Linking Compositional and Functional Predictions to Decipher the Biogeochemical Significance in DFAA Turnover of Abundant Bacterioplankton Lineages in the North Sea

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Site, Sampling and Sample Preparation
2.2. Nucleic Acid Extraction and Sequencing
2.3. Processing of 16S rRNA Data Sets
2.4. Statistical Data Analysis
2.5. Sequence Data Deposition
3. Results and Discussion
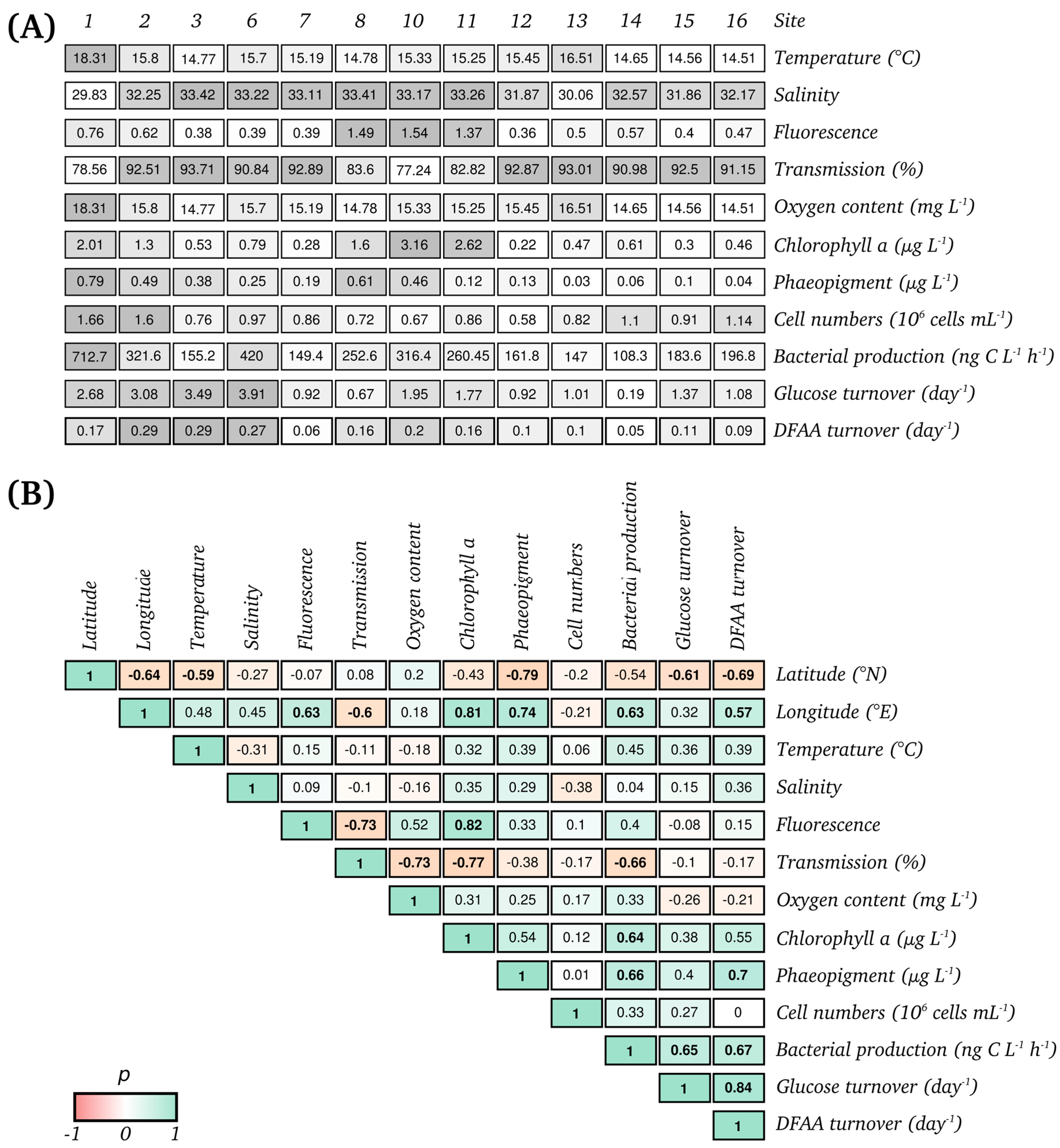
3.1. Biogeochemical and Microbial Characteristics
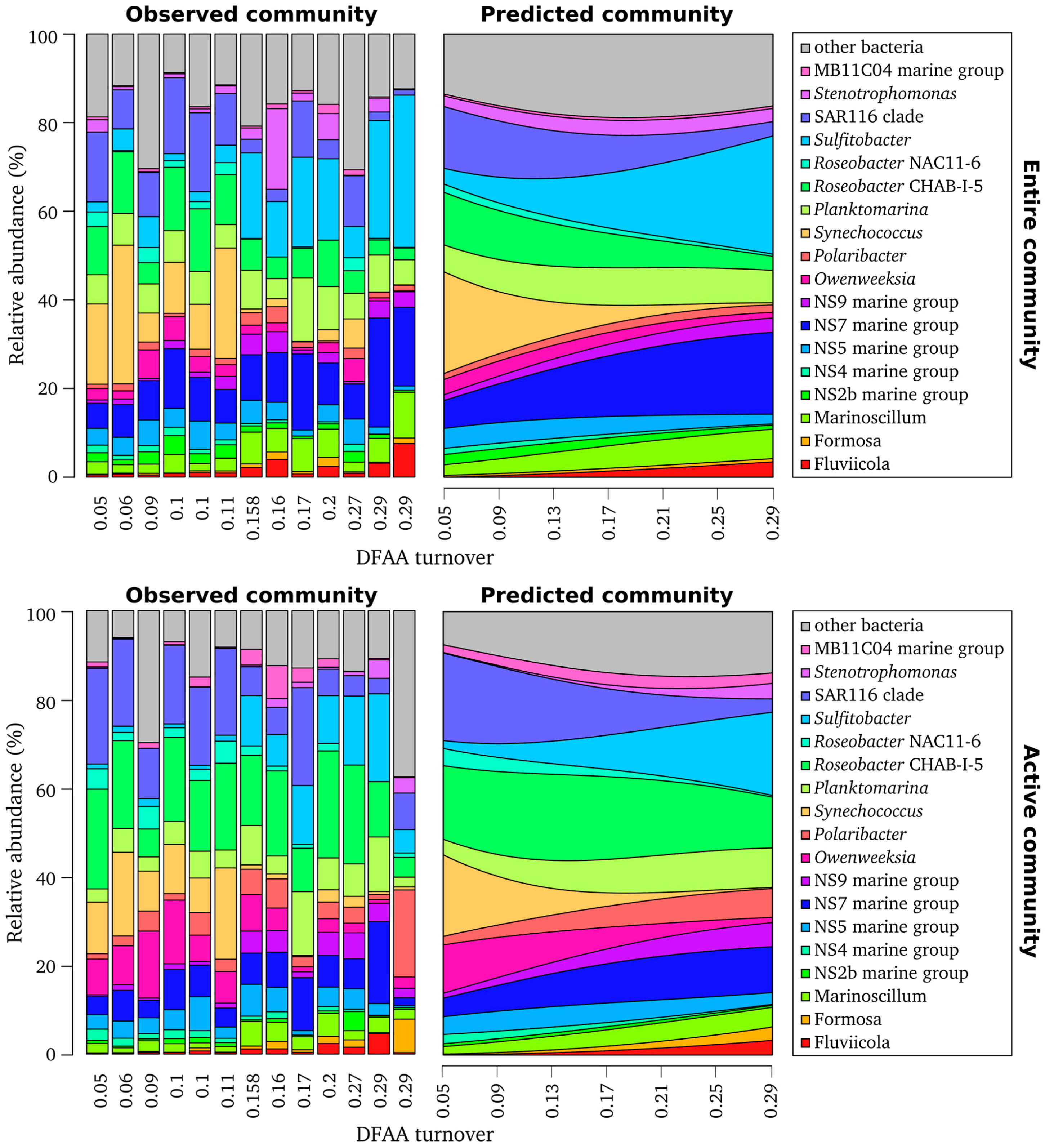
3.2. Composition of Entire and Active Bacterioplankton Community in the North Sea
3.3. Entire and Active Bacterial Communities Displayed Differences in Richness and Community Composition
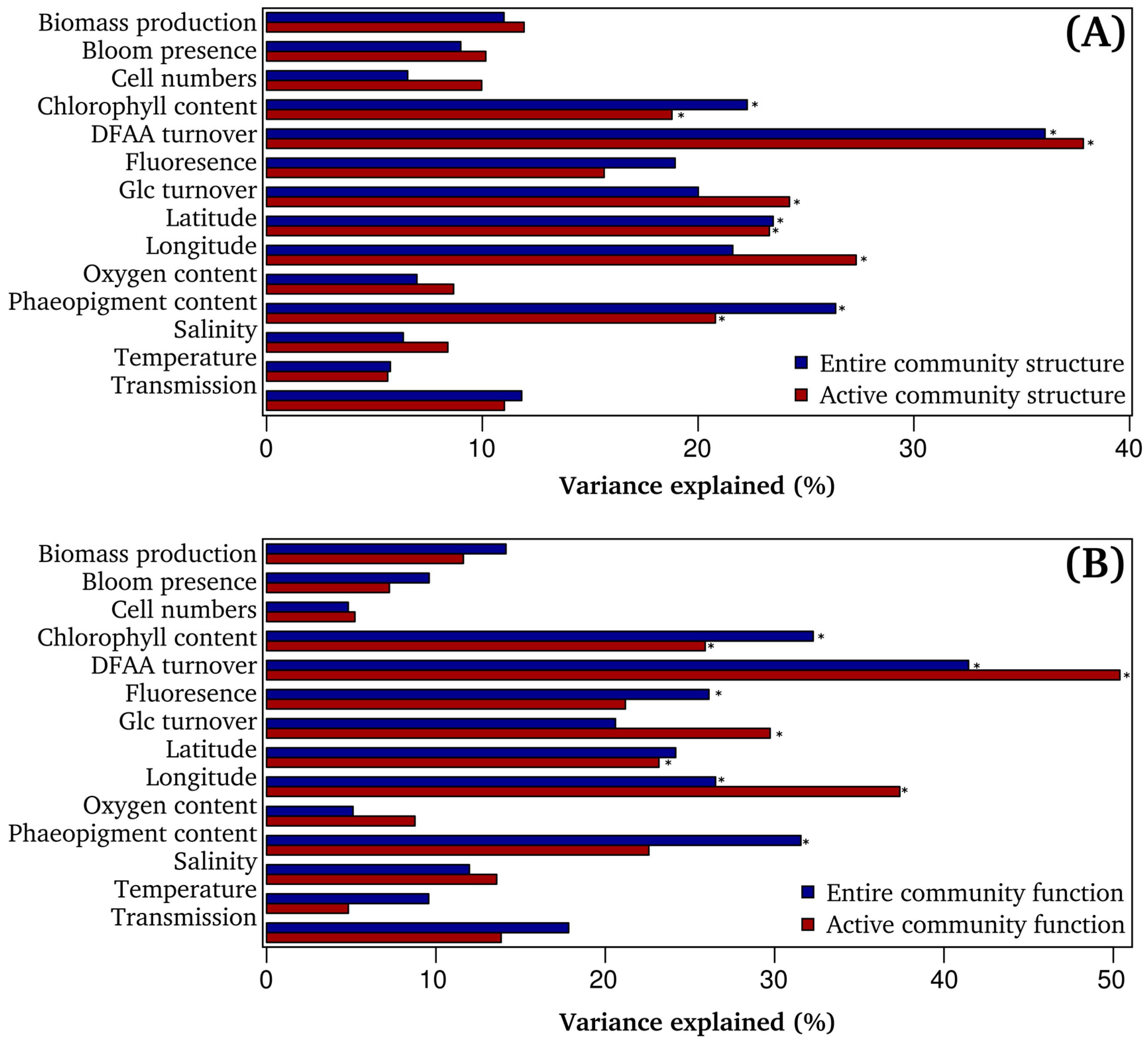
3.4. Bacterioplankton Community Composition and Function are Significantly Correlated to Environmental Properties
3.5. DFAA Turnover as Key Predictor of Bacterioplankton Community Composition
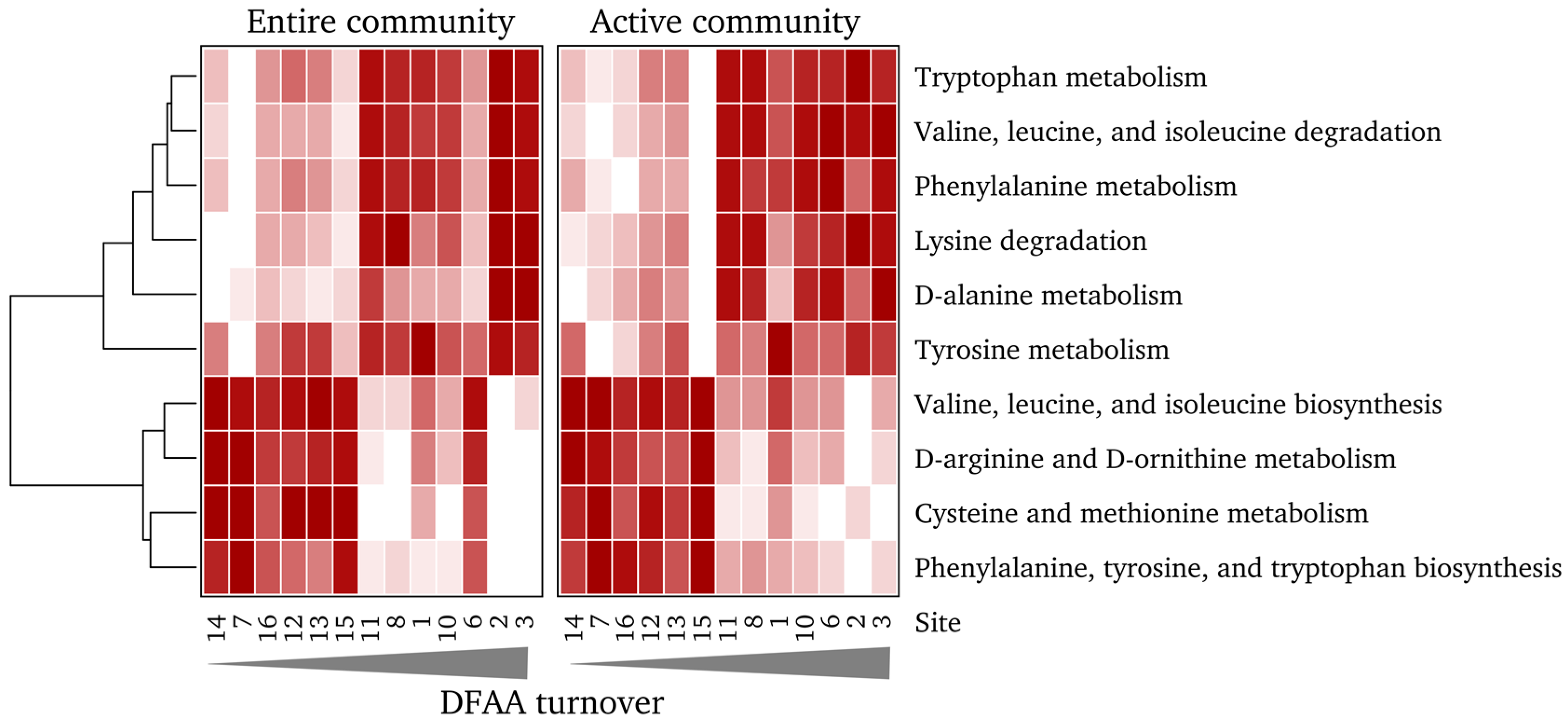
3.6. Community Function is Significantly Linked to Measured DFAA Turnover Rates
3.7. Study Limitations
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Arrigo, K.R. Marine microorganisms and global nutrient cycles. Nature 2005, 437, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Azam, F.; Malfatti, F. Microbial structuring of marine ecosystems. Nat. Rev. Microbiol. 2007, 5, 782–791. [Google Scholar] [CrossRef] [PubMed]
