Early Stage Root-Associated Fungi Show a High Temporal Turnover, but Are Independent of Beech Progeny

 , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
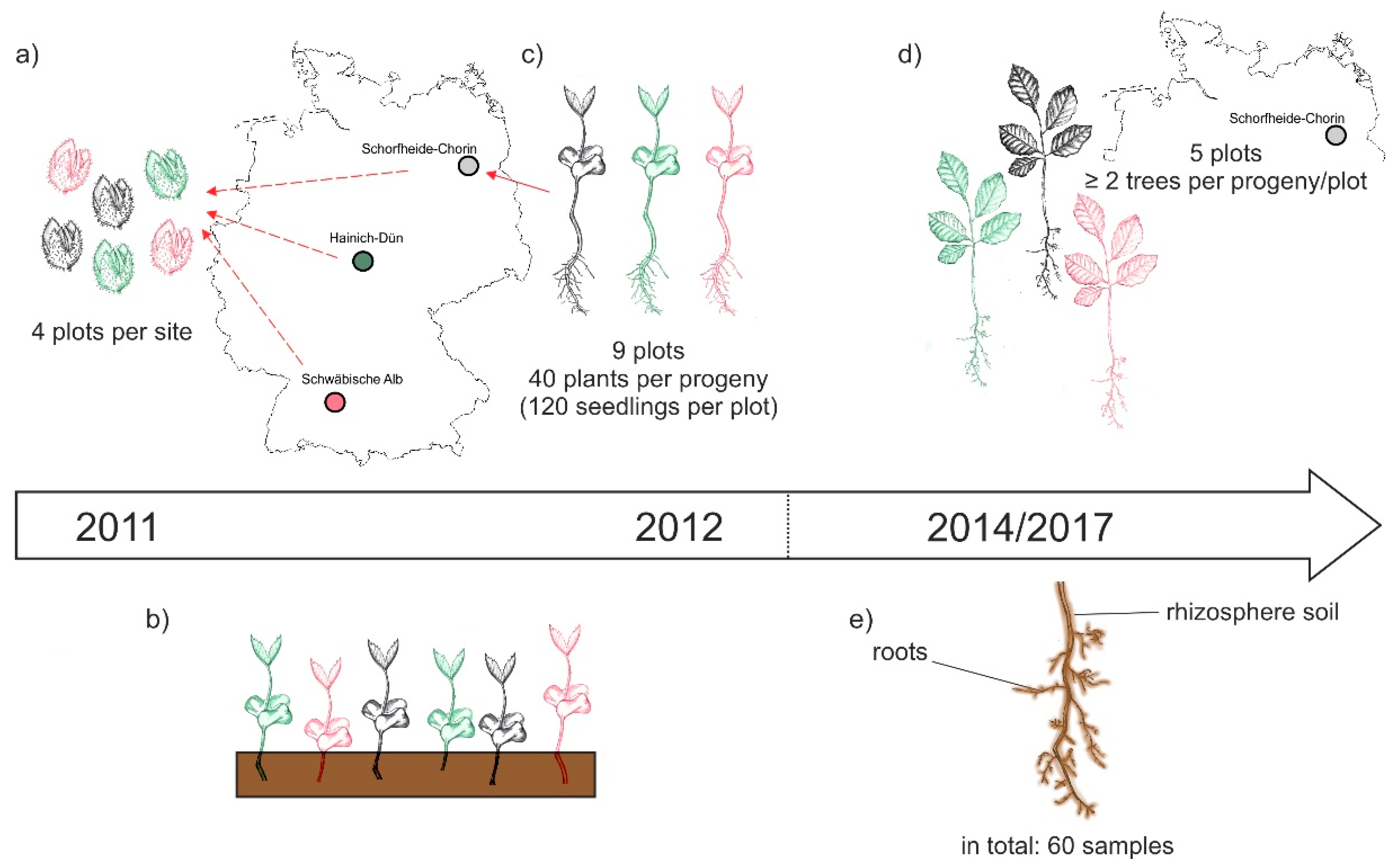
2.1. Study Sites and Experimental Design
2.2. Soil and Root Sampling
2.3. DNA Extraction, Library Preparation, and Illumina Sequencing
2.4. Bioinformatics
2.5. Data Normalization and Statistical Analyses
3. Results
3.1. Sequence Data at a Glance
3.2. Beech Progeny without Impact on Temporal Development on Fungal Diversity
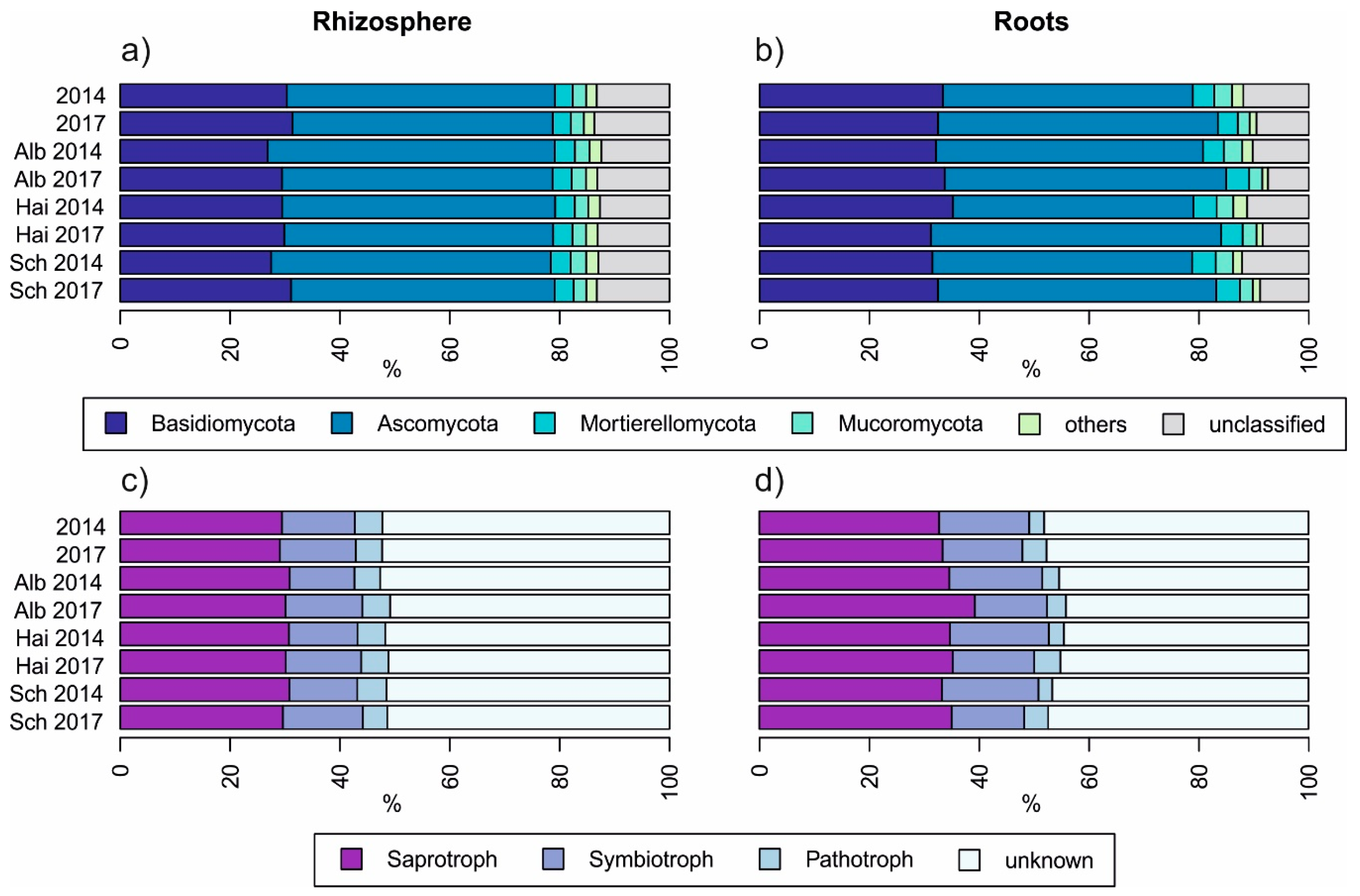
3.3. Beech Progeny without Impact on Temporal Development on Fungal Community Composition
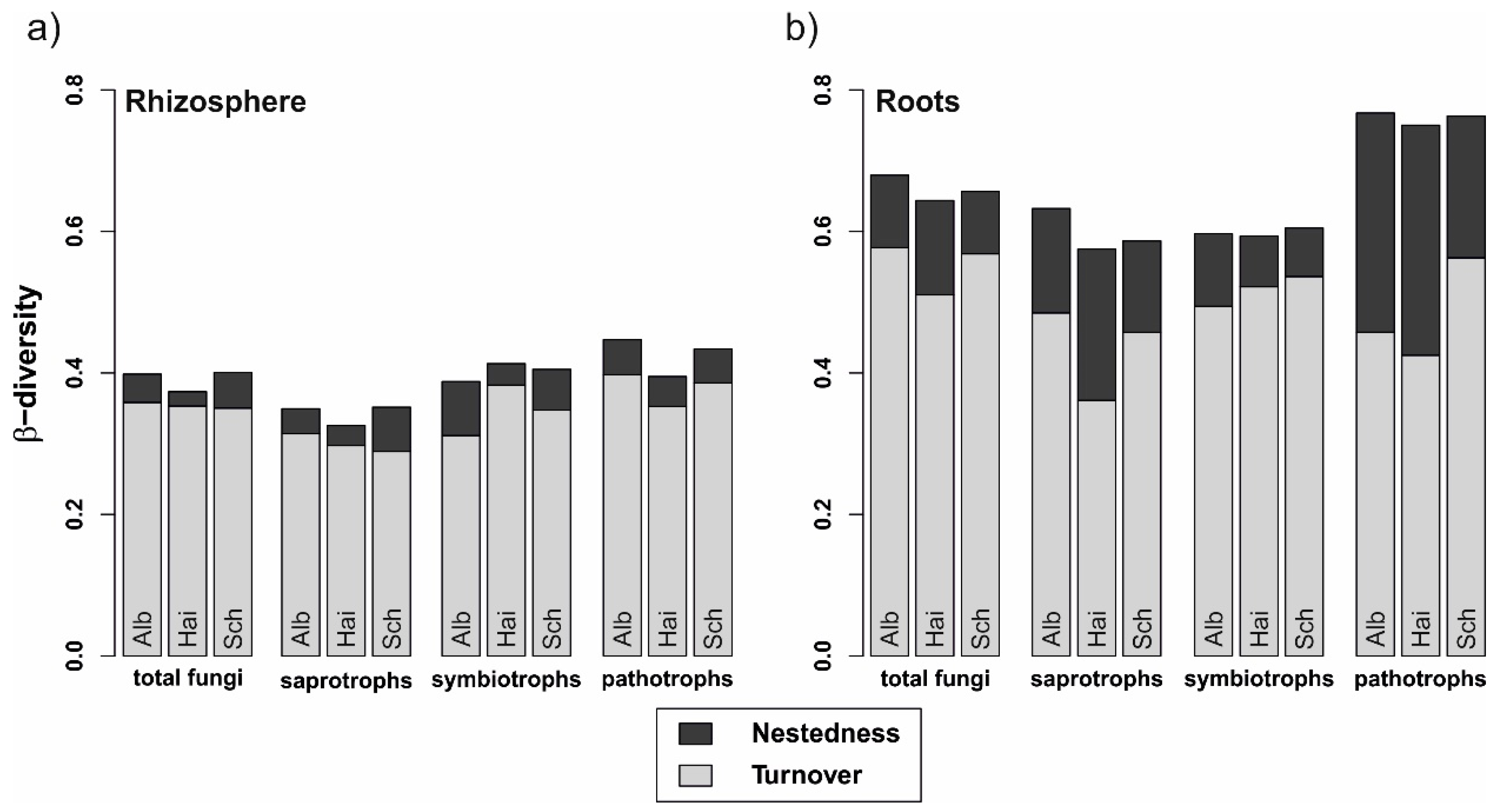
3.4. Fungal Community Turnover Independent of Beech Progeny
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pereira e Silva, M.C.; Semenov, A.V.; Schmitt, H.; van Elsas, J.D.; Salles, J.F. Microbe-mediated processes as indicators to establish the normal operating range of soil functioning. Soil Biol. Biochem. 2013, 57, 995–1002. [Google Scholar] [CrossRef]
- Wardle, D.A.; Bardgett, R.D.; Klironomos, J.N.; Setälä, H.; van der Putten, W.H.; Wall, D.H. Ecological linkages between aboveground and belowground biota. Science 2004, 304, 1629–1633. [Google Scholar] [CrossRef]
- Bulgarelli, D.; Schlaeppi, K.; Spaepen, S.; Ver Loren van Themaat, E.; Schulze-Lefert, P. Structure and functions of the bacterial microbiota of plants. Annu. Rev. Plant Biol. 2013, 64, 807–838. [Google Scholar] [CrossRef] [Green Version]
- Lundberg, D.S.; Lebeis, S.L.; Paredes, S.H.; Yourstone, S.; Gehring, J.; Malfatti, S.; Tremblay, J.; Engelbrektson, A.; Kunin, V.; Del Rio, T.G. Defining the core Arabidopsis thaliana root microbiome. Nature 2012, 488, 86. [Google Scholar] [CrossRef] [Green Version]
- Micallef, S.A.; Shiaris, M.P.; Colón-Carmona, A. Influence of Arabidopsis thaliana accessions on rhizobacterial communities and natural variation in root exudates. J. Exp. Bot. 2009, 60, 1729–1742. [Google Scholar] [CrossRef] [Green Version]
