Metabolic Response of Faecalibacterium prausnitzii to Cell-Free Supernatants from Lactic Acid Bacteria


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Media
2.2. Culture of F. prausnitzii A2-165 with Supernatants of Lactic Acid Bacteria and Bidifobacteria
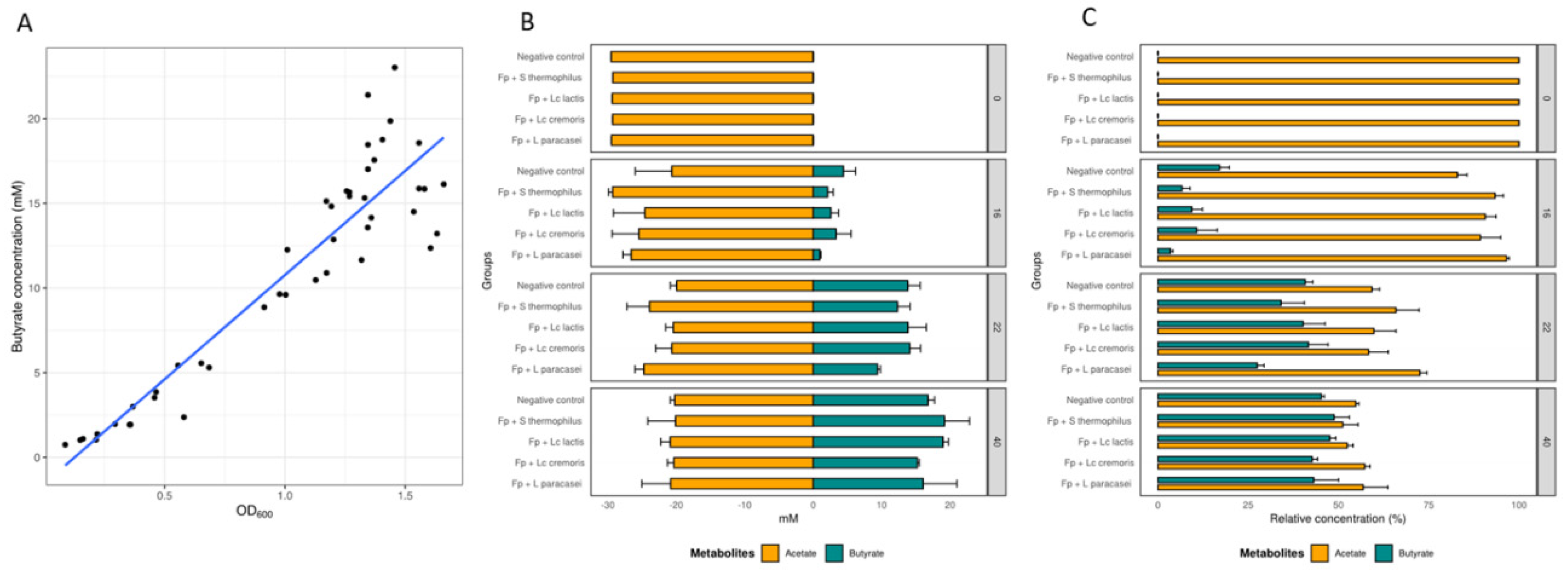
2.3. Analysis of Short Chain Fatty Acids
2.4. RNA Isolation and Sequencing (RNA-seq)
2.5. Differential Expression Analysis
2.6. Functional Annotation
2.7. Statistics and Graphs
2.8. Access to Data and Codes
3. Results and Discussion
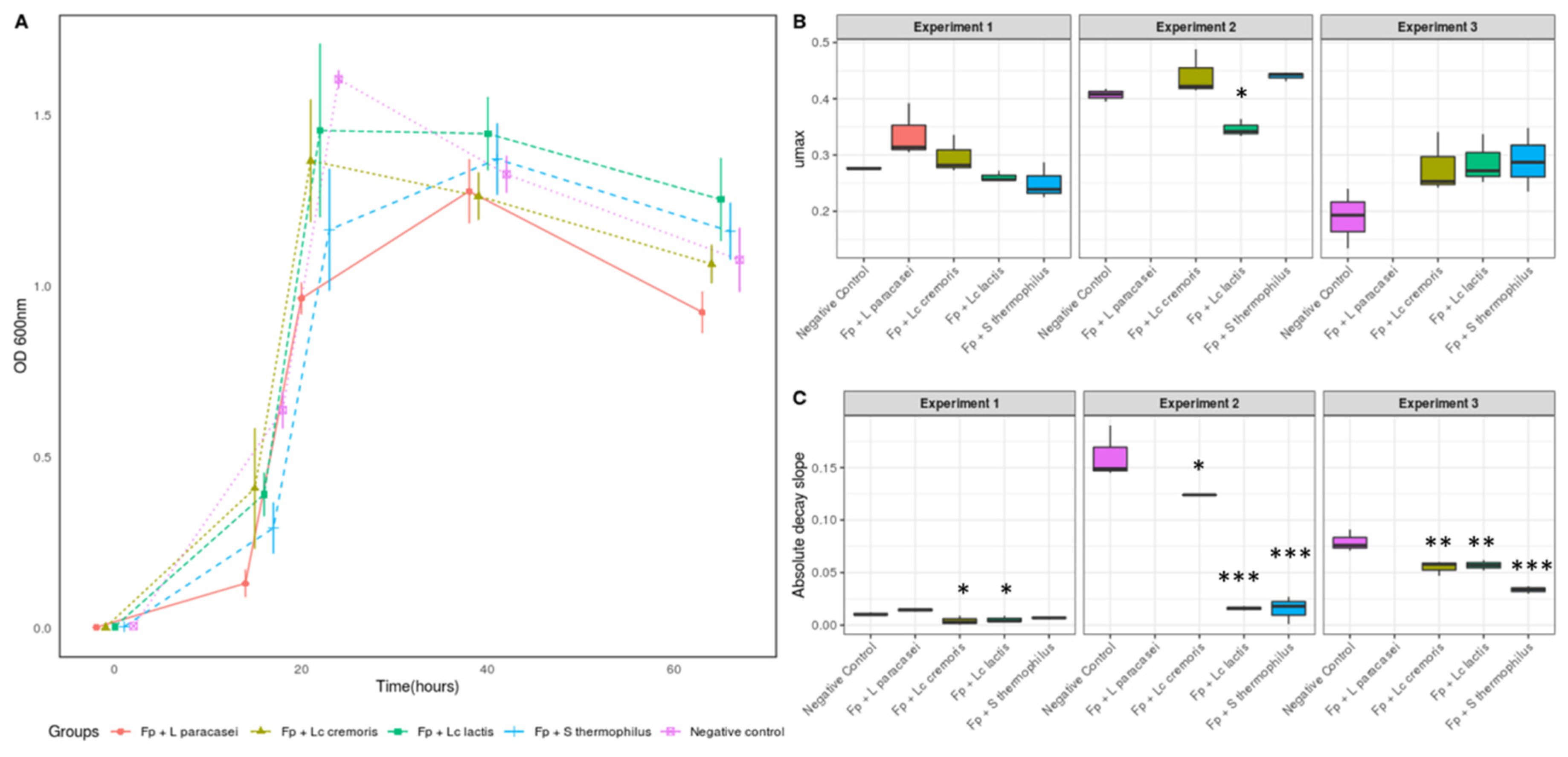
3.1. Selection of Strains Based on the Growth of F. prausnitzii
3.2. Growth and Metabolic Response of F. prausnitzii to Cell-Free Supernatants
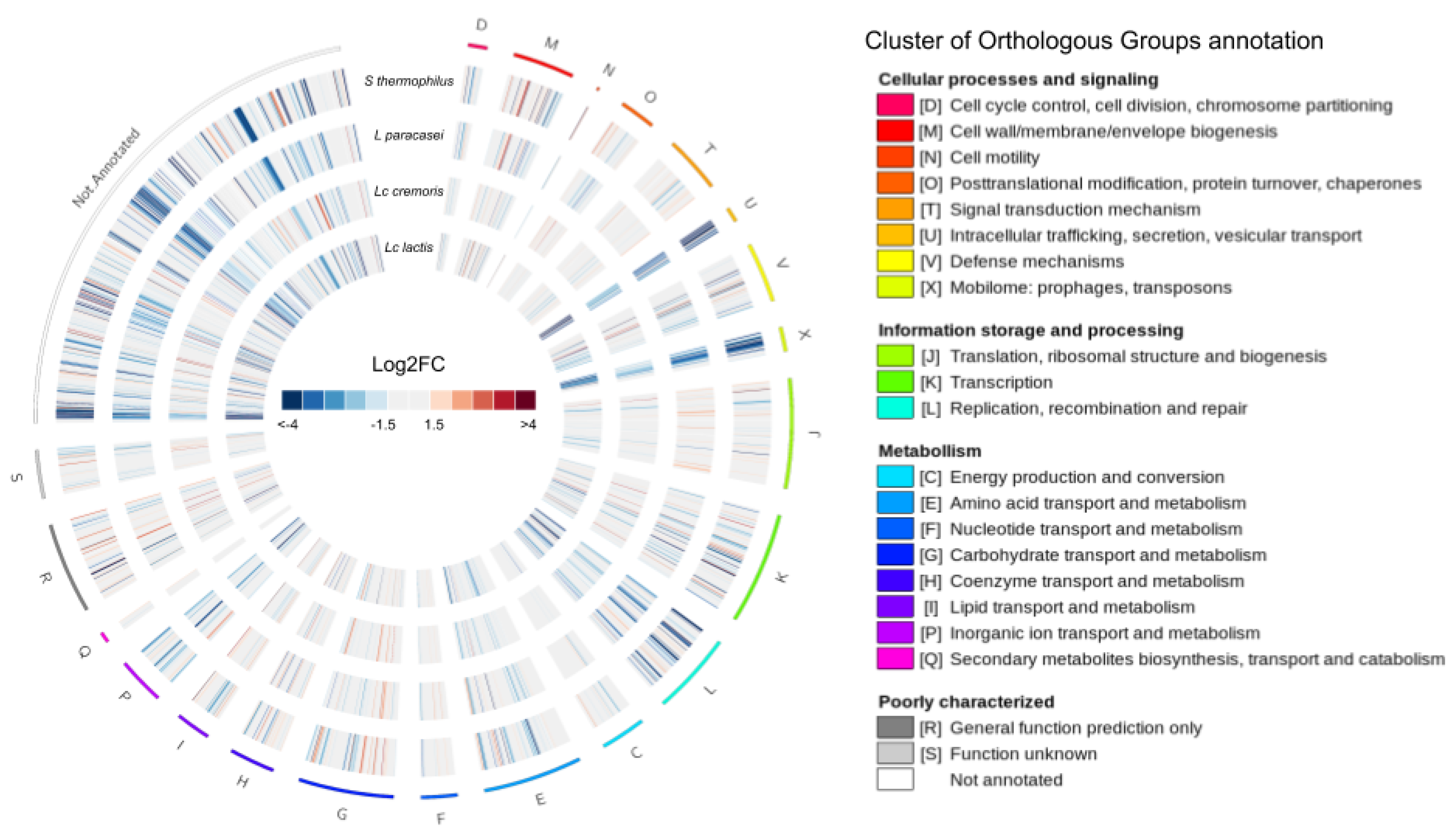
3.3. The Transcriptional Response of F. prausnitzii to Cell-Free Supernatants
3.3.1. Differential Expression
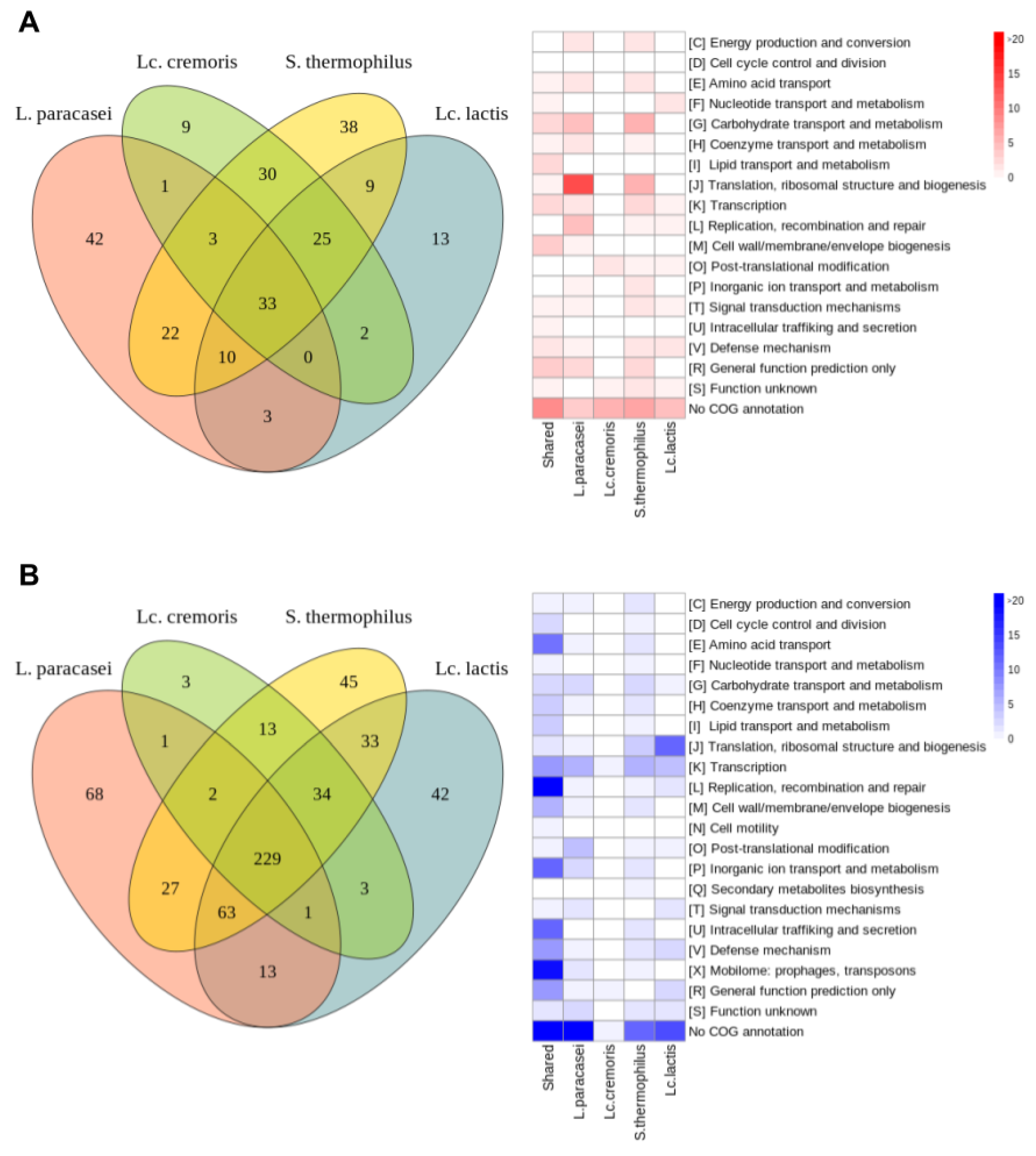
3.3.2. Common Differentially Expressed Genes
- Common upregulated genes
- Common downregulated genes
3.3.3. Specific Differentially Expressed Genes
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamer, H.M.; Jonkers, D.; Venema, K.; Vanhoutvin, S.; Troost, F.J.; Brummer, R.-J. Review article: The role of butyrate on colonic function. Aliment. Pharmacol. Ther. 2008, 27, 104–119. [Google Scholar] [CrossRef] [PubMed]
- Plöger, S.; Stumpff, F.; Penner, G.B.; Schulzke, J.-D.; Gäbel, G.; Martens, H.; Shen, Z.; Günzel, D.; Aschenbach, J.R. Microbial butyrate and its role for barrier function in the gastrointestinal tract. Ann. N. Y. Acad. Sci. 2012, 1258, 52–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokol, H.; Pigneur, B.; Watterlot, L.; Lakhdari, O.; Bermúdez-Humarán, L.G.; Gratadoux, J.-J.; Blugeon, S.; Bridonneau, C.; Furet, J.-P.; Corthier, G.; et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. USA 2008. [Google Scholar] [CrossRef] [Green Version]