- DeLong, E.F. The microbial ocean from genomes to biomes. Nature 2009, 459, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Wemheuer, B.; Güllert, S.; Billerbeck, S.; Giebel, H.-A.; Voget, S.; Simon, M.; Daniel, R. Impact of a phytoplankton bloom on the diversity of the active bacterial community in the southern North Sea as revealed by metatranscriptomic approaches. FEMS Microbiol. Ecol. 2014, 87, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Sunagawa, S.; Coelho, L.P.; Chaffron, S.; Kultima, J.R.; Labadie, K.; Salazar, K.; Djahanschiri, B.; Zeller, G.; Mende, D.R.; Alberti, A.; et al. Structure and function of the global ocean microbiome. Science 2015, 348, 1261359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teeling, H.; Fuchs, B.M.; Bennke, C.M.; Krüger, K.; Chaee, M.; Kappelmann, L.; Reintjes, G.; Waldmann, J.; Quast, C.; Glöckner, F.O.; et al. Recurring patterns in bacterioplankton dynamics during coastal spring algae blooms. eLife 2016, 5, e11888. [Google Scholar] [CrossRef] [PubMed]
- Campbell, B.J.; Kirchman, D.L. Bacterial diversity, community structure and potential growth rates along an estuarine salinity gradient. ISME J. 2013, 7, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhao, Z.; Dai, M.; Jiao, N.; Herndl, G.J. Drivers shaping the diversity and biogeography of total and active bacterial communities in the South China Sea. Mol. Ecol. 2014, 23, 2260–2274. [Google Scholar] [CrossRef] [PubMed]