- Kuzyakov, Y.; Blagodatskaya, E. Microbial hotspots and hot moments in soil: Concept & review. Soil Biol. Biochem. 2015, 83, 184–199. [Google Scholar] [CrossRef]
- Nacke, H.; Goldmann, K.; Schöning, I.; Pfeiffer, B.; Kaiser, K.; Castillo-Villamizar, G.A.; Schrumpf, M.; Buscot, F.; Daniel, R.; Wubet, T. Fine spatial scale variation of soil microbial communities under European beech and Norway spruce. Front. Microbiol. 2016, 7, 2067. [Google Scholar] [CrossRef] [Green Version]
- Goldmann, K.; Boeddinghaus, R.S.; Klemmer, S.; Regan, K.M.; Heintz-Buschart, A.; Fischer, M.; Prati, D.; Piepho, H.-P.; Berner, D.; Marhan, S.; et al. Unraveling spatiotemporal variability of arbuscular mycorrhizal fungi in a temperate grassland plot. Environ. Microbiol. 2019. [Google Scholar] [CrossRef]
- Chaparro, J.M.; Badri, D.V.; Vivanco, J.M. Rhizosphere microbiome assemblage is affected by plant development. ISME J. 2014, 8, 790. [Google Scholar] [CrossRef] [Green Version]
- Dighton, J.; Mason, P. Mycorrhizal dynamics during forest tree development. Dev. Biol. High. Fungi 1985, 117–139. [Google Scholar]
- Pennanen, T.; Liski, J.; Bååth, E.; Kitunen, V.; Uotila, J.; Westman, C.J.; Fritze, H. Structure of the microbial communities in coniferous forest soils in relation to site fertility and stand development stage. Microb. Ecol. 1999, 38, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Wallander, H.; Johansson, U.; Sterkenburg, E.; Brandström Durling, M.; Lindahl, B.D. Production of ectomycorrhizal mycelium peaks during canopy closure in Norway spruce forests. New Phytol. 2010, 187, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Kyaschenko, J.; Clemmensen, K.E.; Hagenbo, A.; Karltun, E.; Lindahl, B.D. Shift in fungal communities and associated enzyme activities along an age gradient of managed Pinus sylvestris stands. ISME J. 2017, 11, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Averill, C.; Cates, L.L.; Dietze, M.C.; Bhatnagar, J.M. Spatial vs. temporal controls over soil fungal community similarity at continental and global scales. ISME J. 2019, 13, 2082–2093. [Google Scholar] [CrossRef] [PubMed]