- Ferreira-Halder, C.V.; Faria, A.V.d.S.; Andrade, S.S. Action and function of Faecalibacterium prausnitzii in health and disease. Best Pract. Res. Clin. Gastroenterol. 2017, 31, 643–648. [Google Scholar] [CrossRef]
- Martín, R.; Chain, F.; Miquel, S.; Lu, J.; Gratadoux, J.-J.; Sokol, H.; Verdu, E.F.; Bercik, P.; Bermúdez-Humarán, L.G.; Langella, P. The commensal bacterium Faecalibacterium prausnitzii is protective in DNBS-induced chronic moderate and severe colitis models. Inflamm. Bowel Dis. 2014, 20, 417–430. [Google Scholar] [CrossRef]
- Martín, R.; Miquel, S.; Chain, F.; Natividad, J.M.; Jury, J.; Lu, J.; Sokol, H.; Theodorou, V.; Bercik, P.; Verdu, E.F.; et al. Faecalibacterium prausnitzii prevents physiological damages in a chronic low-grade inflammation murine model. BMC Microbiol. 2015, 15, 67. [Google Scholar] [CrossRef] [Green Version]
- Breyner, N.M.; Michon, C.; de Sousa, C.S.; Vilas Boas, P.B.; Chain, F.; Azevedo, V.A.; Langella, P.; Chatel, J.M. Microbial anti-inflammatory molecule (MAM) from Faecalibacterium prausnitzii shows a protective effect on DNBS and DSS-induced colitis model in mice through inhibition of NF-κB pathway. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Quévrain, E.; Maubert, M.A.; Michon, C.; Chain, F.; Marquant, R.; Tailhades, J.; Miquel, S.; Carlier, L.; Bermúdez-Humarán, L.G.; Pigneur, B.; et al. Identification of an anti-inflammatory protein from Faecalibacterium prausnitzii, a commensal bacterium deficient in Crohn’s disease. Gut 2016, 65, 415–425. [Google Scholar] [CrossRef] [Green Version]
- Rossi, O.; Khan, M.T.; Schwarzer, M.; Hudcovic, T.; Srutkova, D.; Duncan, S.H.; Stolte, E.H.; Kozakova, H.; Flint, H.J.; Samsom, J.N.; et al. Faecalibacterium prausnitzii strain HTF-F and its extracellular polymeric matrix attenuate clinical parameters in DSS-Induced colitis. PLoS ONE 2015, 10, e0123013. [Google Scholar] [CrossRef] [Green Version]
