Transcriptional Analysis Allows Genome Reannotation and Reveals that Cryptococcus gattii VGII Undergoes Nutrient Restriction during Infection

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. RNA-seq Data
2.3. Reads Alignment and Gene Prediction Refinement
2.4. RNA Isolation and RT-PCR
2.5. BAL Expression Analysis
2.6. Functinal Enrichment Analysis
2.7. Transcriptogram
2.8. KEGG Pathway Mapping
3. Results
3.1. Refinement of C. gattii R265 Genome Annotation
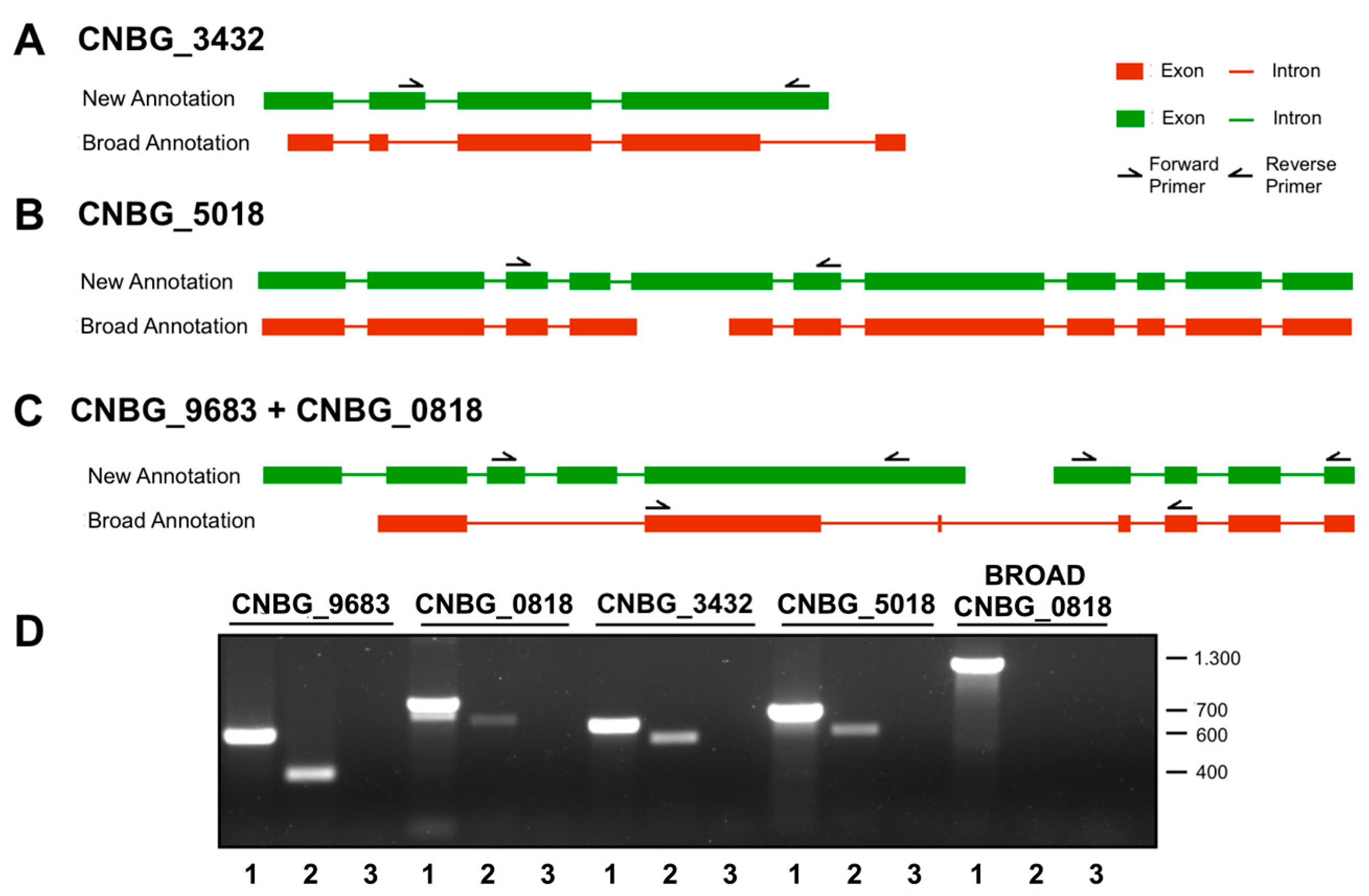
3.2. Experimental Validation of the Gene Models
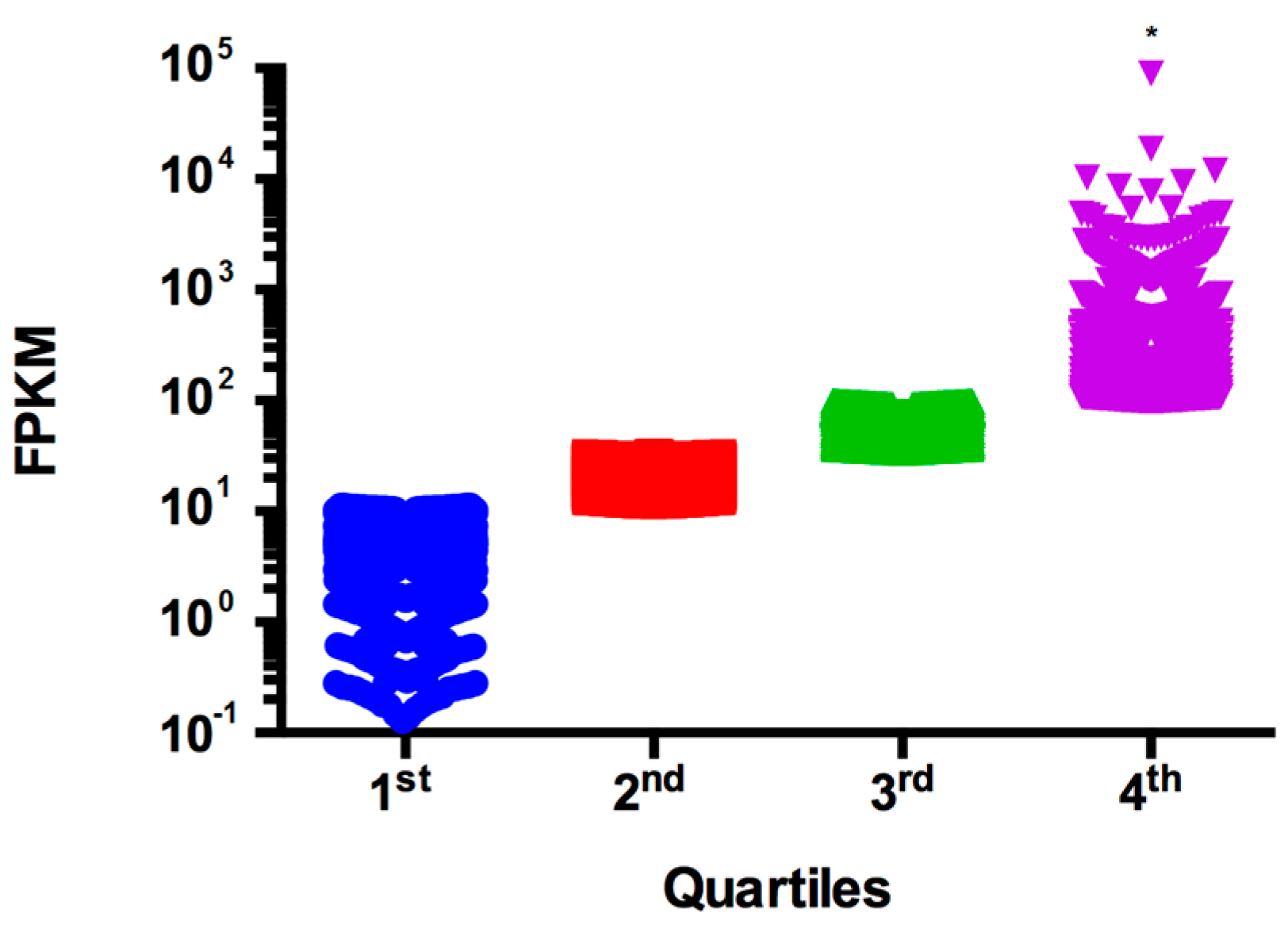
3.3. Transcriptional Profiling of C. gattii Recovered from Murine BAL
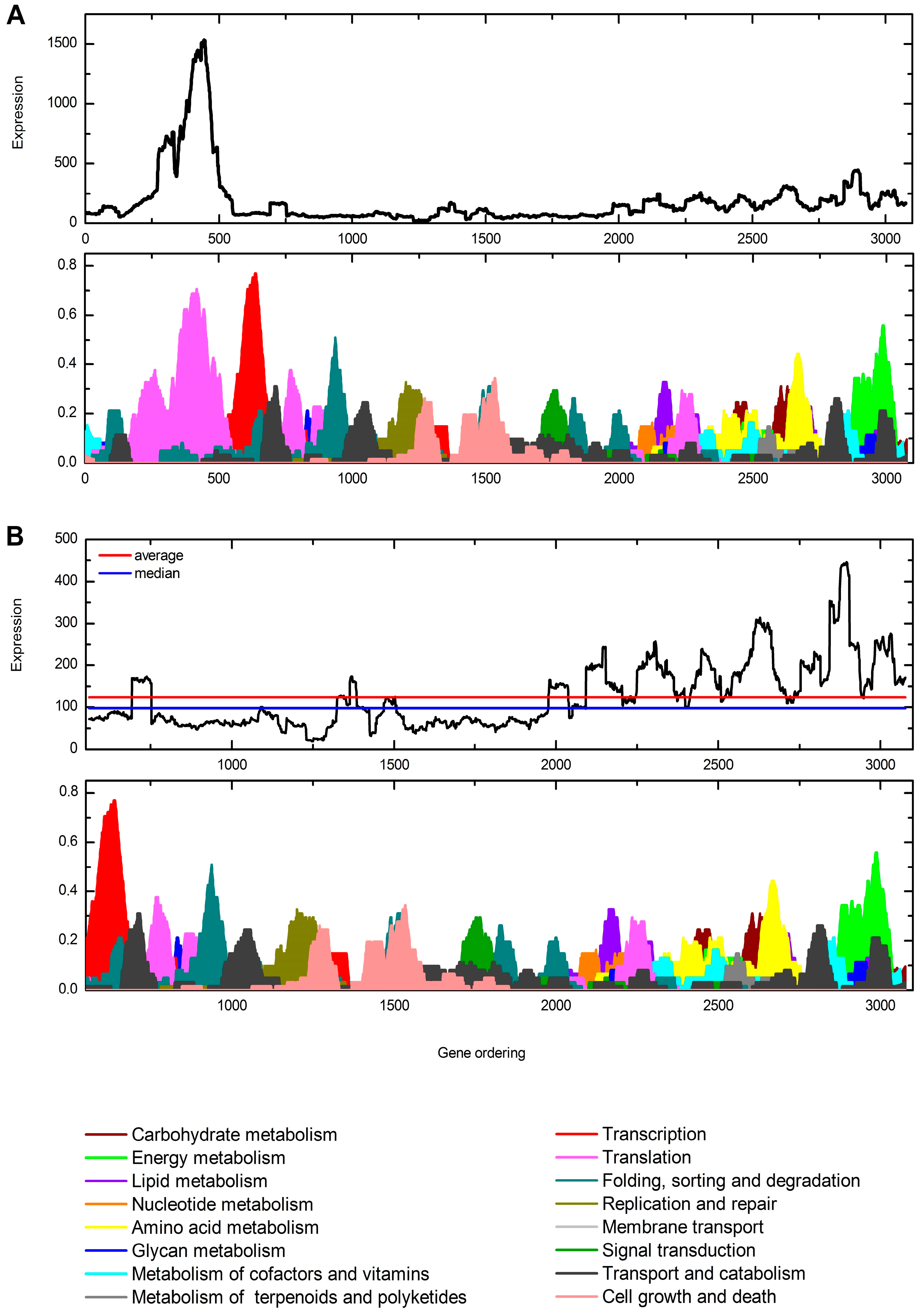
3.4. Functional Profiling of C. gattii Transcriptome Recovered from Murine BAL
3.5. Virulence Gene Expression in C. gattii Recovered from Murine BAL
3.6. Nutrient Uptake Gene Expression in C. gattii Recovered from Murine BAL
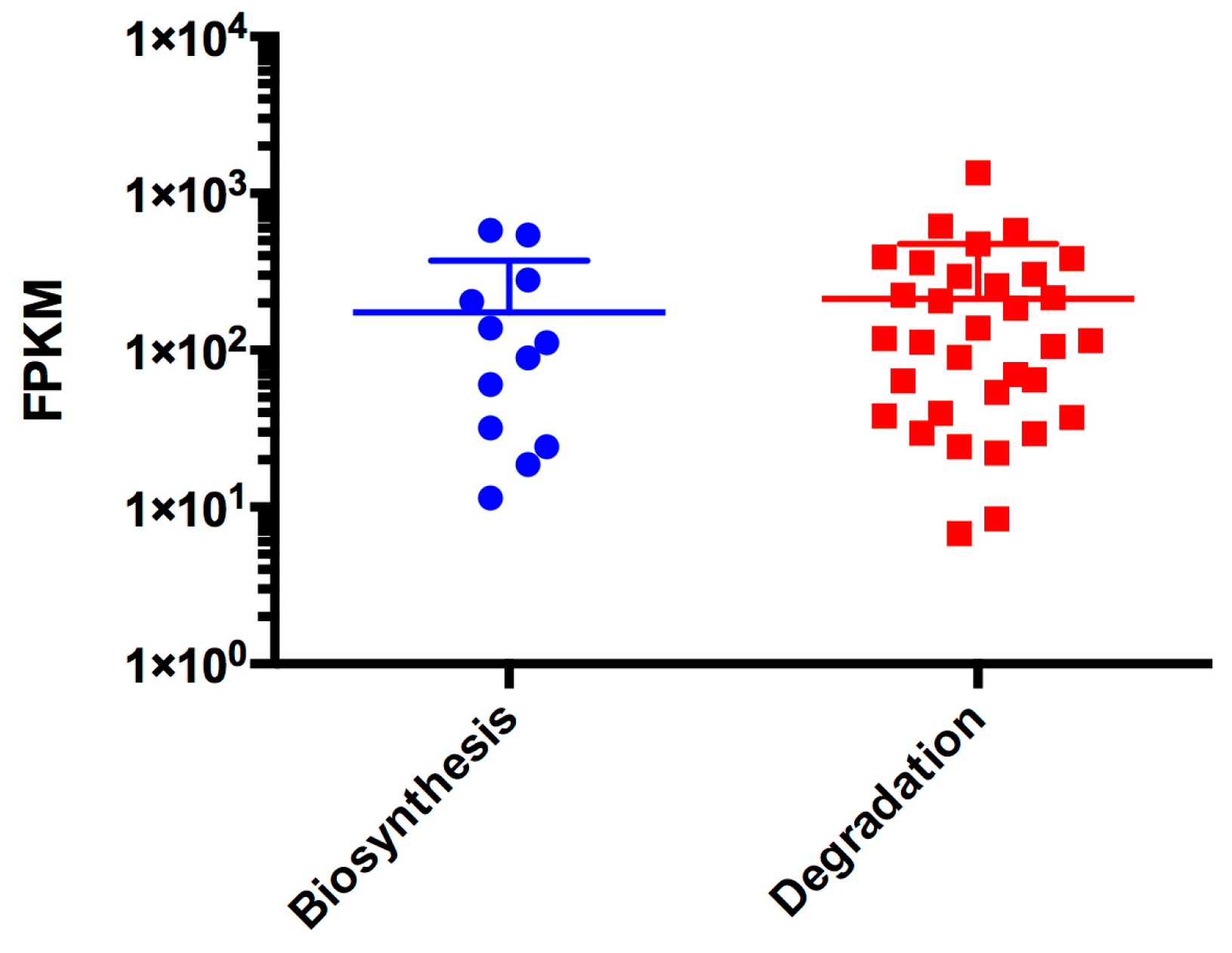
3.7. Nitrogen and Amino Acid Metabolism
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Vallabhaneni, S.; Mody, R.K.; Walker, T.; Chiller, T. The Global Burden of Fungal Diseases. Infect. Dis. Clin. N. Am. 2016, 30, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Rajasingham, R.; Smith, R.M.; Park, B.J.; Jarvis, J.N.; Govender, N.P.; Chiller, T.M.; Denning, D.W.; Loyse, A.; Boulware, D.R. Global burden of disease of HIV-associated cryptococcal meningitis: An updated analysis. Lancet Infect. Dis. 2017. [Google Scholar] [CrossRef]
- Meyer, W.; Gilgado, F.; Ngamskulrungroj, P.; Trilles, L.; Hagen, F.; Castañeda, E.; Boekhout, T. Molecular Typing of the Cryptococcus neoformans/Cryptococcus gattii Species Complex. In Cryptococcus; American Society of Microbiology: Washington, DC, USA, 2011; pp. 327–357. [Google Scholar]
- Chaturvedi, V.; Chaturvedi, S. Cryptococcus gattii: A resurgent fungal pathogen. Trends Microbiol. 2011, 19, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Kidd, S.E.; Hagen, F.; Tscharke, R.L.; Huynh, M.; Bartlett, K.H.; Fyfe, M.; Macdougall, L.; Boekhout, T.; Kwon-Chung, K.J.; Meyer, W. A rare genotype of Cryptococcus gattii caused the cryptococcosis outbreak on Vancouver Island (British Columbia, Canada). Proc. Natl. Acad. Sci. USA 2004, 101, 17258–17263. [Google Scholar] [CrossRef] [PubMed]
