Impact of the Post-Transplant Period and Lifestyle Diseases on Human Gut Microbiota in Kidney Graft Recipients

 ,
,  ,
,  ,
,  , ,
, ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Cohort
2.2. DNA Extraction, First-Step PCR Amplification and Purification
2.3. Second-Step PCR Amplification (Indexing) and Purification
2.4. Bioinformatic and Statistical Analysis
3. Results
3.1. Kidney Transplant Cohort
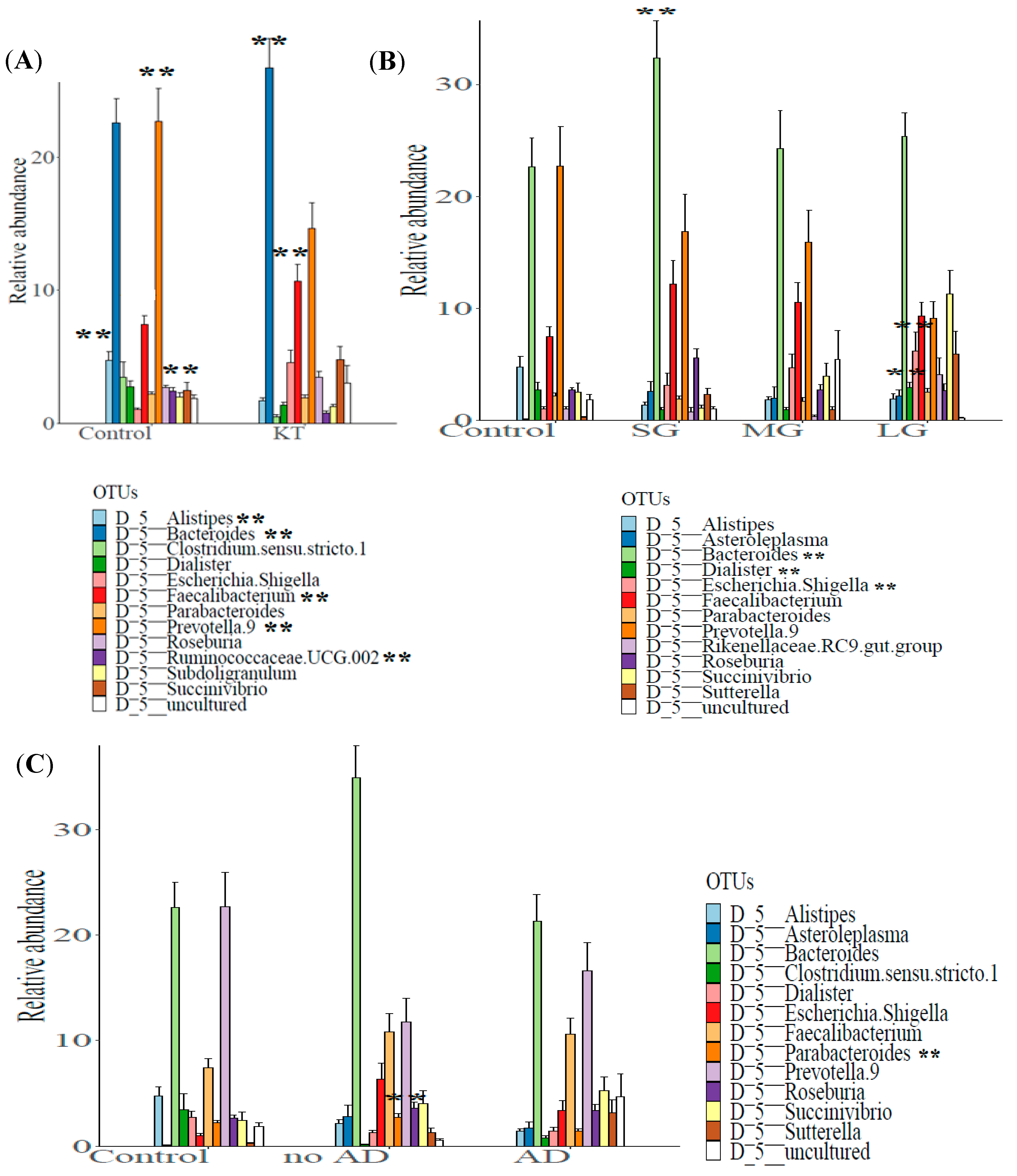
3.2. Structure of the Gut Bacterial Community of the Kidney Transplanted Patients
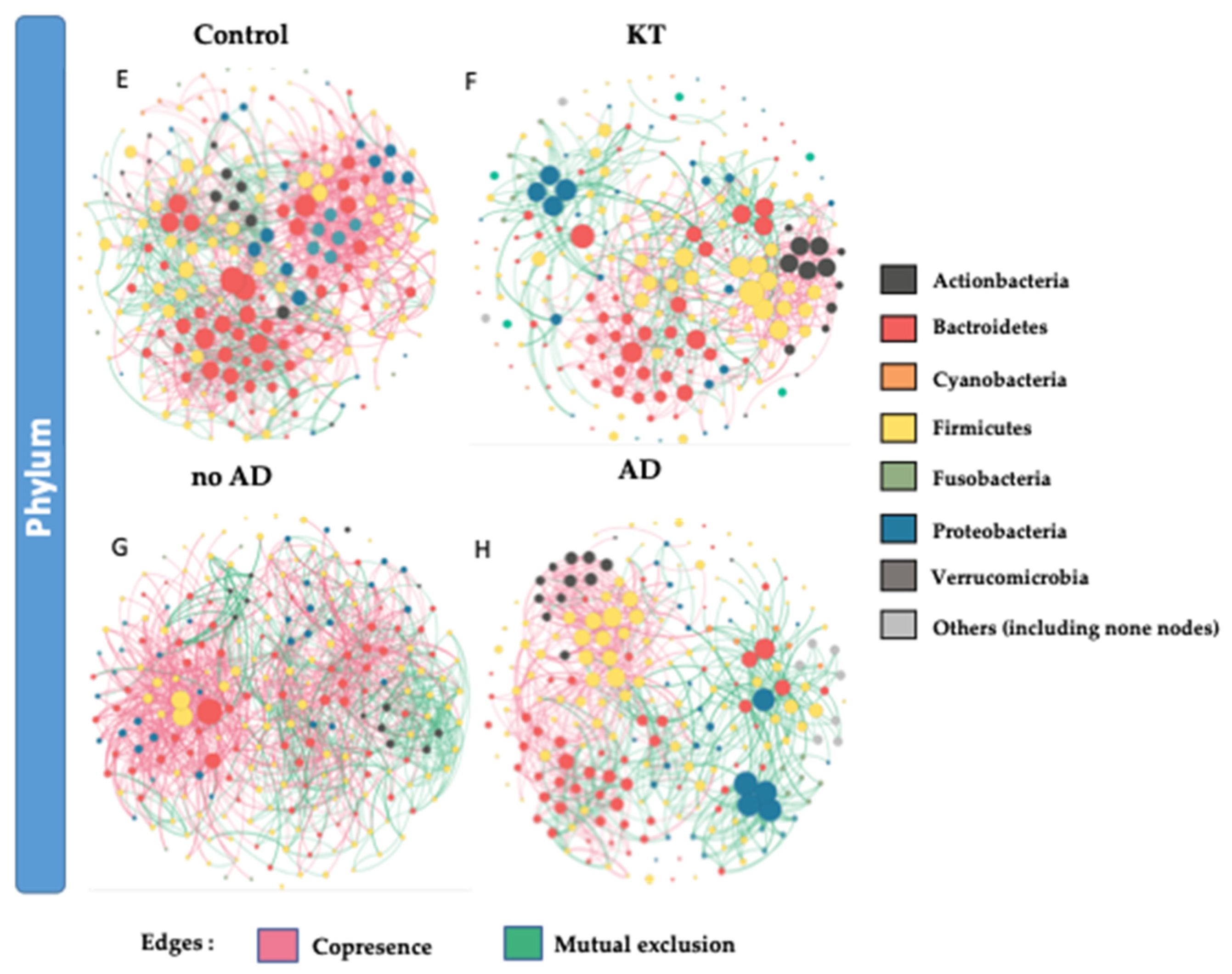
3.3. Bacterial Networking in the Gut
3.4. Analysis of Factors Influencing Gut Bacterial Structure within Kidney Grafted Subjects
3.4.1. Graft Period
3.4.2. Associated Diseases
4. Discussion
4.1. Decrease in Gut Microbiota Richness and Diversity in Kidney Allograft Patients
4.2. Graft Period Impacts Gut Microbial Structure
4.3. Post-Transplant Complications Related to Dysbiosis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability
References
- Bureau, U.C. An Aging World: 2015, Census Bur. Available online: https://www.census.gov/library/publications/2016/demo/P95-16-1.html (accessed on 28 March 2016).
