
Using Network Extracted Ontologies to Identify Novel Genes with Roles in Appressorium Development in the Rice Blast Fungus Magnaporthe oryzae


Abstract
:1. Introduction
2. Materials and Methods
2.1. Genomic and Transcriptomic Data
2.2. Annotation of M. oryzae Genes
2.3. Estimating Expression of M. oryzae Genes
2.4. Generating an Ontology of M. oryzae
2.5. Estimating the Robustness of the M. oryzae Ontology
2.6. Annotating Entities in the Ontology
2.7. Identifying Entities Associated with Appressorium Function
3. Results
3.1. Correlated Expression of M. oryzae Genes
3.2. A M. oryzae Network-Extracted Ontology
3.3. Validation of the Ontology Structure
3.4. Identifying Gene and Entity Function
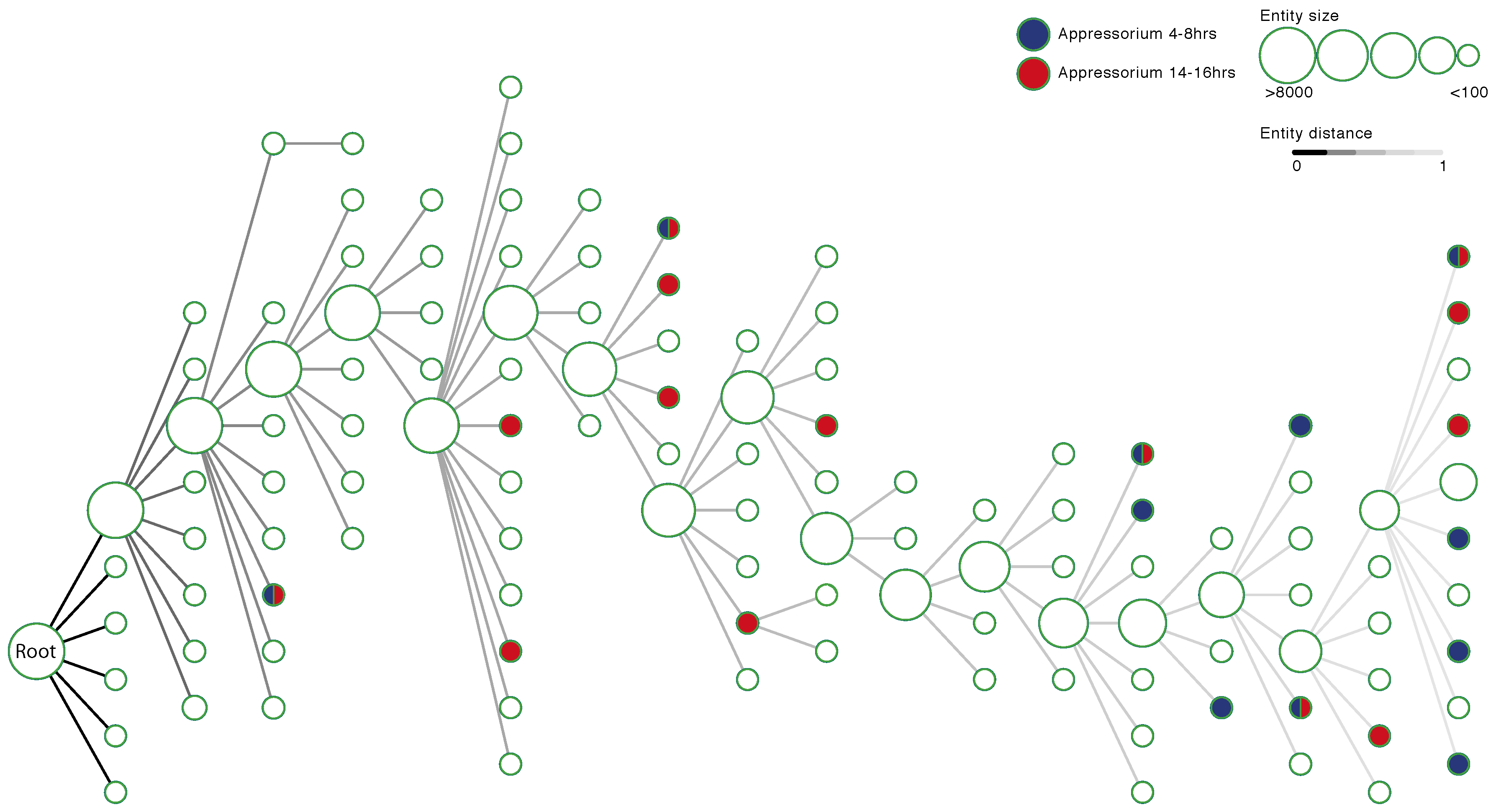
3.5. Identifying Entities Enriched for Appressorium Development Genes
4. Discussion
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Ou, S. Pathogen variability and host resistance in rice blast disease. Annu. Rev. Phytopathol. 1980, 18, 167–187. [Google Scholar] [CrossRef]
- Khush, G.S. What it will take to feed 5.0 billion rice consumers in 2030. Plant Mol. Biol. 2005, 59, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Dean, R.A.; Talbot, N.J.; Ebbole, D.J.; Farman, M.L.; Mitchell, T.K.; Orbach, M.J.; Thon, M.; Kulkarni, R.; Xu, J.R.; Pan, H.; et al. The genome sequence of the rice blast fungus Magnaporthe grisea. Nature 2005, 434, 980–986. [Google Scholar] [CrossRef] [PubMed]
- Dean, R.; Van Kan, J.A.; Pretorius, Z.A.; Hammond-Kosack, K.E.; Di Pietro, A.; Spanu, P.D.; Rudd, J.J.; Dickman, M.; Kahmann, R.; Ellis, J.; et al. The Top 10 fungal pathogens in molecular plant pathology. Mol. Plant Pathol. 2012, 13, 414–430. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.A.; Talbot, N.J. Under pressure: Investigating the biology of plant infection by Magnaporthe oryzae. Nat. Rev. Microbiol. 2009, 7, 185–195. [Google Scholar] [CrossRef]
- De Jong, J.C.; McCormack, B.J.; Smirnoff, N.; Talbot, N.J. Glycerol generates turgor in rice blast. Nature 1997, 389, 244. [Google Scholar] [CrossRef]
- Saunders, D.G.; Aves, S.J.; Talbot, N.J. Cell Cycle–Mediated Regulation of Plant Infection by the Rice Blast Fungus. Plant Cell 2010, 22, 497–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veneault-Fourrey, C.; Barooah, M.; Egan, M.; Wakley, G.; Talbot, N.J. Autophagic fungal cell death is necessary for infection by the rice blast fungus. Science 2006, 312, 580–583. [Google Scholar] [CrossRef] [PubMed]