- Bakker, M.G.; Schlatter, D.C.; Otto-Hanson, L.; Kinkel, L.L. Diffuse symbioses: Roles of plant–plant, plant–microbe and microbe–microbe interactions in structuring the soil microbiome. Mol. Ecol. 2014, 23, 1571–1583. [Google Scholar] [CrossRef]
- Baldrian, P. Forest microbiome: Diversity, complexity and dynamics. FEMS Microbiol. Rev. 2017, 41, 109–130. [Google Scholar] [CrossRef] [Green Version]
- Unuk, T.; Martinović, T.; Finžgar, D.; Šibanc, N.; Grebenc, T.; Kraigher, H. Root-associated fungal communities from two phenologically contrasting Silver Fir (Abies alba Mill.) groups of trees. Front. Plant Sci. 2019, 10, 214. [Google Scholar] [CrossRef] [Green Version]
- Cregger, M.A.; Veach, A.M.; Yang, Z.K.; Crouch, M.J.; Vilgalys, R.; Tuskan, G.A.; Schadt, C.W. The Populus holobiont: Dissecting the effects of plant niches and genotype on the microbiome. Microbiome 2018, 6, 31. [Google Scholar] [CrossRef]
- Pérez-Izquierdo, L.; Zabal-Aguirre, M.; Flores-Rentería, D.; González-Martínez, S.C.; Buée, M.; Rincón, A. Functional outcomes of fungal community shifts driven by tree genotype and spatial-temporal factors in Mediterranean pine forests. Environ. Microbiol. 2017, 19, 1639–1652. [Google Scholar] [CrossRef]
- Latham, R.E.; Ricklefs, R.E. Continental comparisons of temperate-zone tree species diversity. Species Divers. Ecol. Communities 1993, 26, 294–314. [Google Scholar]
- Leuschner, C.; Meier, I.C.; Hertel, D. On the niche breadth of Fagus sylvatica: Soil nutrient status in 50 Central European beech stands on a broad range of bedrock types. Ann. Forest Sci. 2006, 63, 355–368. [Google Scholar] [CrossRef] [Green Version]