- Wolfe, R.A.; Ashby, V.B.; Milford, E.L.; Ojo, A.O.; Ettenger, R.E.; Agodoa, L.Y.; Held, P.J.; Port, F.K. Comparison of Mortality in All Patients on Dialysis, Patients on Dialysis Awaiting Transplantation, and Recipients of a First Cadaveric Transplant. N. Engl. J. Med. 1999, 341, 1725–1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaston, R.S.; Alveranga, D.Y.; Becker, B.N.; Distant, D.A.; Held, P.J.; Bragg-Gresham, J.L.; Humar, A.; Ting, A.; Wynn, J.J.; Leichtman, A.B. Kidney and pancreas transplantation. Arab. Archaeol. Epigr. 2003, 3, 64–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.H.; Skeans, M.A.; Israni, A.K. Current Status of Kidney Transplant Outcomes: Dying to Survive. Adv. Chronic Kidney Dis. 2016, 23, 281–286. [Google Scholar] [CrossRef] [PubMed]
- National Kidney Foundation. K/DOQI clinical practice guidelines for chronic kidney disease: Evaluation, classification, and stratification. Am. J. Kidney Dis. 2002, 39 (Suppl. 1), S1–S266. [Google Scholar]
- Peabody, J.W.; Billings, P.; Valdenor, C.; Demko, Z.; Moshkevich, S.; Paculdo, D.; Tran, M. Variation in Assessing Renal Allograft Rejection: A National Assessment of Nephrology Practice. Int. J. Nephrol. 2019, 2019, 5303284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkataramanan, R.; Swaminathan, A.; Prasad, T.; Jain, A.B.; Zuckerman, S.; Warty, V.; McMichael, J.; Lever, J.; Burckart, G.; Starzl, T. Clinical Pharmacokinetics of Tacrolimus. Clin. Pharmacokinet. 1995, 29, 404–430. [Google Scholar] [CrossRef]
- Oh, P.L.; Martínez, I.; Sun, Y.; Walter, J.; Peterson, D.; Mercer, D.F. Characterization of the Ileal Microbiota in Rejecting and Nonrejecting Recipients of Small Bowel Transplants. Arab. Archaeol. Epigr. 2011, 12, 753–762. [Google Scholar] [CrossRef]
- Lee, J.R.; Muthukumar, T.; Dadhania, D.; Toussaint, N.C.; Ling, L.; Pamer, E.; Suthanthiran, M. Gut Microbial Community Structure and Complications After Kidney Transplantation. Transplantation 2014, 98, 697–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, K.; Goto, N.; Futamura, K.; Okada, M.; Yamamoto, T.; Tsujita, M.; Hiramitsu, T.; Narumi, S.; Tominaga, Y.; Watarai, Y. Death and kidney allograft dysfunction after bacteremia. Clin. Exp. Nephrol. 2015, 20, 309–315. [Google Scholar] [CrossRef]
- Lee, J.R.; Muthukumar, T.; Dadhania, D.; Taur, Y.; Jenq, R.R.; Toussaint, N.C.; Ling, L.; Pamer, E.; Suthanthiran, M. Gut Microbiota and Tacrolimus Dosing in Kidney Transplantation. PLoS ONE 2015, 10, e0122399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lun, H.; Yang, W.; Zhao, S.; Jiang, M.; Xu, M.; Liu, F.; Wang, Y. Altered gut microbiota and microbial biomarkers associated with chronic kidney disease. Microbiologyopen 2018, 8, e00678. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2012, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. SEARCH_16S: A new algorithm for identifying 16S ribosomal RNA genes in contigs and chromosomes. Bioinformatics 2017, 124131. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. UNOISE2: Improved error-correction for Illumina 16S and ITS amplicon sequencing. Bioinformatics 2016, 081257. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. SINTAX: A simple non-Bayesian taxonomy classifier for 16S and ITS sequences. Bioinformatics 2016, 074161. [Google Scholar] [CrossRef] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Ssekagiri, A.T.; Sloan, W.; Ijaz, U.Z. microbiomeSeq: An R package for analysis of microbial communities in an environmental context. In Proceedings of the ISCB Africa ASBCB Conference, Kumasi, Ghana, 11–15 November 2017; Available online: https://github.com/umerijaz/microbiomeSeq (accessed on 1 September 2020). [CrossRef]