- Byrnes, E.J.; Marr, K.A. The Outbreak of Cryptococcus gattii in Western North America: Epidemiology and Clinical Issues. Curr. Infect. Dis. Rep. 2011, 13, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Byrnes, E.J.; Li, W.; Lewit, Y.; Ma, H.; Voelz, K.; Ren, P.; Carter, D.A.; Chaturvedi, V.; Bildfell, R.J.; May, R.C.; et al. Emergence and pathogenicity of highly virulent Cryptococcus gattii genotypes in the northwest United States. PLoS Pathog. 2010, 6, e1000850. [Google Scholar] [CrossRef] [PubMed]
- Leopold Wager, C.M.; Hole, C.R.; Wozniak, K.L.; Wormley, F.L., Jr. Cryptococcus and Phagocytes: Complex Interactions that Influence Disease Outcome. Front. Microbiol. 2016, 7, 105. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Sun, D.; Shi, M. Dancing cheek to cheek: Cryptococcus neoformans and phagocytes. Springerplus 2015, 4, 410. [Google Scholar] [CrossRef] [PubMed]
- DeLeon-Rodriguez, C.M.; Casadevall, A. Cryptococcus neoformans: Tripping on acid in the phagolysosome. Front. Microbiol. 2016, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Johnston, S.A.; May, R.C. Cryptococcus interactions with macrophages: Evasion and manipulation of the phagosome by a fungal pathogen. Cell Microbiol. 