- Kreyling, J.; Buhk, C.; Backhaus, S.; Hallinger, M.; Huber, G.; Huber, L.; Jentsch, A.; Konnert, M.; Thiel, D.; Wilmking, M.; et al. Local adaptations to frost in marginal and central populations of the dominant forest tree Fagus sylvatica L. as affected by temperature and extreme drought in common garden experiments. Ecol. Evol. 2014, 4, 594–605. [Google Scholar] [CrossRef] [Green Version]
- Perez, G.; Aubert, M.; Decaëns, T.; Trap, J.; Chauvat, M. Home-Field Advantage: A matter of interaction between litter biochemistry and decomposer biota. Soil Biol. Biochem. 2013, 67, 245–254. [Google Scholar] [CrossRef]
- Purahong, W.; Kahl, T.; Krüger, D.; Buscot, F.; Hoppe, B. Home-field Advantage in wood decomposition is mainly mediated by fungal community shifts at “home” versus “away”. Microb. Ecol. 2019, 78, 725–736. [Google Scholar] [CrossRef]
- Lauber, C.L.; Strickland, M.S.; Bradford, M.A.; Fierer, N. The influence of soil properties on the structure of bacterial and fungal communities across land-use types. Soil Biol. Biochem. 2008, 40, 2407–2415. [Google Scholar] [CrossRef]
- Brundrett, M.C. Coevolution of roots and mycorrhizas of land plants. New Phytol. 2002, 154, 275–304. [Google Scholar] [CrossRef]
- McLaughlin, D.J.; Spatafora, J.W. The Mycota 7. Systematics and Evolution Part A; Springer: Heidelberg, Germany, 2014. [Google Scholar]
- Purahong, W.; Wubet, T.; Krüger, D.; Buscot, F. Molecular evidence strongly supports deadwood-inhabiting fungi exhibiting unexpected tree species preferences in temperate forests. ISME J. 2018, 12, 289. [Google Scholar] [CrossRef]
- Van der Heijden, M.G.A.; Martin, F.M.; Selosse, M.; Sanders, I.R. Mycorrhizal ecology and evolution: The past, the present, and the future. New Phytol. 2015, 205, 1406–1423. [Google Scholar] [CrossRef]
- Awad, A.; Majcherczyk, A.; Schall, P.; Schröter, K.; Schöning, I.; Schrumpf, M.; Ehbrecht, M.; Boch, S.; Kahl, T.; Bauhus, J.; et al. Ectomycorrhizal and saprotrophic soil fungal biomass are driven by different factors and vary among broadleaf and coniferous temperate forests. Soil Biol. Biochem. 2019, 131, 9–18. [Google Scholar] [CrossRef]