- Nordstokke, D.; Stelnicki, A.M. Pairwise Comparisons. In Encyclopedia of Quality of Life and Well-Being Research; Springer: Berlin/Heidelberg, Germany, 2014; pp. 4575–4576. [Google Scholar]
- Anderson, M.J. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001, 26, 32–46. [Google Scholar] [CrossRef]
- Steele, J.A.; Countway, P.D.; Xia, L.; Vigil, P.D.; Beman, J.M.; Kim, D.Y.; Chow, C.-E.T.; Sachdeva, R.; Jones, A.C.; Schwalbach, M.S.; et al. Marine bacterial, archaeal and protistan association networks reveal ecological linkages. ISME J. 2011, 5, 1414–1425. [Google Scholar] [CrossRef]
- Zhou, J.; Deng, Y.; Luo, F.; He, Z.; Yang, Y. Phylogenetic Molecular Ecological Network of Soil Microbial Communities in Response to Elevated CO2. mBio 2011, 2, e00122-11. [Google Scholar] [CrossRef] [Green Version]
- Faust, K.; Raes, J. CoNet app: Inference of biological association networks using Cytoscape. F1000 Res. 2016, 5, 1519. [Google Scholar] [CrossRef]
- Hollander, M.; Wolfe, D.A.; Chicken, E. Nonparametric Statistical Methods; Wiley: Hoboken, NJ, USA, 2015. [Google Scholar]
- Fricke, W.F.; Maddox, C.; Song, Y.; Bromberg, J.S. Human Microbiota Characterization in the Course of Renal Transplantation. Arab. Archaeol. Epigr. 2013, 14, 416–427. [Google Scholar] [CrossRef]
- Li, F.; Wang, M.; Wang, J.; Li, R.; Zhang, Y. Alterations to the Gut Microbiota and Their Correlation With Inflammatory Factors in Chronic Kidney Disease. Front. Cell. Infect. Microbiol. 2019, 9, 206. [Google Scholar] [CrossRef] [Green Version]
- Ye, G.; Zhou, M.; Yu, L.; Ye, J.; Yao, L.; Shi, L. Gut microbiota in renal transplant recipients, patients with chronic kidney disease and healthy subjects. J. South Med. Univ. 2018, 38, 1401–1408. [Google Scholar]
- Xiao, J.; Peng, Z.; Liao, Y.; Sun, H.; Chen, W.; Chen, X.; Wei, Z.; Yang, C.; Nüssler, A.K.; Liu, J.; et al. Organ transplantation and gut microbiota: Current reviews and future challenges. Am. J. Transl. Res. 2018, 10, 3330–3344. [Google Scholar]
- Le Chatelier, E.; MetaHIT Consortium; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.-M.; et al. Richness of human gut microbiome correlates with metabolic markers. Nat. Cell Biol. 2013, 500, 541–546. [Google Scholar] [CrossRef]
- Doycheva, I.; Leise, M.D.; Watt, K.D. The Intestinal Microbiome and the Liver Transplant Recipient. Transplantation 2016, 100, 61–68. [Google Scholar] [CrossRef]
- La Manna, G.; Capelli, I.; Gasperoni, L.; Comai, G.; Ravaioli, M.; Marchetti, A.; Rucci, P.; D’Arcangelo, G.L.; Faenza, S.; Pinna, A.D.; et al. Long Term Outcomes of Kidney Transplant: Characteristics of Recipients with 20 or More Years of Graft Survival. J. Med. Surg. Pathol. 2016, 1. [Google Scholar] [CrossRef] [Green Version]
- Taur, Y.; Xavier, J.B.; Lipuma, L.; Ubeda, C.; Goldberg, J.; Gobourne, A.; Lee, Y.J.; Dubin, K.A.; Socci, N.D.; Viale, A.; et al. Intestinal Domination and the Risk of Bacteremia in Patients Undergoing Allogeneic Hematopoietic Stem Cell Transplantation. Clin. Infect. Dis. 2012, 55, 905–914. [Google Scholar] [CrossRef]
- Huttenhower, C.; Gevers, D.; Knight, R.; Abubucker, S.; Badger, J.H.; Chinwalla, A.T.; Creasy, H.H.; Earl, A.M.; Fitzgerald, M.G.; Fulton, R.; et al. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Mahmoodpoor, F.; Saadat, Y.R.; Barzegari, A.; Ardalan, M.R.; Vahed, S.Z. The impact of gut microbiota on kidney function and pathogenesis. Biomed. Pharmacother. 2017, 93, 412–419. [Google Scholar] [CrossRef]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nat. Cell Biol. 2012, 490, 55–60. [Google Scholar] [CrossRef]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Human gut microbes associated with obesity. Nat. Cell Biol. 2006, 444, 1022–1023. [Google Scholar] [CrossRef]