- Mock, T.; Daines, S.J.; Geider, R.; Collins, S.; Metodiev, M.; Millar, A.J.; Moulton, V.; Lenton, T.M. Bridging the gap between omics and earth system science to better understand how environmental change impacts marine microbes. Glob. Chang. Biol. 2016, 22, 61–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stingl, U.; Desiderio, R.A.; Cho, J.C.; Vergin, K.L.; Giovannoni, S.J. The SAR92 clade: An abundant coastal clade of culturable marine bacteria possessing proteorhodopsin. Appl. Environ. Microbiol. 2007, 73, 2290–2296. [Google Scholar] [CrossRef] [PubMed]
- Giebel, H.-A.; Kalhoefer, D.; Gahl-Janssen, R.; Choo, Y.-J.; Lee, K.; Cho, J.-C.; Tindall, B.J.; Rhiel, E.; Beardsley, C.; Aydogmus, Ö.; et al. Planktomarina temperata gen. nov., sp. nov., belonging to the globally distributed RCA cluster of the marine Roseobacter clade, isolated from the German Wadden Sea. Int. J. Syst. Evol. Microbiol. 2013, 63, 4207–4217. [Google Scholar] [CrossRef] [PubMed]
- Tsementzi, D.; Wu, J.; Deutsch, S.; Nath, S.; Rodriguez-R, L.M.; Burns, A.S.; Ranjan, P.; Sarode, N.; Malmstrom, R.R.; Padilla, C.C.; et al. SAR11 bacteria linked to ocean anoxia and nitrogen loss. Nature 2016, 536, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Teeling, H.; Fuchs, B.; Becher, D.; Klockow, C.; Gardebrecht, A.; Bennke, C.M.; Kassabgy, M.; Huang, S.; Mann, A.J.; Waldmann, J.; et al. Substrate-controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom. Science 2012, 336, 608–611. [Google Scholar] [CrossRef] [PubMed]
- Lucas, J.; Wichels, A.; Teeling, H.; Chafee, M.; Scharfe, M.; Gerdts, G. Annual dynamics of North Sea bacterioplankton: Seasonal variability superimposes short-term variation. FEMS Microbiol. Ecol. 2015, 91, fiv099. [Google Scholar] [CrossRef] [PubMed]
