubMed]
- Naeimpoor, F.; Mavituna, F. Metabolic Flux Analysis in Streptomyces coelicolor under Various Nutrient Limitations. Metab. Eng. 2000, 2, 140–148. [Google Scholar] [CrossRef]
- Borodina, I.; Siebring, J.; Zhang, J.; Smith, C.P.; van Keulen, G.; Dijkhuizen, L.; Nielsen, J. Antibiotic Overproduction in Streptomyces coelicolor A3(2) Mediated by Phosphofructokinase Deletion. J. Biol. Chem. 2008, 283, 25186–25199. [Google Scholar] [CrossRef] [PubMed]
- Borodina, I.; Krabben, P.; Nielsen, J. Genome-scale analysis of Streptomyces coelicolor A3 (2) metabolism. Genome Res. 2005, 15, 820–829. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.A.; Laing, E.; Allenby, N.; Bucca, G.; Brenner, V.; Harrison, M.; Kierzek, A.M.; Smith, C.P. Metabolic and evolutionary insights into the closely-related species Streptomyces coelicolor and Streptomyces lividans deduced from high-resolution comparative genomic hybridization. BMC Genom. 2010, 11, 682. [Google Scholar] [CrossRef]
- Bum Kim, H.; Smith, C.P.; Micklefield, J.; Mavituna, F. Metabolic flux analysis for calcium dependent antibiotic (CDA) production in Streptomyces coelicolor. Metab. Eng. 2004, 6, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.T.; Merlo, M.E.; Hodgson, D.A.; Wellington, E.M.H.; Takano, E.; Breitling, R. Metabolic modeling and analysis of the metabolic switch in Streptomyces coelicolor. BMC Genom. 2010, 11, 202. [Google Scholar] [CrossRef]
- Gómez-Ríos, D.; López-Agudelo, V.A.; Ramírez-Malule, H.; Neubauer, P.; Junne, S.; Ochoa, S.; Ríos-Estepa, R. A Genome-scale insight into the effect of shear stress during the fed-batch production of clavulanic acid by Streptomyces clavuligerus. Microorganisms 2020, 8, 1255. [Google Scholar] [CrossRef]
- Kim, M.; Sang, Y.J.; Kim, J.; Kim, J.-N.; Kim, M.W.; Kim, B.-G. Reconstruction of a high-quality metabolic model enables the identification of gene overexpression targets for enhanced antibiotic production in Streptomyces coelicolor A3(2). Biotechnol. J. 2014, 9, 1185–1194. [Google Scholar] [CrossRef]
- Cavallieri, A.P.; Baptista, A.S.; Leite, C.A.; da Costa Araujo, M.L.G. A case study in flux balance analysis: Lysine, a cephamycin C precursor, can also increase clavulanic acid production. Biochem. Eng. J. 2016, 112, 42–53. [Google Scholar] [CrossRef]
- Harir, M.; Bendif, H.; Bellahcene, M.; Fortas, Z.; Pogni, R. Streptomyces secondary metabolites. Basic Biol. Appl. Actinobacteria 2018, 6, 99–122. [Google Scholar]
- Demain, A.L. Regulation of secondary metabolism in fungi. Pure Appl. Chem. 1986, 58, 219–226. [Google Scholar] [CrossRef]
- González-Lergier, J.; Broadbelt, L.J.; Hatzimanikatis, V. Theoretical considerations and computational analysis of the complexity in polyketide synthesis pathways. J. Am. Chem. Soc. 2005, 127, 9930–9938. [Google Scholar] [CrossRef] [PubMed]
- Paradise, E.M.; Kirby, J.; Chan, R.; Keasling, J.D. Redirection of flux through the FPP branch-point in Saccharomyces cerevisiae by down-regulating squalene synthase. Biotechnol. Bioeng. 2008, 100, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.S.; Ramakrishna, R.; Palsson, B.O. Characterizing the metabolic phenotype: A phenotype phase plane analysis. Biotechnol. Bioeng. 2002, 77, 27–36. [Google Scholar] [CrossRef]