- Saunders, D.G.; Dagdas, Y.F.; Talbot, N.J. Spatial uncoupling of mitosis and cytokinesis during appressorium-mediated plant infection by the rice blast fungus Magnaporthe oryzae. Plant Cell 2010, 22, 2417–2428. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, M.J.; Talbot, N.J. Genome-wide functional analysis reveals that infection-associated fungal autophagy is necessary for rice blast disease. Proc. Natl. Acad. Sci. USA 2009, 106, 15967–15972. [Google Scholar] [CrossRef] [PubMed]
- Adachi, K.; Hamer, J.E. Divergent cAMP signaling pathways regulate growth and pathogenesis in the rice blast fungus Magnaporthe grisea. Plant Cell 1998, 10, 1361–1373. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.R.; Hamer, J.E. MAP kinase and cAMP signaling regulate infection structure formation and pathogenic growth in the rice blast fungus Magnaporthe grisea. Genes Dev. 1996, 10, 2696–2706. [Google Scholar] [CrossRef] [PubMed]
- Thines, E.; Weber, R.W.; Talbot, N.J. MAP kinase and protein kinase A–dependent mobilization of triacylglycerol and glycogen during appressorium turgor generation by Magnaporthe grisea. Plant Cell 2000, 12, 1703–1718. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Y.; Thornton, C.R.; Kershaw, M.J.; Debao, L.; Talbot, N.J. The glyoxylate cycle is required for temporal regulation of virulence by the plant pathogenic fungus Magnaporthe grisea. Mol. Microbiol. 2003, 47, 1601–1612. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Y.; Soanes, D.M.; Kershaw, M.J.; Talbot, N.J. Functional analysis of lipid metabolism in Magnaporthe grisea reveals a requirement for peroxisomal fatty acid β-oxidation during appressorium-mediated plant infection. Mol. Plant-Microbe Interact. 2007, 20, 475–491. [Google Scholar] [CrossRef]
- Gowda, M.; Venu, R.C.; Raghupathy, M.B.; Nobuta, K.; Li, H.; Wing, R.; Stahlberg, E.; Couglan, S.; Haudenschild, C.D.; Dean, R.; et al. Deep and comparative analysis of the mycelium and appressorium transcriptomes of Magnaporthe grisea using MPSS, RL-SAGE, and oligoarray methods. BMC Genom. 2006, 7, 1. [Google Scholar] [CrossRef] [PubMed]
- Mathioni, S.M.; Beló, A.; Rizzo, C.J.; Dean, R.A.; Donofrio, N.M. Transcriptome profiling of the rice blast fungus during invasive plant infection and in vitro stresses. BMC Genom. 2011, 12, 1. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Chung, H.; Lee, G.W.; Koh, S.K.; Chae, S.K.; Lee, Y.H. Genome-wide analysis of hypoxia-responsive genes in the rice blast fungus, Magnaporthe oryzae. PLoS ONE 2015, 10, e0134939. [Google Scholar] [CrossRef]