- Voget, S.; Wemheuer, B.; Brinkhoff, T.; Vollmers, J.; Dietrich, S.; Giebel, H.-A.; Beardsley, C.; Sardemann, C.; Bakenhus, I.; Billerbeck, S.; et al. Adaptation of an abundant Roseobacter RCA organism to pelagic systems revealed by genomic and transcriptomic analyses. ISME J. 2015, 9, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Wemheuer, B.; Wemheuer, F.; Hollensteiner, J.; Meyer, F.-D.; Voget, S.; Daniel, R. The green impact: Bacterioplankton response towards a phytoplankton spring bloom in the southern North Sea assessed by comparative metagenomic and metatranscriptomic approaches. Front. Microbiol. 2015, 6, 805. [Google Scholar] [CrossRef] [PubMed]
- Zubkov, M.V.; Fuchs, B.M.; Archer, S.D.; Kiene, R.P.; Amann, R.; Burkill, P.H. Linking the composition of bacterioplankton to rapid turnover of dissolved dimethylsulphoniopropionate in an algal bloom in the North Sea. Environ. Microbiol. 2001, 3, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Giebel, H.-A.; Kalhoefer, D.; Lemke, A.; Thole, S.; Gahl-Janssen, R.; Simon, M.; Brinkhoff, T. Distribution of Roseobacter RCA and SAR11 lineages in the North Sea and characteristics of an abundant RCA isolate. ISME J. 2011, 5, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Billerbeck, S.; Wemheuer, B.; Voget, S.; Poehlein, A.; Giebel, H.-A.; Brinkhoff, T.; Gram, L.; Jeffrey, W.H.; Daniel, R.; Meinhard, S. Biogeography and environmental genomics of the Roseobacter group affiliated pelagic CHAB-I-5 lineage. Nat. Microbiol. 2016, 1, 16063. [Google Scholar] [CrossRef] [PubMed]