- Miquel, S.; Martín, R.; Rossi, O.; Bermúdez-Humarán, L.G.; Chatel, J.M.; Sokol, H.; Thomas, M.; Wells, J.M.; Langella, P. Faecalibacterium prausnitzii and human intestinal health. Curr. Opin. Microbiol. 2013, 16, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Collij, V.; Jaeger, M.; van den Munckhof, I.C.L.; Vich Vila, A.; Kurilshikov, A.; Gacesa, R.; Sinha, T.; Oosting, M.; Joosten, L.A.B.; et al. Gut microbial co-abundance networks show specificity in inflammatory bowel disease and obesity. Nat. Commun. 2020, 11, 4018. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Siles, M.; Duncan, S.H.; Garcia-Gil, L.J.; Martinez-Medina, M. Faecalibacterium prausnitzii: From microbiology to diagnostics and prognostics. ISME J. 2017, 11, 841–852. [Google Scholar] [CrossRef] [PubMed]
- Martín, R.; Miquel, S.; Benevides, L.; Bridonneau, C.; Robert, V.; Hudault, S.; Chain, F.; Berteau, O.; Azevedo, V.; Chatel, J.M.; et al. Functional characterization of novel Faecalibacterium prausnitzii strains isolated from healthy volunteers: A step forward in the use of F. prausnitzii as a next-generation probiotic. Front. Microbiol. 2017, 8, 1226. [Google Scholar] [CrossRef]
- Lopez-Siles, M.; Khan, T.M.; Duncan, S.H.; Harmsen, H.J.M.; Garcia-Gil, L.J.; Flint, H.J. Cultured representatives of two major phylogroups of human colonic Faecalibacterium prausnitzii can utilize pectin, uronic acids, and host-derived substrates for growth. Appl. Environ. Microbiol. 2012, 78, 420–428. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, C.B.; Shkoporov, A.N.; Sutton, T.D.S.; Chaplin, A.V.; Velayudhan, V.; Ross, R.P.; Hill, C. Comparative analysis of Faecalibacterium prausnitzii genomes shows a high level of genome plasticity and warrants separation into new species-level taxa. BMC Genom. 2018, 19, 931. [Google Scholar] [CrossRef] [Green Version]