- Edwards, J.S.; Ibarra, R.U.; Palsson, B.O. In silico predictions of Escherichia coli metabolic capabilities are consistent with experimental data. Nat. Biotech. 2001, 19, 125–130. [Google Scholar] [CrossRef]
- Duarte, N.C.; Palsson, B.Ø.; Fu, P. Integrated analysis of metabolic phenotypes in Saccharomyces cerevisiae. BMC Genom. 2004, 5, 63. [Google Scholar] [CrossRef] [PubMed]
- Burgard, A.P.; Pharkya, P.; Maranas, C.D. Optknock: A bilevel programming framework for identifying gene knockout strategies for microbial strain optimization. Biotechnol. Bioeng. 2003, 84, 647–657. [Google Scholar] [CrossRef]
- Pharkya, P.; Burgard, A.P.; Maranas, C.D. Exploring the overproduction of amino acids using the bilevel optimization framework OptKnock. Biotechnol. Bioeng. 2003, 84, 887–899. [Google Scholar] [CrossRef]
- Fong, S.S.; Burgard, A.P.; Herring, C.D.; Knight, E.M.; Blattner, F.R.; Maranas, C.D.; Palsson, B.O. In silico design and adaptive evolution of Escherichia coli for production of lactic acid. Biotechnol. Bioeng. 2005, 91, 643–648. [Google Scholar] [CrossRef]
- Tepper, N.; Shlomi, T. Predicting metabolic engineering knockout strategies for chemical production: Accounting for competing pathways. Bioinformatics 2010, 26, 536–543. [Google Scholar] [CrossRef]
- Pharkya, P.; Maranas, C.D. An optimization framework for identifying reaction activation/inhibition or elimination candidates for overproduction in microbial systems. Metab. Eng. 2006, 8, 1–13. [Google Scholar] [CrossRef]
- Kim, J.; Reed, J.L. OptORF: Optimal metabolic and regulatory perturbations for metabolic engineering of microbial strains. BMC Syst. Biol. 2010, 4, 53. [Google Scholar] [CrossRef]
- Rocha, I.; Maia, P.; Evangelista, P.; Vilaça, P.; Soares, S.; Pinto, J.P.; Nielsen, J.; Patil, K.R.; Ferreira, E.C.; Rocha, M. OptFlux: An open-source software platform for in silico metabolic engineering. BMC Syst. Biol. 2010, 4, 45. [Google Scholar] [CrossRef] [PubMed]
- Pharkya, P.; Burgard, A.P.; Maranas, C.D. OptStrain: A computational framework for redesign of microbial production systems. Genome Res. 2004, 14, 2367–2376. [Google Scholar] [CrossRef] [PubMed]
- Purdy, H.M.; Pfleger, B.F.; Reed, J.L. Introduction of NADH-dependent nitrate assimilation in Synechococcus sp. PCC 7002 improves photosynthetic production of 2-methyl-1-butanol and isobutanol. Metab. Eng. 2022, 69, 87–97. [Google Scholar] [CrossRef]
- Malci, K.; Santibanez, R.; Jonguitud-Borrego, N.; Santoyo-Garcia, J.H.; Kherkoven, E.J.; Rios Solis, L. Improved Production of Taxol® Precursors in S. cerevisiae using Combinatorial in silico Design and Metabolic Engineering. bioRxiv 2023. bioRxiv:2023-06. [Google Scholar]
- Zakrzewski, P.; Medema, M.H.; Gevorgyan, A.; Kierzek, A.M.; Breitling, R.; Takano, E. MultiMetEval: Comparative and Multi-Objective Analysis of Genome-Scale Metabolic Models. PLoS ONE 2012, 7, e51511. [Google Scholar] [CrossRef]