- Aßhauer, K.P.; Wemheuer, B.; Daniel, R.; Meinicke, P. Tax4Fun: Predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics 2015, 31, 2882–2884. [Google Scholar] [CrossRef]
- McQuatters-Gollop, A.; Raitsos, D.E.; Edwards, M.; Pradhan, Y.; Mee, L.D.; Lavender, S.J.; Attrill, M.J. A long-term chlorophyll data set reveals regime shift in North Sea phytoplankton biomass unconnected to nutrient trends. Limnol. Oceanogr. 2007, 52, 635–648. [Google Scholar] [CrossRef]
- Osterholz, H.; Singer, G.; Wemheuer, B.; Daniel, R.; Simon, M.; Niggemann, J.; Dittmar, T. Deciphering associations between dissolved organic molecules and bacterial communities in a pelagic marine system. ISME J. 2016, 10, 1717–1730. [Google Scholar] [CrossRef] [PubMed]
- Wiltshire, K.H.; Manly, B.F.J. The warming trend at Helgoland Roads, North Sea: Phytoplankton response. Helgol. Mar. Res. 2004, 58, 269–273. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria, 2016. Available online: http://www.R-project.org/ (accessed on 17 August 2015).
- Brownrigg, R. Mapdata: Extra Map Databases. R Package Version 2.2-6. 2016. Available online: https://cran.r-project.org/web/packages/mapdata/ (accessed on 17 January 2016).
- Minka, T.P.; Deckmyn, A. Maps: Draw Geographical Maps. R Package Version 3.1.1. 2016. Available online: https://cran.r-project.org/web/packages/maps/ (accessed on 30 July 2016).