- Heinken, A.; Khan, M.T.; Paglia, G.; Rodionov, D.A.; Harmsen, H.J.M.; Thiele, I. Functional metabolic map of Faecalibacterium prausnitzii, a beneficial human gut microbe. J. Bacteriol. 2014, 196, 3289–3302. [Google Scholar] [CrossRef] [Green Version]
- Duncan, S.H.; Holtrop, G.; Lobley, G.E.; Calder, A.G.; Stewart, C.S.; Flint, H.J. Contribution of acetate to butyrate formation by human faecal bacteria. Br. J. Nutr. 2004, 91, 915–923. [Google Scholar] [CrossRef] [Green Version]
- Rios-Covian, D.; Gueimonde, M.; Duncan, S.H.; Flint, H.J.; de los Reyes-Gavilan, C.G. Enhanced butyrate formation by cross-feeding between Faecalibacterium prausnitzii and Bifidobacterium adolescentis. Fems Microbiol. Lett. 2015, 362. [Google Scholar] [CrossRef] [Green Version]
- Chung, W.S.F.; Meijerink, M.; Zeuner, B.; Holck, J.; Louis, P.; Meyer, A.S.; Wells, J.M.; Flint, H.J.; Duncan, S.H. Prebiotic potential of pectin and pectic oligosaccharides to promote anti-inflammatory commensal bacteria in the human colon. Fems Microbiol. Ecol. 2017, 93. [Google Scholar] [CrossRef]
- Chung, W.S.F.; Walker, A.W.; Vermeiren, J.; Sheridan, P.O.; Bosscher, D.; Garcia-Campayo, V.; Parkhill, J.; Flint, H.J.; Duncan, S.H. Impact of carbohydrate substrate complexity on the diversity of the human colonic microbiota. FEMS Microbiol. Ecol. 2018, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichardt, N.; Vollmer, M.; Holtrop, G.; Farquharson, F.M.; Wefers, D.; Bunzel, M.; Duncan, S.H.; Drew, J.E.; Williams, L.M.; Milligan, G.; et al. Specific substrate-driven changes in human faecal microbiota composition contrast with functional redundancy in short-chain fatty acid production. ISME J. 2018, 12, 610–622. [Google Scholar] [CrossRef] [PubMed]
- Gurry, T.; Dannenberg, P.H.; Finlayson, S.G.; Hughes, T.K.; Macias-Trevino, C.; Owusu-Boaitey, K.; Shomorony, A.; Tuang, S.L.; Valenstein, M.L.; Wang, K.K.; et al. Predictability and persistence of prebiotic dietary supplementation in a healthy human cohort. Sci. Rep. 2018, 8, 12699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derrien, M.; van Hylckama Vlieg, J.E.T. Fate, activity, and impact of ingested bacteria within the human gut microbiota. Trends Microbiol. 2015, 23, 354–366. [Google Scholar] [CrossRef] [Green Version]