2013, 15, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Tucker, S.C.; Casadevall, A. Replication of Cryptococcus neoformans in macrophages is accompanied by phagosomal permeabilization and accumulation of vesicles containing polysaccharide in the cytoplasm. Proc. Natl. Acad. Sci. USA 2002, 99, 3165–3170. [Google Scholar] [CrossRef] [PubMed]
- Potrykus, J.; Ballou, E.R.; Childers, D.S.; Brown, A.J. Conflicting interests in the pathogen-host tug of war: Fungal micronutrient scavenging versus mammalian nutritional immunity. PLoS Pathog. 2014, 10, e1003910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kehl-Fie, T.E.; Chitayat, S.; Hood, M.I.; Damo, S.; Restrepo, N.; Garcia, C.; Munro, K.A.; Chazin, W.J.; Skaar, E.P. Nutrient metal sequestration by calprotectin inhibits bacterial superoxide defense, enhancing neutrophil killing of Staphylococcus aureus. Cell Host Microbe 2011, 10, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Cheng, P.Y.; Sham, A.; Perfect, J.R.; Kronstad, J.W. Metabolic adaptation in Cryptococcus neoformans during early murine pulmonary infection. Mol. Microbiol. 2008, 69, 1456–1475. [Google Scholar] [CrossRef] [PubMed]