- Simon, M.; Azam, F. Protein content and protein synthesis rates of planktonic marine bacteria. Mar. Ecol. Prog. Ser. 1989, 51, 201–213. [Google Scholar] [CrossRef]
- Osterholz, H.; Niggemann, J.; Giebel, H.-A.; Simon, M.; Dittmar, T. Inefficient microbial production of refractory dissolved organic matter in the ocean. Nat. Commun. 2015, 6, 7422. [Google Scholar] [CrossRef] [PubMed]
- Schneider, D.; Wemheuer, F.; Pfeiffer, B.; Wemheuer, B. Extraction of total DNA and RNA from marine filter samples and generation of a cDNA as universal template for marker gene studies. In Metagenomics: Methods and Protocols, 2nd ed.; Streit, W.R., Daniel, R., Eds.; Springer: New York, NY, USA, 2017; pp. 13–22. ISBN 978-1-4939-6691-2. [Google Scholar]
- Muyzer, G.; Teske, A.; Wirsen, C.O.; Jannasch, H.W. Phylogenetic relationships of Thiomicrospira species and their identification in deep-sea hydrothermal vent samples by denaturing gradient gel electrophoresis of 16S rDNA fragments. Arch. Microbiol. 1995, 164, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Wemheuer, B.; Wemheuer, F.; Daniel, R. RNA-based assessment of diversity and composition of active archaeal communities in the German Bight. Archaea 2012, 2012, 695826. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Gonzalez Peña, A.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O'Hara, R.B.; Simpson, G.L.; Solymos, P.; Stevens, H.M.H.; Wagner, H.H. Vegan: Community Ecology Package. R Package Version 2.4-0. Available online: https://cran.r-project.org/web/packages/vegan/ (accessed on 18 June 2016).