- Soanes, D.M.; Chakrabarti, A.; Paszkiewicz, K.H.; Dawe, A.L.; Talbot, N.J. Genome-wide transcriptional profiling of appressorium development by the rice blast fungus Magnaporthe oryzae. PLoS Pathog. 2012, 8, e1002514. [Google Scholar] [CrossRef] [PubMed]
- Nagalakshmi, U.; Wang, Z.; Waern, K.; Shou, C.; Raha, D.; Gerstein, M.; Snyder, M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 2008, 320, 1344–1349. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; O’Malley, R.C.; Tonti-Filippini, J.; Gregory, B.D.; Berry, C.C.; Millar, A.H.; Ecker, J.R. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 2008, 133, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Cloonan, N.; Forrest, A.R.; Kolle, G.; Gardiner, B.B.; Faulkner, G.J.; Brown, M.K.; Taylor, D.F.; Steptoe, A.L.; Wani, S.; Bethel, G.; et al. Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat. Methods 2008, 5, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Kramer, M.; Dutkowski, J.; Yu, M.; Bafna, V.; Ideker, T. Inferring gene ontologies from pairwise similarity data. Bioinformatics 2014, 30, i34–i42. [Google Scholar] [CrossRef] [PubMed]
- Ames, R.M.; MacPherson, J.I.; Pinney, J.W.; Lovell, S.C.; Robertson, D.L. Modular Biological Function Is Most Effectively Captured by Combining Molecular Interaction Data Types. PLoS ONE 2013, 8, e62670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutkowski, J.; Kramer, M.; Surma, M.; Balakrishnan, R.; Cherry, J.; Krogan, N.; Ideker, T. A gene ontology inferred from molecular networks. Nat. Biotechnol. 2013, 31, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Kersey, P.J.; Allen, J.E.; Armean, I.; Boddu, S.; Bolt, B.J.; Carvalho-Silva, D.; Christensen, M.; Davis, P.; Falin, L.J.; Grabmueller, C.; et al. Ensembl Genomes 2016: More genomes, more complexity. Nucleic Acids Res. 2016, 44, D574–D580. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed]
- Penn, T.J.; Wood, M.E.; Soanes, D.M.; Csukai, M.; Corran, A.J.; Talbot, N.J. Protein kinase C is essential for viability of the rice blast fungus Magnaporthe oryzae. Mol. Microbiol. 2015, 98, 403–419. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.; Blake, J.; Botstein, D.; Butler, H.; Cherry, J.; Davis, A.; Dolinski, K.; Dwight, S.; Eppig, J.; et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Soanes, D.E.; University of Exeter, Exeter, U.K. Personal Communication, 2015.