- Derengowski, L.d.S.; Paes, H.C.; Albuquerque, P.; Tavares, A.H.; Fernandes, L.; Silva-Pereira, I.; Casadevall, A. The transcriptional response of Cryptococcus neoformans to ingestion by Acanthamoeba castellanii and macrophages provides insights into the evolutionary adaptation to the mammalian host. Eukaryot. Cell 2013, 12, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Ngamskulrungroj, P.; Chang, Y.; Sionov, E.; Kwon-Chung, K.J. The primary target organ of Cryptococcus gattii is different from that of Cryptococcus neoformans in a murine model. MBio 2012, 3, e00103-12. [Google Scholar] [CrossRef] [PubMed]
- Babraham Bioinformatics. FastQC: A Quality Control Tool for High throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 1 July 2017).
- Hannon Lab. FASTX-Toolkit. Available online: http://hannonlab.cshl.edu/fastx_toolkit (accessed on 1 July 2017).
- De Oliveira Schneider, R.; de Souza Süffert Fogaça, N.; Kmetzsch, L.; Schrank, A.; Vainstein, M.H.; Staats, C.C. Zap1 regulates zinc homeostasis and modulates virulence in Cryptococcus gattii. PLoS ONE 2012, 7, e43773. [Google Scholar]
- Broad Institute. Cryptococcus Gattii R265 Genome and Annotation. Available online: http://archive.broadinstitute.org/ftp/pub/annotation/fungi/cryptococcus_gattii/genomes/cryptococcus_gattii_r265 (accessed on 1 July 2015).