- Ritz, C.; Streibig, J.C. Bioassay Analysis using R. J. Stat. Softw. 2005, 12. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 1995, 57, 289–300. [Google Scholar]
- Chen, J.; Bittinger, K.; Charlson, E.S.; Hoffmann, C.; Lewis, J.; Wu, G.D.; Collman, R.G.; Bushman, F.D.; Li, H. Associating microbiome composition with environmental covariates using generalized UniFrac distances. Bioinformatics 2012, 28, 2106–2113. [Google Scholar] [CrossRef] [PubMed]
- Venables, W.N.; Ripley, B.D. Modern Applied Statistics with S, 4th ed.; Springer: New York, NY, USA, 2002; ISBN 0-387-95457-0. [Google Scholar]
- Wietz, M.; Gram, L.; Jørgensen, B.; Schramm, A. Latitudinal patterns in the abundance of major marine bacterioplankton groups. Aquat. Microb. Ecol. 2010, 61, 179–189. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, Y.; Jiao, N.; Stepanauskas, R.; Luo, H. Ecological genomics of the uncultivated marine Roseobacter lineage CHAB-I-5. Appl. Environ. Microbiol. 2016, 82, 2100–2111. [Google Scholar] [CrossRef] [PubMed]
- Klindworth, A.; Mann, A.J.; Huang, S.; Wichels, A.; Quast, C.; Waldmann, J.; Teeling, H.; Glöckner, F.O. Diversity and activity of marine bacterioplankton during a diatom bloom in the North Sea assessed by total RNA and pyrotag sequencing. Mar. Genom. 2014, 18, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.M.; Rappe, M.S.; Connon, S.A.; Vergin, K.L.; Siebold, W.A.; Carlson, C.A. SAR11 clade dominates ocean surface bacterioplankton communities. Nature 2002, 420, 806–810. [Google Scholar] [CrossRef] [PubMed]
- Schattenhofer, M.; Fuchs, B.M.; Amann, R.; Zubkov, M.V.; Tarran, G.A.; Pernthaler, J. Latitudinal distribution of prokaryotic picoplankton populations in the Atlantic Ocean. Environ. Microbiol. 2009, 11, 2078–2093. [Google Scholar] [CrossRef] [PubMed]
- Alderkamp, A.C.; Sintes, E.; Herndl, G.J. Abundance and activity of major groups of prokaryotic plankton in the coastal North Sea during spring and summer. Aquat. Microb. Ecol. 2006, 45, 237–246. [Google Scholar] [CrossRef]
- De Corte, D.; Sintes, E.; Yokokawa, T.; Herndl, G.J. Comparison between MICRO–CARD–FISH and 16S rRNA gene clone libraries to assess the active versus total bacterial community in the coastal Arctic. Environ. Microbiol. Rep. 2013, 5, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Blazewicz, S.J.; Barnard, R.L.; Daly, R.A.; Firestone, M.K. Evaluating rRNA as an indicator of microbial activity in environmental communities: Limitations and uses. ISME J. 2013, 7, 2061–2068. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.C.; Nielsen, A.K.; Molin, S.; Hammer, K.; Kilstrup, M. Changes in rRNA levels during stress invalidates results from mRNA blotting: Fluorescence in situ rRNA hybridization permits renormalization for estimation of cellular mRNA levels. J. Bacteriol. 2001, 183, 4747–4751. [Google Scholar] [CrossRef] [PubMed]
- Moeseneder, M.M.; Arrieta, J.M.; Herndl, G.J. A comparison of DNA- and RNA-based clone libraries from the same marine bacterioplankton community. FEMS Microbiol. Ecol. 2005, 51, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Gentile, G.; Giuliano, L.; D'Auria, G.; Smedile, F.; Azzaro, M.; De Domenico, M.; Yakimov, M.M. Study of bacterial communities in Antarctic coastal waters by a combination of 16S rRNA and 16S rDNA sequencing. Environ. Microbiol. 2006, 8, 2150–2161. [Google Scholar] [CrossRef] [PubMed]