- Vasiliauskas, R.; Menkis, A.; Finlay, R.D.; Stenlid, J. Wood-decay fungi in fine living roots of conifer seedlings. New Phytol. 2007, 174, 441–446. [Google Scholar] [CrossRef]
- Fischer, M.; Bossdorf, O.; Gockel, S.; Hänsel, F.; Hemp, A.; Hessenmöller, D.; Korte, G.; Nieschulze, J.; Pfeiffer, S.; Prati, D.; et al. Implementing large-scale and long-term functional biodiversity research: The Biodiversity Exploratories. Basic Appl. Ecol. 2010, 11, 473–485. [Google Scholar] [CrossRef]
- Goldmann, K.; Schöning, I.; Buscot, F.; Wubet, T. Forest management type influences diversity and community composition of soil fungi across temperate forest ecosystems. Front. Microbiol. 2015, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajendra, K.C.; Seifert, S.; Prinz, K.; Gailing, O.; Finkeldey, R. Subtle human impacts on neutral genetic diversity and spatial patterns of genetic variation in European beech (Fagus sylvatica). For. Ecol. Manag. 2014, 319, 138–149. [Google Scholar] [CrossRef]
- Nguyen, D.Q.; Pena, R.; Polle, A. Impact of ectomycorrhizal community composition and soil treatment on inorganic nitrogen nutrition and performance of beech (Fagus sylvatica L.) provenances. Trees 2017, 31, 1891–1904. [Google Scholar] [CrossRef]
- Toju, H.; Tanabe, A.S.; Yamamoto, S.; Sato, H. High-coverage ITS primers for the DNA-based identification of Ascomycetes and Basidiomycetes in environmental samples. PLoS ONE 2012, 7, e40863. [Google Scholar] [CrossRef] [Green Version]
- White, T.; Brans, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR Protoc. 1990, 18, 315–322. [Google Scholar]
- Ihrmark, K.; Bödeker, I.T.; Cruz-Martinez, K.; Friberg, H.; Kubartova, A.; Schenck, J.; Strid, Y.; Stenlid, J.; Brandstrom-Durling, M.; Clemmensen, K.E.; et al. New primers to amplify the fungal ITS2 region—Evaluation by 454-sequencing of artificial and natural communities. FEMS Microbiol. Ecol. 2012, 82, 666–677. [Google Scholar] [CrossRef]