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed]
- Testa, A.C.; Hane, J.K.; Ellwood, S.R.; Oliver, R.P. CodingQuarry: Highly accurate hidden Markov model gene prediction in fungal genomes using RNA-seq transcripts. BMC Genom. 2015, 16, 170. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Stajich, J.E.; Harris, T.; Brunk, B.P.; Brestelli, J.; Fischer, S.; Harb, O.S.; Kissinger, J.C.; Li, W.; Nayak, V.; Pinney, D.F.; et al. FungiDB: An integrated functional genomics database for fungi. Nucleic Acids Res. 2012, 40, D675–D681. [Google Scholar] [CrossRef] [PubMed]
- Rybarczyk-Filho, J.L.; Castro, M.A.A.; Dalmolin, R.J.S.; Moreira, J.C.F.; Brunnet, L.G.; De Almeida, R.M.C. Towards a genome-wide transcriptogram: The Saccharomyces cerevisiae case. Nucleic Acids Res. 2010, 39, 3005–3016. [Google Scholar] [CrossRef] [PubMed]
- String Database. Protein Network Data from Cryptococcus gattii WM276. Available online: https://string-db.org/cgi/download.pl?UserId=6Sz4kwKfQN72&sessionId=DlfdC4uHXzBV&species_text=Cryptococcus+gattii+WM276 (accessed on 1 July 2017).
- KEGG Pathways. Cryptococcus gattii WM276. Available online: http://www.kegg.jp/kegg-bin/search_pathway_text?map=cgi&keyword=&mode=1&viewImage=true (accessed on 1 July 2017).
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Pant, G.; Bhavnasi, Y.K.; Blanchard, S.G.; Brouwer, C. Pathview Web: User friendly pathway visualization and data integration. Nucleic Acids Res. 2017, 175, 1292–1302. [Google Scholar] [CrossRef] [PubMed]
- Urban, M.; Cuzick, A.; Rutherford, K.; Irvine, A.; Pedro, H.; Pant, R.; Sadanadan, V.; Khamari, L.; Billal, S.; Mohanty, S.; et al. PHI-base: A new interface and further additions for the multi-species pathogen–host interactions database. Nucleic Acids Res. 2017, 45, D604–D610. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, F.M.; Piffer, A.C.; Schneider, R.D.O.; Ribeiro, N.S.; Garcia, A.W.A.; Schrank, A.; Kmetzsch, L.; Vainstein, M.H.; Staats, C.C. Alterations of zinc homeostasis in response to Cryptococcus neoformans in a murine macrophage cell line. Future Microbiol. 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Jung, W.H.; Sham, A.; Lian, T.; Singh, A.; Kosman, D.J.; Kronstad, J.W. Iron source preference and regulation of iron uptake in Cryptococcus neoformans. PLoS Pathog. 2008, 4, e45. [Google Scholar] [CrossRef] [PubMed]
- Caza, M.; Kronstad, J.W. Shared and distinct mechanisms of iron acquisition by bacterial and fungal pathogens of humans. Front. Cell. Infect. Microbiol. 2013, 3, 80. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.; Yang, D.; Maeng, S.; Lee, K.; So, Y.; Hong, J.; Choi, J.; Byun, H.; Kim, H.; Bang, S.; et al. Systematic functional profiling of transcription factor networks in Cryptococcus neoformans. Nat. Commun. 2015, 6, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Waalwijk, C.; De Wit, P.J.G.M.; Tang, D.; Van Der Lee, T. RNA-Seq analysis reveals new gene models and alternative splicing in the fungal pathogen Fusarium graminearum. BMC Genom. 2013, 14, 21. [Google Scholar] [CrossRef] [PubMed]
- Schliebner, I.; Becher, R.; Hempel, M.; Deising, H.B.; Horbach, R. New gene models and alternative splicing in the maize pathogen Colletotrichum graminicola revealed by RNA-Seq analysis. BMC. Genom. 2014, 15, 842. [Google Scholar] [CrossRef] [PubMed]
- Janbon, G.; Ormerod, K.L.; Paulet, D.; Byrnes, E.J.; Yadav, V.; Chatterjee, G.; Mullapudi, N.; Hon, C.C.; Billmyre, R.B.; Brunel, F.; et al. Analysis of the Genome and Transcriptome of Cryptococcus neoformans var. grubii Reveals Complex RNA Expression and Microevolution Leading to Virulence Attenuation. PLoS Genet. 2014, 10, e1004261. [Google Scholar] [CrossRef] [PubMed]