- Lemke, A.; Lunau, M.; Badewien, T.H.; Simon, M. Short-term and seasonal dynamics of bacterial biomass production and amino acid turnover in the water column of an intertidal ecosystem, the Wadden Sea. Aquat. Microb. Ecol. 2010, 61, 205–218. [Google Scholar] [CrossRef]
- Simon, M.; Rosenstock, B. Different coupling of dissolved amino acid, protein, and carbohydrate turnover to heterotrophic picoplankton production in the Southern Ocean in austral summer and fall. Limnol. Oceanogr. 2007, 52, 85–95. [Google Scholar] [CrossRef]
- Ducklow, H.W.; Kirchman, D.L.; Quinby, H.L.; Carlson, C.A.; Dam, H.G. Stocks and dynamics of bacterioplankton carbon during the spring bloom in the eastern North Atlantic Ocean. Deep Sea Res. Part II 1993, 40, 245–263. [Google Scholar] [CrossRef]
- Rich, J.; Gosselin, M.; Sherr, E.; Sherr, B.; Kirchman, D.L. High bacterial production, uptake and concentrations of dissolved organic matter in the Central Arctic Ocean. Deep Sea Res. Part II 1997, 44, 1645–1663. [Google Scholar] [CrossRef]
- Keil, R.G.; Kirchman, D.L. Contribution of dissolved free amino acids and ammonium to the nitrogen requirements of heterotrophic bacterioplankton. Mar. Ecol. Prog. Ser. 1991, 73, 1–10. [Google Scholar] [CrossRef]
- Fuhrman, J.A.; Cram, J.A.; Needham, D.M. Marine microbial community dynamics and their ecological interpretation. Nat. Rev. Microbiol. 2015, 13, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, K.; Wemheuer, B.; Korolkow, V.; Wemheuer, F.; Nacke, H.; Schöning, I.; Schrumpf, M.; Daniel, R. Driving forces of soil bacterial community structure, diversity, and function in temperate grasslands and forests. Sci. Rep. 2016, 6, 33696. [Google Scholar] [CrossRef] [PubMed]
- Sarmento, H.; Romera-Castillo, C.; Lindh, M.; Pinhassi, J.; Sala, M.M.; Gasol, J.M.; Marrase, C.; Taylor, G.T. Phytoplankton species-specific release of dissolved free amino acids and their selective consumption by bacteria. Limnol. Oceanogr. 2013, 58, 1123–1135. [Google Scholar] [CrossRef] [Green Version]






© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wemheuer, B.; Wemheuer, F.; Meier, D.; Billerbeck, S.; Giebel, H.-A.; Simon, M.; Scherber, C.; Daniel, R. Linking Compositional and Functional Predictions to Decipher the Biogeochemical Significance in DFAA Turnover of Abundant Bacterioplankton Lineages in the North Sea. Microorganisms 2017, 5, 68. https://doi.org/10.3390/microorganisms5040068
Wemheuer B, Wemheuer F, Meier D, Billerbeck S, Giebel H-A, Simon M, Scherber C, Daniel R. Linking Compositional and Functional Predictions to Decipher the Biogeochemical Significance in DFAA Turnover of Abundant Bacterioplankton Lineages in the North Sea. Microorganisms. 2017; 5(4):68. https://doi.org/10.3390/microorganisms5040068
Chicago/Turabian StyleWemheuer, Bernd, Franziska Wemheuer, Dimitri Meier, Sara Billerbeck, Helge-Ansgar Giebel, Meinhard Simon, Christoph Scherber, and Rolf Daniel. 2017. "Linking Compositional and Functional Predictions to Decipher the Biogeochemical Significance in DFAA Turnover of Abundant Bacterioplankton Lineages in the North Sea" Microorganisms 5, no. 4: 68. https://doi.org/10.3390/microorganisms5040068
APA StyleWemheuer, B., Wemheuer, F., Meier, D., Billerbeck, S., Giebel, H. -A., Simon, M., Scherber, C., & Daniel, R. (2017). Linking Compositional and Functional Predictions to Decipher the Biogeochemical Significance in DFAA Turnover of Abundant Bacterioplankton Lineages in the North Sea. Microorganisms, 5(4), 68. https://doi.org/10.3390/microorganisms5040068