- Navid, A.; Jiao, Y.; Wong, S.E.; Pett-Ridge, J. System-level analysis of metabolic trade-offs during anaerobic photoheterotrophic growth in Rhodopseudomonas palustris. BMC Bioinform. 2019, 20, 233. [Google Scholar] [CrossRef] [PubMed]
- Gowen, C.M.; Fong, S.S. Exploring Biodiversity for Cellulosic Biofuel Production. Chem. Biodivers. 2010, 7, 1086–1097. [Google Scholar] [CrossRef]
- Nazem-Bokaee, H.; Gopalakrishnan, S.; Ferry, J.G.; Wood, T.K.; Maranas, C.D. Assessing methanotrophy and carbon fixationfor biofuel production by Methanosarcina acetivorans. Microb. Cell Factories 2016, 15, 10. [Google Scholar] [CrossRef]
- Dash, S.; Mueller, T.J.; Venkataramanan, K.P.; Papoutsakis, E.T.; Maranas, C.D. Capturing the response of Clostridium acetobutylicum to chemical stressors using a regulated genome-scale metabolic model. Biotechnol. Biofuels 2014, 7, 144. [Google Scholar] [CrossRef] [PubMed]
- Shabestary, K.; Hudson, E.P. Computational metabolic engineering strategies for growth-coupled biofuel production by Synechocystis. Metab. Eng. Commun. 2016, 3, 216–226. [Google Scholar] [CrossRef]
- Fatma, Z.; Hartman, H.; Poolman, M.G.; Fell, D.A.; Srivastava, S.; Shakeel, T.; Yazdani, S.S. Model-assisted metabolic engineering of Escherichia coli for long chain alkane and alcohol production. Metab. Eng. 2018, 46, 1–12. [Google Scholar] [CrossRef]
- McKinlay, J.B.; Harwood, C.S. Photobiological production of hydrogen gas as a biofuel. Curr. Opin. Biotechnol. 2010, 21, 244–251. [Google Scholar] [CrossRef]
- Huang, J.J.; Heiniger, E.K.; McKinlay, J.B.; Harwood, C.S. Production of hydrogen gas from light and the inorganic electron donor thiosulfate by Rhodopseudomonas palustris. Appl. Environ. Microbiol. 2010, 76, 7717–7722. [Google Scholar] [CrossRef]
- Oh, Y.-K.; Seol, E.-H.; Kim, M.-S.; Park, S. Photoproduction of hydrogen from acetate by a chemoheterotrophic bacterium Rhodopseudomonas palustris P4. Int. J. Hydrogen Energy 2004, 29, 1115–1121. [Google Scholar] [CrossRef]
- Nagrath, D.; Avila-Elchiver, M.; Berthiaume, F.; Tilles, A.W.; Messac, A.; Yarmush, M.L. Soft Constraints-Based Multiobjective Framework for Flux Balance Analysis. Metab. Eng. 2010, 12, 429. [Google Scholar] [CrossRef] [PubMed]
- Messac, A.; Ismail-Yahaya, A.; Mattson, C.A. The normalized normal constraint method for generating the Pareto frontier. Struct. Multidiscip. Optim. 2003, 25, 86–98. [Google Scholar] [CrossRef]
- Heirendt, L.; Arreckx, S.; Pfau, T.; Mendoza, S.N.; Richelle, A.; Heinken, A.; Haraldsdóttir, H.S.; Wachowiak, J.; Keating, S.M.; Vlasov, V.; et al. Creation and analysis of biochemical constraint-based models using the COBRA Toolbox v. 3.0. Nat. Protoc. 2019, 14, 639–702. [Google Scholar] [CrossRef]
- Shurin, J.B.; Abbott, R.L.; Deal, M.S.; Kwan, G.T.; Litchman, E.; McBride, R.C.; Mandal, S.; Smith, V.H. Industrial-strength ecology: Trade-offs and opportunities in algal biofuel production. Ecol. Lett. 2013, 16, 1393–1404. [Google Scholar] [CrossRef]
- Lindemann, S.R.; Bernstein, H.C.; Song, H.-S.; Fredrickson, J.K.; Fields, M.W.; Shou, W.; Johnson, D.R.; Beliaev, A.S. Engineering microbial consortia for controllable outputs. ISME J. 2016, 10, 2077–2084. [Google Scholar] [CrossRef]