- Bruno, V.M.; Wang, Z.; Marjani, S.L.; Euskirchen, G.M.; Martin, J.; Sherlock, G.; Snyder, M. Comprehensive annotation of the transcriptome of the human fungal pathogen Candida albicans using RNA-seq. Genome Res. 2010, 20, 1451–1458. [Google Scholar] [CrossRef] [PubMed]
- Haas, A. The Phagosome: Compartment with a License to Kill. Traffic 2007, 8, 311–330. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Kraus, P.R.; Boily, M.J.; Heitman, J. Cryptococcus neoformans gene expression during murine macrophage infection. Eukaryot. Cell 2005, 4, 1420–1433. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Toffaletti, D.L.; Tenor, J.L.; Litvintseva, A.P.; Fang, C.; Mitchell, T.G.; McDonald, T.R.; Nielsen, K.; Boulware, D.R.; Bicanic, T.; et al. The Cryptococcus neoformans transcriptome at the site of human meningitis. MBio 2014, 5, e01087-13. [Google Scholar] [CrossRef] [PubMed]
- Russel Lee, I.; Chow, E.W.L.; Morrow, C.A.; Djordjevic, J.T.; Fraser, J.A. Nitrogen metabolite repression of metabolism and virulence in the human fungal pathogen Cryptococcus neoformans. Genetics 2011, 188, 309–323. [Google Scholar]
- Kmetzsch, L.; Staats, C.C.; Simon, E.; Fonseca, F.L.; Oliveira, D.L.; Joffe, L.S.; Rodrigues, J.; Lourenco, R.F.; Gomes, S.L.; Nimrichter, L.; et al. The GATA-type transcriptional activator Gat1 regulates nitrogen uptake and metabolism in the human pathogen Cryptococcus neoformans. Fungal Genet. Biol. 2011, 48, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Ngamskulrungroj, P.; Chang, Y.; Roh, J.; Kwon-Chung, K.J. Differences in nitrogen metabolism between Cryptococcus neoformans and C. gattii, the two etiologic agents of cryptococcosis. PLoS ONE 2012, 7, e34258. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, J.C.; Lin, X.; Nielsen, K.; Heitman, J. Amt2 permease is required to induce ammonium-responsive invasive growth and mating in Cryptococcus neoformans. Eukaryot. Cell 2008, 7, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, T.S.; Andhare, R.; Cooper, T.G. Nitrogen catabolite repression of DAL80 expression depends on the relative levels of Gat1p and Ure2p production in Saccharomyces cerevisiae. J. Biol. Chem. 2000, 275, 14408–14414. [Google Scholar] [CrossRef] [PubMed]
- Luzzani, C.; Cardillo, S.B.; Moretti, M.B.; García, S.C. New insights into the regulation of the Saccharomyces cerevisiae UGA54 gene: Two parallel pathways participate in carbon-regulated transcription. Microbiology 2007, 153, 3677–3684. [Google Scholar] [CrossRef] [PubMed]
- Cardillo, S.B.; Moretti, M.B.; García, S.C. Uga3 and Uga35/Dal81 transcription factors regulate UGA4 transcription in response to γ-Aminobutyric acid and Leucine. Eukaryot. Cell 2010, 9, 1262–1271. [Google Scholar] [CrossRef] [PubMed]
- Martho, K.F.C.; De Melo, A.T.; Takahashi, J.P.F.; Guerra, J.M.; Da Silva Santos, D.C.; Purisco, S.U.; Melhem, M.D.S.C.; Dos Anjos Fazioli, R.; Phanord, C.; Sartorelli, P.; et al. Amino acid permeases and virulence in Cryptococcus neoformans. PLoS ONE 2016, 11, e0163919. [Google Scholar] [CrossRef] [PubMed]
- Liew, K.L.; Jee, J.M.; Yap, I.; Yong, P.V.C. In vitro analysis of metabolites secreted during infection of lung epithelial cells by Cryptococcus neoformans. PLoS ONE 2016, 11, e0153356. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, M.C.; Bender, J.A.; Fink, G.R. Transcriptional Response of Candida albicans upon Internalization by Macrophages Transcriptional Response of Candida albicans upon Internalization by Macrophages. Eukaryot. Cell 2004, 3, 1076–1087. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Name | Benjamini Corrected p-Value |