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; et al. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B (Methodological) 1995, 57, 289–300. [Google Scholar]
- Meng, S.; Brown, D.E.; Ebbole, D.J.; Torto-Alalibo, T.; Oh, Y.Y.; Deng, J.; Mitchell, T.K.; Dean, R.A. Gene Ontology annotation of the rice blast fungus, Magnaporthe oryzae. BMC Microbiol. 2009, 9, S8. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.; Donofrio, N.; Pan, H.; Coughlan, S.; Brown, D.E.; Meng, S.; Mitchell, T.; Dean, R.A. Transcriptome analysis reveals new insight into appressorium formation and function in the rice blast fungus Magnaporthe oryzae. Genome Biol. 2008, 9, 1. [Google Scholar] [CrossRef]
- Seong, K.; Li, L.; Hou, Z.; Tracy, M.; Kistler, H.C.; Xu, J.R. Cryptic promoter activity in the coding region of the HMG-CoA reductase gene in Fusarium graminearum. Fungal Genet. Biol. 2006, 43, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Carvunis, A.R.; Ideker, T. Siri of the Cell: What Biology Could Learn from the iPhone. Cell 2014, 157, 534–538. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.N.; Kroken, S.; Chou, D.Y.; Robbertse, B.; Yoder, O.; Turgeon, B.G. Functional analysis of all nonribosomal peptide synthetases in Cochliobolus heterostrophus reveals a factor, NPS6, involved in virulence and resistance to oxidative stress. Eukaryot. Cell 2005, 4, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Oide, S.; Moeder, W.; Krasnoff, S.; Gibson, D.; Haas, H.; Yoshioka, K.; Turgeon, B.G. NPS6, encoding a nonribosomal peptide synthetase involved in siderophore-mediated iron metabolism, is a conserved virulence determinant of plant pathogenic ascomycetes. Plant Cell 2006, 18, 2836–2853. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Brown, M.S. Regulation of the mevalonate pathway. Nature 1990, 343, 425. [Google Scholar] [CrossRef] [PubMed]
- Hirai, N.; Yoshida, R.; Todoroki, Y.; Ohigashi, H. Biosynthesis of abscisic acid by the non-mevalonate pathway in plants, and by the mevalonate pathway in fungi. Biosci. Biotechnol. Biochem. 2000, 64, 1448–1458. [Google Scholar] [CrossRef] [PubMed]
- Siewers, V.; Kokkelink, L.; Smedsgaard, J.; Tudzynski, P. Identification of an abscisic acid gene cluster in the grey mold Botrytis cinerea. Appl. Environ. Microbiol. 2006, 72, 4619–4626. [Google Scholar] [CrossRef] [PubMed]
- Spence, C.A.; Lakshmanan, V.; Donofrio, N.; Bais, H.P. Crucial roles of abscisic acid biogenesis in virulence of rice blast fungus Magnaporthe oryzae. Frontiers Plant Sci. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Dutkowski, J.; Ono, K.; Kramer, M.; Yu, M.; Pratt, D.; Demchak, B.; Ideker, T. NeXO Web: The NeXO ontology database and visualization platform. Nucleic Acids Res. 2013. p. gkt1192. [Google Scholar] [CrossRef] [PubMed]
- McKinney, J.D.; zu Bentrup, K.H.; Muñoz-Elías, E.J.; Miczak, A.; Chen, B.; Chan, W.T.; Swenson, D.; Sacchettini, J.C.; Jacobs, W.R.; Russell, D.G. Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature 2000, 406, 735–738. [Google Scholar] [CrossRef]