- Belenguer, A.; Duncan, S.H.; Calder, A.G.; Holtrop, G.; Louis, P.; Lobley, G.E.; Flint, H.J. Two routes of metabolic cross-feeding between Bifidobacterium adolescentis and butyrate-producing anaerobes from the human gut. Appl. Environ. Microbiol. 2006, 72, 3593–3599. [Google Scholar] [CrossRef] [Green Version]
- Rivière, A.; Selak, M.; Lantin, D.; Leroy, F.; De Vuyst, L. Bifidobacteria and butyrate-producing colon acteria: Importance and strategies for their stimulation in the human gut. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Castro-Bravo, N.; Wells, J.M.; Margolles, A.; Ruas-Madiedo, P. Interactions of surface exopolysaccharides from Bifidobacterium and Lactobacillus within the intestinal environment. Front. Microbiol. 2018, 9, 2426. [Google Scholar] [CrossRef] [Green Version]
- Bello, F.D.; Walter, J.; Hertel, C.; Hammes, W.P. In vitro study of prebiotic properties of levan-type exopolysaccharides from Lactobacilli and non-digestible carbohydrates using denaturing gradient gel electrophoresis. Syst. Appl. Microbiol. 2001, 24, 232–237. [Google Scholar] [CrossRef]
- Soto-Martin, E.C.; Warnke, I.; Farquharson, F.M.; Christodoulou, M.; Horgan, G.; Derrien, M.; Faurie, J.M.; Flint, H.J.; Duncan, S.H.; Louis, P. Vitamin biosynthesis by human gut butyrate-producing bacteria and cross-feeding in synthetic microbial communities. mBio 2020, 11. [Google Scholar] [CrossRef]
- Kareem, K.Y.; Hooi Ling, F.; Teck Chwen, L.; May Foong, O.; Anjas Asmara, S. Inhibitory activity of postbiotic produced by strains of Lactobacillus plantarum using reconstituted media supplemented with inulin. Gut Pathog. 2014, 6, 23. [Google Scholar] [CrossRef] [Green Version]