- Yang, T.; Santisteban, M.M.; Rodriguez, V.; Vermali, R.; Ahmari, N.; Carvajal, J.M.; Zadeh, M.; Gong, M.; Qi, Y.; Zubcevic, J.; et al. Gut Dysbiosis Is Linked to Hypertension. Hypertension 2015, 65, 1331–1340. [Google Scholar] [CrossRef] [Green Version]
- Larsen, N.; Vogensen, F.K.; Berg, F.W.J.V.D.; Nielsen, D.S.; Andreasen, A.S.; Pedersen, B.K.; Abu Al-Soud, W.; Sørensen, S.J.; Hansen, L.H.; Jakobsen, M. Gut Microbiota in Human Adults with Type 2 Diabetes Differs from Non-Diabetic Adults. PLoS ONE 2010, 5, e9085. [Google Scholar] [CrossRef] [PubMed]
- Rizzatti, G.; Lopetuso, L.R.; Gibiino, G.; Binda, C.; Gasbarrini, A. Proteobacteria: A Common Factor in Human Diseases. BioMed Res. Int. 2017, 2017, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Barka, E.A.; Vatsa, P.; Sanchez, L.; Gaveau-Vaillant, N.; Jacquard, C.; Klenk, H.-P.; Clément, C.; Ouhdouch, Y.; Van Wezel, G.P. Taxonomy, Physiology, and Natural Products of Actinobacteria. Microbiol. Mol. Biol. Rev. 2015, 80, 1–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.R.; Magruder, M.; Zhang, L.; Westblade, L.F.; Satlin, M.J.; Robertson, A.; Edusei, E.; Crawford, C.; Ling, L.; Taur, Y.; et al. Gut microbiota dysbiosis and diarrhea in kidney transplant recipients. Arab. Archaeol. Epigr. 2018, 19, 488–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.; Zhou, L.; Guo, H.; Xu, Y.; Xu, Y. The role of short-chain fatty acids in kidney injury induced by gut-derived inflammatory response. Metab. Clin. Exp. 2017, 68, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Onal, E.M.; Afsar, B.; Covic, A.; Vaziri, N.D.; Kanbay, M. Gut microbiota and inflammation in chronic kidney disease and their roles in the development of cardiovascular disease. Hypertens. Res. 2018, 42, 123–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, S.; Xie, S.; Lv, D.; Wang, P.; He, H.; Zhang, T.; Zhou, Y.; Lin, Q.; Zhou, H.; Jiang, J.; et al. Alteration of the gut microbiota in Chinese population with chronic kidney disease. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrade-Oliveira, V.; Amano, M.T.; Correa-Costa, M.; Castoldi, A.; Felizardo, R.J.; De Almeida, D.C.; Bassi, E.J.; Moraes-Vieira, P.M.; Hiyane, M.I.; Rodas, A.C.; et al. Gut Bacteria Products Prevent AKI Induced by Ischemia-Reperfusion. J. Am. Soc. Nephrol. 2015, 26, 1877–1888. [Google Scholar] [CrossRef]
- Wilmes, D.; Coche, E.; Rodriguez-Villalobos, H.; Kanaan, N. Bacterial pneumonia in kidney transplant recipients. Respir. Med. 2018, 137, 89–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basha, J.A.; Kiel, M.; Görlich, D.; Schütte-Nütgen, K.; Witten, A.; Pavenstadt, H.J.; Kahl, B.C.; Dobrindt, U.; Reuter, S. Phenotypic and Genotypic Characterization of Escherichia coli Causing Urinary Tract Infections in Kidney-Transplanted Patients. J. Clin. Med. 2019, 8, 988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turunç, V.; Eroglu, A.; Cihandide, E.; Tabandeh, B.; Orug, T.; Guven, B. Escherichia Coli–Related Necrotizing Fasciitis After Renal Transplantation: A Case Report. Transplant. Proc. 2015, 47, 1518–1521. [Google Scholar] [CrossRef] [PubMed]
- Hiippala, K.; Kainulainen, V.; Kalliomäki, M.; Arkkila, P.; Satokari, R. Mucosal Prevalence and Interactions with the Epithelium Indicate Commensalism of Sutterella spp. Front. Microbiol. 2016, 7, 1706. [Google Scholar] [CrossRef] [Green Version]