- Podolsky, I.A.; Seppälä, S.; Lankiewicz, T.S.; Brown, J.L.; Swift, C.L.; O’Malley, M.A. Harnessing nature’s anaerobes for biotechnology and bioprocessing. Annu. Rev. Chem. Biomol. Eng. 2019, 10, 105–128. [Google Scholar] [CrossRef]
- Swift, C.L.; Brown, J.L.; Seppälä, S.; O’Malley, M.A. Co-cultivation of the anaerobic fungus Anaeromyces robustus with Methanobacterium bryantii enhances transcription of carbohydrate active enzymes. J. Ind. Microbiol. Biotechnol. 2019, 46, 1427–1433. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.; Qiao, K.; Edgar, S.; Stephanopoulos, G. Distributing a metabolic pathway among a microbial consortium enhances production of natural products. Nat. Biotechnol. 2015, 33, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Navid, A.; Stewart, B.; McKinlay, J.; Thelen, M.; Pett-Ridge, J. Syntrophic metabolism of a co-culture containing Clostridium cellulolyticum and Rhodopseudomonas palustris for hydrogen production. Int. J. Hydrogen Energy 2012, 37, 11719–11726. [Google Scholar] [CrossRef]
- Zomorrodi, A.R.; Maranas, C.D. OptCom: A multi-level optimization framework for the metabolic modeling and analysis of microbial communities. PLoS Comput. Biol. 2012, 8, e1002363. [Google Scholar] [CrossRef] [PubMed]
- Zomorrodi, A.R.; Islam, M.M.; Maranas, C.D. d-OptCom: Dynamic multi-level and multi-objective metabolic modeling of microbial communities. ACS Synth. Biol. 2014, 3, 247–257. [Google Scholar] [CrossRef]
- Shoaie, S.; Ghaffari, P.; Kovatcheva-Datchary, P.; Mardinoglu, A.; Sen, P.; Pujos-Guillot, E.; De Wouters, T.; Juste, C.; Rizkalla, S.; Chilloux, J.; et al. Quantifying diet-induced metabolic changes of the human gut microbiome. Cell Metab. 2015, 22, 320–331. [Google Scholar] [CrossRef]
- Ding, S.; Cai, P.; Yuan, L.; Tian, Y.; Tu, W.; Zhang, D.; Cheng, X.; Sun, D.; Chen, J.; Hu, Q.N. CF-Targeter: A rational biological cell factory targeting platform for biosynthetic target chemicals. ACS Synth. Biol. 2019, 8, 2280–2286. [Google Scholar] [CrossRef]
- Zur, H.; Ruppin, E.; Shlomi, T. iMAT: An integrative metabolic analysis tool. Bioinformatics 2010, 26, 3140–3142. [Google Scholar] [CrossRef]
- Becker, S.A.; Palsson, B.O. Context-specific metabolic networks are consistent with experiments. PLoS Comput. Biol. 2008, 4, e1000082. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.A.; Papin, J.A. Functional integration of a metabolic network model and expression data without arbitrary thresholding. Bioinformatics 2011, 27, 541–547. [Google Scholar] [CrossRef]
- Chandrasekaran, S.; Price, N.D. Probabilistic integrative modeling of genome-scale metabolic and regulatory networks in Escherichia coli and Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2010, 107, 17845. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Wallqvist, A.; Reifman, J. Modeling phenotypic metabolic adaptations of Mycobacterium tuberculosis H37Rv under hypoxia. PLoS Comput. Biol. 2012, 8, e1002688. [Google Scholar] [CrossRef] [PubMed]