- Mariam, S.H.; Zegeye, N.; Tariku, T.; Andargie, E.; Endalafer, N.; Aseffa, A. Potential of cell-free supernatants from cultures of selected lactic acid bacteria and yeast obtained from local fermented foods as inhibitors of Listeria monocytogenes, Salmonella spp. and Staphylococcus aureus. BMC Res. Notes 2014, 7, 606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zacharof, M.P.; Lovitt, R.W. Bacteriocins produced by lactic acid bacteria a review article. Apcbee Procedia 2012, 2, 50–56. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 3. [Google Scholar] [CrossRef]
- Kopylova, E.; Noé, L.; Touzet, H. SortMeRNA: Fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 2012, 28, 3211–3217. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics (Oxford, England) 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Galperin, M.Y.; Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 2015, 43, D261–D269. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.K.; Xu, Y.; Yin, Y. dbCAN2: A meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [Green Version]
- Cornuault, J.K.; Petit, M.-A.; Mariadassou, M.; Benevides, L.; Moncaut, E.; Langella, P.; Sokol, H.; De Paepe, M. Phages infecting Faecalibacterium prausnitzii belong to novel viral genera that help to decipher intestinal viromes. Microbiome 2018, 6, 65. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fehlbaum, S.; Chassard, C.; Schwab, C.; Voolaid, M.; Fourmestraux, C.; Derrien, M.; Lacroix, C. In vitro study of Lactobacillus paracasei CNCM I-1518 in healthy and Clostridioides difficile colonized elderly gut microbiota. Front. Nutr. 2019, 6. [Google Scholar] [CrossRef] [PubMed]
- D’Hoe, K.; Vet, S.; Faust, K.; Moens, F.; Falony, G.; Gonze, D.; Lloréns-Rico, V.; Gelens, L.; Danckaert, J.; De Vuyst, L.; et al. Integrated culturing, modeling and transcriptomics uncovers complex interactions and emergent behavior in a three-species synthetic gut community. eLife 2018, 7, e37090. [Google Scholar] [CrossRef]