|---|---|---|
| GO:1901566 | Organonitrogen compound biosynthetic process | 8.59 × 10−11 |
| GO:1901564 | Organonitrogen compound metabolic process | 3.73 × 10−10 |
| GO:0006412 | Translation | 9.05 × 10−10 |
| GO:0043043 | Peptide biosynthetic process | 1.53 × 10−9 |
| GO:0043604 | Amide biosynthetic process | 2.40 × 10−9 |
| GO:0006518 | Peptide metabolic process | 2.65 × 10−9 |
| GO:0043603 | Cellular amide metabolic process | 6.16×10−9 |
| GO:0044271 | Cellular nitrogen compound biosynthetic process | 4.24 × 10−6 |
| GO:0055114 | Oxidation-reduction process | 5.89 × 10−6 |
| GO:0008152 | Metabolic process | 6.56 × 10−6 |
| GENE | DESCRIPTION | FPKM BAL |
|---|---|---|
| CNBG_0332 | Ammonium permease 1 (AMT1) | 415.458 |
| CNBG_6023 | Ammonium permease 2 (AMT2) | 40.3613 |
| CNBG_1602 | Gamma-aminobutyric acid transporter | 677.554 |
| CNBG_3901 | Gamma-aminobutyric acid transporter | 268.427 |
| CNBG_4571 | Gamma-aminobutyric acid transporter | 61.0116 |
| CNBG_4665 | Gamma-aminobutyric acid transporter | 4.20125 |
| CNBG_4156 | Choline transporter | 74.1659 |
| CNBG_5513 | l-methione transporter | 11.9407 |
| CNBG_4785 | General amino acid transporter (AAP2) | 441.806 |
| CNBG_1371 | General amino acid transporter (AAP4) | 416.72 |
| CNBG_9416 | General amino acid transporter (AAP1) | 363.575 |
| CNBG_6051 | General amino acid transporter (AAP6) | 32.0951 |
| CNBG_1350 | Gamma-aminobutyric acid transporter (AAP8) | 18.3341 |
| CNBG_2012 | Neutral amino acid permease | 258.968 |
| CNBG_1852 | Neutral amino acid permease | 194.189 |
| CNBG_2927 | Ure2p | 21.9955 |
| CNBG_4137 | Bwc2 | 148.2437 |
| CNBG_9614 | Cir1 | 6.505 |
| CNBG_0368 | Gat1 | 29.7171 |
| CNBG_3885 | Gat201 | 340.642 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferrareze, P.A.G.; Streit, R.S.A.; Santos, P.R.d.; Santos, F.M.d.; Almeida, R.M.C.d.; Schrank, A.; Kmetzsch, L.; Vainstein, M.H.; Staats, C.C. Transcriptional Analysis Allows Genome Reannotation and Reveals that Cryptococcus gattii VGII Undergoes Nutrient Restriction during Infection. Microorganisms 2017, 5, 49. https://doi.org/10.3390/microorganisms5030049
Ferrareze PAG, Streit RSA, Santos PRd, Santos FMd, Almeida RMCd, Schrank A, Kmetzsch L, Vainstein MH, Staats CC. Transcriptional Analysis Allows Genome Reannotation and Reveals that Cryptococcus gattii VGII Undergoes Nutrient Restriction during Infection. Microorganisms. 2017; 5(3):49. https://doi.org/10.3390/microorganisms5030049
Chicago/Turabian StyleFerrareze, Patrícia Aline Gröhs, Rodrigo Silva Araujo Streit, Patricia Ribeiro dos Santos, Francine Melise dos Santos, Rita Maria Cunha de Almeida, Augusto Schrank, Livia Kmetzsch, Marilene Henning Vainstein, and Charley Christian Staats. 2017. "Transcriptional Analysis Allows Genome Reannotation and Reveals that Cryptococcus gattii VGII Undergoes Nutrient Restriction during Infection" Microorganisms 5, no. 3: 49. https://doi.org/10.3390/microorganisms5030049
APA StyleFerrareze, P. A. G., Streit, R. S. A., Santos, P. R. d., Santos, F. M. d., Almeida, R. M. C. d., Schrank, A., Kmetzsch, L., Vainstein, M. H., & Staats, C. C. (2017). Transcriptional Analysis Allows Genome Reannotation and Reveals that Cryptococcus gattii VGII Undergoes Nutrient Restriction during Infection. Microorganisms, 5(3), 49. https://doi.org/10.3390/microorganisms5030049