- Di Filippo, M.; Pescini, D.; Galuzzi, B.G.; Bonanomi, M.; Gaglio, D.; Mangano, E.; Consolandi, C.; Alberghina, L.; Vanoni, M.; Damiani, C. INTEGRATE: Model-based multi-omics data integration to characterize multi-level metabolic regulation. PLoS Comput. Biol. 2022, 18, e1009337. [Google Scholar] [CrossRef]
- Hadadi, N.; Pandey, V.; Chiappino-Pepe, A.; Morales, M.; Gallart-Ayala, H.; Mehl, F.; Ivanisevic, J.; Sentchilo, V.; Meer, J.R.V.D. Mechanistic insights into bacterial metabolic reprogramming from omics-integrated genome-scale models. NPJ Syst. Biol. Appl. 2020, 6, 1. [Google Scholar] [CrossRef]
- Machado, D.; Herrgård, M. Systematic evaluation of methods for integration of transcriptomic data into constraint-based models of eetabolism. PLoS Comput. Biol. 2014, 10, e1003580. [Google Scholar] [CrossRef]
- Cruz, F.; Faria, J.P.; Rocha, M.; Rocha, I.; Dias, O. A review of methods for the reconstruction and analysis of integrated genome-scale models of metabolism and regulation. Biochem. Soc. Trans. 2020, 48, 1889–1903. [Google Scholar] [CrossRef]
- Wendering, P.; Nikoloski, Z. Model-driven insights into the effects of temperature on metabolism. Biotechnol. Adv. 2023, 67, 108203. [Google Scholar] [CrossRef]
- Schellenberger, J.; Que, R.; Fleming, R.M.T.; Thiele, I.; Orth, J.D.; Feist, A.M.; Zielinski, D.C.; Bordbar, A.; Lewis, N.E.; Rahmanian, S.; et al. Quantitative prediction of cellular metabolism with constraint-based models: The COBRA Toolbox v2. 0. Nat. Protoc. 2011, 6, 1290–1307. [Google Scholar] [CrossRef]
- Becker, S.A.; Feist, A.M.; Mo, M.L.; Hannum, G.; Palsson, B.O.; Herrgard, M.J. Quantitative prediction of cellular metabolism with constraint-based models: The COBRA Toolbox. Nat. Protoc. 2007, 2, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, R.; Schilling, C.H. The effects of alternate optimal solutions in constraint-based genome-scale metabolic models. Metab. Eng. 2003, 5, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Feist, A.M.; Henry, C.S.; Reed, J.L.; Krummenacker, M.; Joyce, A.R.; Karp, P.D.; Broadbelt, L.J.; Hatzimanikatis, V.; Palsson, B.Ø. A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol. Syst. Biol. 2007, 3, 121. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Griesemer, M.; Navid, A. Uses of Multi-Objective Flux Analysis for Optimization of Microbial Production of Secondary Metabolites. Microorganisms 2023, 11, 2149. https://doi.org/10.3390/microorganisms11092149
Griesemer M, Navid A. Uses of Multi-Objective Flux Analysis for Optimization of Microbial Production of Secondary Metabolites. Microorganisms. 2023; 11(9):2149. https://doi.org/10.3390/microorganisms11092149
Chicago/Turabian StyleGriesemer, Marc, and Ali Navid. 2023. "Uses of Multi-Objective Flux Analysis for Optimization of Microbial Production of Secondary Metabolites" Microorganisms 11, no. 9: 2149. https://doi.org/10.3390/microorganisms11092149
APA StyleGriesemer, M., & Navid, A. (2023). Uses of Multi-Objective Flux Analysis for Optimization of Microbial Production of Secondary Metabolites. Microorganisms, 11(9), 2149. https://doi.org/10.3390/microorganisms11092149