- Venturelli, O.S.; Carr, A.V.; Fisher, G.; Hsu, R.H.; Lau, R.; Bowen, B.P.; Hromada, S.; Northen, T.; Arkin, A.P. Deciphering microbial interactions in synthetic human gut microbiome communities. Mol. Syst. Biol. 2018, 14, e8157. [Google Scholar] [CrossRef]
- Foditsch, C.; Santos, T.M.A.; Teixeira, A.G.V.; Pereira, R.V.V.; Dias, J.M.; Gaeta, N.; Bicalho, R.C. Isolation and characterization of Faecalibacterium prausnitzii from calves and piglets. PLoS ONE 2015, 9, e116465. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Jeong, Y.; Kang, S.; You, H.J.; Ji, G.E. Co-Culture with Bifidobacterium catenulatum improves the growth, gut colonization, and butyrate production of Faecalibacterium prausnitzii: In vitro and in vivo studies. Microorganisms 2020, 8, 8. [Google Scholar] [CrossRef]
- Chia, L.W.; Hornung, B.V.H.; Aalvink, S.; Schaap, P.J.; de Vos, W.M.; Knol, J.; Belzer, C. Deciphering the trophic interaction between Akkermansia muciniphila and the butyrogenic gut commensal Anaerostipes caccae using a metatranscriptomic approach. Antonie Van Leeuwenhoek 2018, 111, 859–873. [Google Scholar] [CrossRef] [Green Version]
- Crost, E.H.; Le Gall, G.; Laverde-Gomez, J.A.; Mukhopadhya, I.; Flint, H.J.; Juge, N. Mechanistic insights into the cross-feeding of Ruminococcus gnavus and Ruminococcus bromii on host and dietary carbohydrates. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Vermassen, A.; Leroy, S.; Talon, R.; Provot, C.; Popowska, M.; Desvaux, M. Cell wall hydrolases in bacteria: Insight on the diversity of cell wall amidases, glycosidases and peptidases toward peptidoglycan. Front. Microbiol. 2019, 10. [Google Scholar] [CrossRef]
- Reith, J.; Mayer, C. Peptidoglycan turnover and recycling in Gram-positive bacteria. Appl. Microbiol. Biotechnol. 2011, 92, 1. [Google Scholar] [CrossRef] [PubMed]