- Lorenz, M.C.; Fink, G.R. The glyoxylate cycle is required for fungal virulence. Nature 2001, 412, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.b.; Chen, G.Q.; Min, H.; Lin, F.C. MoFLP1, encoding a novel fungal fasciclin-like protein, is involved in conidiation and pathogenicity in Magnaporthe oryzae. J. Zhejiang Univ. Sci. B 2009, 10, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Park, S.W.; Bae, J.S.; Kim, K.S.; Park, S.H.; Lee, B.H.; Choi, J.Y.; Park, J.Y.; Ha, S.W.; Kim, Y.L.; Kwon, T.H.; et al. Beta ig-h3 promotes renal proximal tubular epithelial cell adhesion, migration and proliferation through the interaction with α3β1 integrin. Exp. Mol. Med. 2004, 36, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Nishi, N.; Nomizu, M. Characterization of a fasciclin I-like protein with cell attachment activity from sea urchin (Strongylocentrotus intermedius) ovaries. Arch. Biochem. Biophys. 2004, 424, 1–10. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Zhang, Y.; Chen, H.; Zhang, Z.; Peng, Y.L. The prediction of protein-protein interaction networks in rice blast fungus. BMC Genom. 2008, 9, 519. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entity ID | Up-Regulated Genes | Entity Size | ||
|---|---|---|---|---|
| CLX0008732 | 16 | 88 | 0.007 | 0.016 |
| CLX0008701 | 8 | 18 | 8.776 × 10 | 3.510 × 10 |
| CLX0008738 | 30 | 183 | 0.001 | 0.004 |
| CLX0008733 | 29 | 91 | 1.268 × 10 | 2.537 × 10 |
| CLX0008723 | 15 | 61 | 2.959 × 10 | 9.864 × 10 |
| CLX0008717 | 16 | 44 | 7.793 × 10 | 5.195 × 10 |
| CLX0008704 | 6 | 23 | 0.014 | 0.029 |
| CLX0008709 | 11 | 25 | 4.678 × 10 | 2.339 × 10 |
| CLX0008712 | 8 | 31 | 0.005 | 0.013 |
| CLX0008718 | 18 | 48 | 9.465 × 10 | 9.465 × 10 |
| CLX0008721 | 11 | 58 | 0.018 | 0.032 |
| Entity ID | Up-Regulated Genes | Entity Size | ||
|---|---|---|---|---|
| CLX0008666 | 5 | 7 | 3.535 × 10 | 0.001 |
| CLX0008638 | 3 | 5 | 0.012 | 0.030 |
| CLX0008664 | 5 | 7 | 3.535 × 10 | 0.001 |
| CLX0008726 | 34 | 69 | 9.521 × 10 | 2.951 × 10 |
| CLX0008701 | 6 | 18 | 0.012 | 0.030 |
| CLX0008703 | 14 | 22 | 1.022 × 10 | 1.584 × 10 |
| CLX0008734 | 20 | 99 | 0.011 | 0.030 |
| CLX0008695 | 6 | 13 | 0.001 | 0.008 |
| CLX0008731 | 19 | 85 | 0.005 | 0.021 |
| CLX0008733 | 25 | 91 | 4.043 × 10 | 3.133 × 10 |
| CLX0008722 | 14 | 59 | 0.007 | 0.022 |
| CLX0008712 | 9 | 31 | 0.006 | 0.022 |
| CLX0008718 | 11 | 48 | 0.021 | 0.048 |
| CLX0008721 | 23 | 58 | 4.601 × 10 | 4.754 × 10 |
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ames, R.M. Using Network Extracted Ontologies to Identify Novel Genes with Roles in Appressorium Development in the Rice Blast Fungus Magnaporthe oryzae. Microorganisms 2017, 5, 3. https://doi.org/10.3390/microorganisms5010003
Ames RM. Using Network Extracted Ontologies to Identify Novel Genes with Roles in Appressorium Development in the Rice Blast Fungus Magnaporthe oryzae. Microorganisms. 2017; 5(1):3. https://doi.org/10.3390/microorganisms5010003
Chicago/Turabian StyleAmes, Ryan M. 2017. "Using Network Extracted Ontologies to Identify Novel Genes with Roles in Appressorium Development in the Rice Blast Fungus Magnaporthe oryzae" Microorganisms 5, no. 1: 3. https://doi.org/10.3390/microorganisms5010003
APA StyleAmes, R. M. (2017). Using Network Extracted Ontologies to Identify Novel Genes with Roles in Appressorium Development in the Rice Blast Fungus Magnaporthe oryzae. Microorganisms, 5(1), 3. https://doi.org/10.3390/microorganisms5010003