- Schöps, R.; Goldmann, K.; Herz, K.; Lentendu, G.; Schöning, I.; Bruelheide, H.; Wubet, T.; Buscot, F. Land-use intensity rather than plant functional identity shapes bacterial and fungal rhizosphere communities. Front. Microbiol. 2018, 9, 2711. [Google Scholar] [CrossRef]
- Masella, A.P.; Bartram, A.K.; Truszkowski, J.M.; Brown, D.G.; Neufeld, J.D. PANDAseq: Paired-end assembler for illumina sequences. BMC Bioinform. 2012, 13, 31. [Google Scholar] [CrossRef] [Green Version]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyer, F.; Mercier, C.; Bonin, A.; Le Bras, Y.; Taberlet, P.; Coissac, E. obitools: A unix-inspired software package for DNA metabarcoding. Mol. Ecol. Resour. 2016, 16, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson-Palme, J.; Ryberg, M.; Hartmann, M.; Branco, S.; Wang, Z.; Godhe, A.; De Wit, P.; Sánchez-García, M.; Ebersberger, I.; de Sousa, F.; et al. Improved software detection and extraction of ITS1 and ITS2 from ribosomal ITS sequences of fungi and other eukaryotes for analysis of environmental sequencing data. Methods Ecol. Evol. 2013, 4, 914–919. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. Peer J. 2016, 4, e2584. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, R.H.; Larsson, K.H.; Taylor, A.F.S.; Bengtsson-Palme, J.; Jeppesen, T.S.; Schigel, D.; Kennedy, P.; Picard, K.; Glockner, F.O.; Tedersoo, L.; et al. The UNITE database for molecular identification of fungi: Handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.H.; Song, Z.; Bates, S.T.; Branco, S.; Tedersoo, L.; Menke, J.; Schilling, J.S.; Kennedy, P.G. FUNGuild: An open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 2016, 20, 241–248. [Google Scholar] [CrossRef]