- Scheffers, D.-J.; Pinho, M.G. Bacterial cell wall synthesis: New insights from localization studies. Microbiol. Mol. Biol. Rev. 2005, 69, 585–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miquel, S.; Martín, R.; Bridonneau, C.; Robert, V.; Sokol, H.; Bermúdez-Humarán, L.G.; Thomas, M.; Langella, P. Ecology and metabolism of the beneficial intestinal commensal bacterium Faecalibacterium prausnitzii. Gut Microbes 2014, 5, 146–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jantzen, E.; Hofstad, T. Fatty acids of Fusobacterium species: Taxonomic implications. J. Gen. Microbiol. 1981, 123, 163–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mills, S.; Shanahan, F.; Stanton, C.; Hill, C.; Coffey, A.; Ross, R.P. Movers and shakers: Influence of bacteriophages in shaping the mammalian gut microbiota. Gut Microbes 2013, 4, 4–16. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lebas, M.; Garault, P.; Carrillo, D.; Codoñer, F.M.; Derrien, M. Metabolic Response of Faecalibacterium prausnitzii to Cell-Free Supernatants from Lactic Acid Bacteria. Microorganisms 2020, 8, 1528. https://doi.org/10.3390/microorganisms8101528
Lebas M, Garault P, Carrillo D, Codoñer FM, Derrien M. Metabolic Response of Faecalibacterium prausnitzii to Cell-Free Supernatants from Lactic Acid Bacteria. Microorganisms. 2020; 8(10):1528. https://doi.org/10.3390/microorganisms8101528
Chicago/Turabian StyleLebas, Mathilde, Peggy Garault, Daniel Carrillo, Francisco M. Codoñer, and Muriel Derrien. 2020. "Metabolic Response of Faecalibacterium prausnitzii to Cell-Free Supernatants from Lactic Acid Bacteria" Microorganisms 8, no. 10: 1528. https://doi.org/10.3390/microorganisms8101528
APA StyleLebas, M., Garault, P., Carrillo, D., Codoñer, F. M., & Derrien, M. (2020). Metabolic Response of Faecalibacterium prausnitzii to Cell-Free Supernatants from Lactic Acid Bacteria. Microorganisms, 8(10), 1528. https://doi.org/10.3390/microorganisms8101528