- Ticinesi, A.; Nouvenne, A.; Meschi, T. Gut microbiome and kidney stone disease: Not just an Oxalobacter story. Kidney Int. 2019, 96, 25–27. [Google Scholar] [CrossRef]
- Nellore, A.; Fishman, J.A. The Microbiome, Systemic Immune Function, and Allotransplantation. Clin. Microbiol. Rev. 2015, 29, 191–199. [Google Scholar] [CrossRef] [Green Version]
- Rousee, J.M.; Bermond, D.; Piemont, Y.; Tournoud, C.; Heller, R.; Kehrli, P.; Harlay, M.L.; Monteil, H.; Jaulhac, B. Dialister pneumosintes Associated with Human Brain Abscesses. J. Clin. Microbiol. 2002, 40, 3871–3873. [Google Scholar] [CrossRef] [Green Version]
- Boente, R.F.; Ferreira, L.Q.; Falcão, L.S.; Miranda, K.R.; Guimarães, P.L.; Santos-Filho, J.; Vieira, J.M.; Barroso, D.E.; Emond, J.-P.; Ferreira, E.O.; et al. Detection of resistance genes and susceptibility patterns in Bacteroides and Parabacteroides strains. Anaerobe 2010, 16, 190–194. [Google Scholar] [CrossRef]
- Karlsson, F.H.; Tremaroli, V.; Nookaew, I.; Bergström, G.; Behre, C.J.; Fagerberg, B.; Nielsen, J.B.; Bäckhed, F. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nat. Cell Biol. 2013, 498, 99–103. [Google Scholar] [CrossRef]
- Chiu, C.-M.; Huang, W.-C.; Weng, S.-L.; Tseng, H.-C.; Liang, C.; Wang, W.-C.; Yang, T.; Yang, T.-L.; Weng, C.-T.; Chang, T.-H.; et al. Systematic Analysis of the Association between Gut Flora and Obesity through High-Throughput Sequencing and Bioinformatics Approaches. BioMed Res. Int. 2014, 2014, 1–10. [Google Scholar] [CrossRef]
- Bhat, M.; Pasini, E.; Das, A.; Baciu, C.; Angeli, M.; Humar, A.; Watt, K.D.; Allard, J. Diabetogenic Effects of Immunosuppression. Transplantation 2020, 104, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.T.; Taur, Y.; Walkup, J.T. Gut Microbiota and Autism: Key Concepts and Findings. J. Autism Dev. Disord. 2017, 47, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Southern, P.M. Bacteremia Due to Succinivibrio dextrinosolvens: Report of a Case. Am. J. Clin. Pathol. 1975, 64, 540–543. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xu, S.; Ren, Z.; Jiang, J.; Zheng, S. Gut microbiota and allogeneic transplantation. J. Transl. Med. 2015, 13, 275. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Souai, N.; Zidi, O.; Mosbah, A.; Kosai, I.; Manaa, J.E.; Mokhtar, N.B.; Asimakis, E.; Stathopoulou, P.; Cherif, A.; Tsiamis, G.; et al. Impact of the Post-Transplant Period and Lifestyle Diseases on Human Gut Microbiota in Kidney Graft Recipients. Microorganisms 2020, 8, 1724. https://doi.org/10.3390/microorganisms8111724
Souai N, Zidi O, Mosbah A, Kosai I, Manaa JE, Mokhtar NB, Asimakis E, Stathopoulou P, Cherif A, Tsiamis G, et al. Impact of the Post-Transplant Period and Lifestyle Diseases on Human Gut Microbiota in Kidney Graft Recipients. Microorganisms. 2020; 8(11):1724. https://doi.org/10.3390/microorganisms8111724
Chicago/Turabian StyleSouai, Nessrine, Oumaima Zidi, Amor Mosbah, Imen Kosai, Jameleddine El Manaa, Naima Bel Mokhtar, Elias Asimakis, Panagiota Stathopoulou, Ameur Cherif, George Tsiamis, and et al. 2020. "Impact of the Post-Transplant Period and Lifestyle Diseases on Human Gut Microbiota in Kidney Graft Recipients" Microorganisms 8, no. 11: 1724. https://doi.org/10.3390/microorganisms8111724
APA StyleSouai, N., Zidi, O., Mosbah, A., Kosai, I., Manaa, J. E., Mokhtar, N. B., Asimakis, E., Stathopoulou, P., Cherif, A., Tsiamis, G., & Kouidhi, S. (2020). Impact of the Post-Transplant Period and Lifestyle Diseases on Human Gut Microbiota in Kidney Graft Recipients. Microorganisms, 8(11), 1724. https://doi.org/10.3390/microorganisms8111724