- R Development Core Team. R: A Language and Environment for Statistical Computing; Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Oksanen, J.; Blanchet, F.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.; O’Hara, R.; Simpson, G.; Solymos, P.; et al. vegan: Community Ecology Package. R Package Version 2.5-2. 2019. Available online: https://CRAN.R-project.org/package=vegan (accessed on 8 February 2020).
- Baselga, A.; Orme, C.D.L. betapart: An R package for the study of beta diversity. Methods Ecol. Evol. 2012, 3, 808–812. [Google Scholar] [CrossRef]
- Weiner, J. Riverplot: Sankey or Ribbon Plots. R Package Version 0.6.2017. Available online: https://CRAN.R-project.org/package=riverplot (accessed on 8 February 2020).
- Goldmann, K.; Schröter, K.; Pena, R.; Schöning, I.; Schrumpf, M.; Buscot, F.; Polle, A.; Wubet, T. Divergent habitat filtering of root and soil fungal communities in temperate beech forests. Sci. Rep. 2016, 6, 31439. [Google Scholar] [CrossRef]
- Vandenkoornhuyse, P.; Quaiser, A.; Duhamel, M.; Le Van, A.; Dufresne, A. The importance of the microbiome of the plant holobiont. New Phytol. 2015, 206, 1196–1206. [Google Scholar] [CrossRef]
- Danielsen, L.; Thürmer, A.; Meinicke, P.; Buee, M.; Morin, E.; Martin, F.; Pilate, G.; Daniel, R.; Polle, A.; Reich, M. Fungal soil communities in a young transgenic poplar plantation form a rich reservoir for fungal root communities. Ecol. Evol. 2012, 2, 1935–1948. [Google Scholar] [CrossRef]
- Lang, C.; Finkeldey, R.; Polle, A. Spatial patterns of ectomycorrhizal assemblages in a monospecific forest in relation to host tree genotype. Front. Plant Sci. 2013, 4, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, C.; Montoya, L.; Xu, L.; Madera, M.; Hollingsworth, J.; Purdom, E.; Hutmacher, R.B.; Dahlberg, J.A.; Coleman-Derr, D.; Lemaux, P.G.; et al. Strong succession in arbuscular mycorrhizal fungal communities. ISME J. 2019, 13, 214–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, M.D.; Durall, D.M.; Cairney, J.W. Ectomycorrhizal fungal communities in young forest stands regenerating after clearcut logging. New Phytol. 2003, 157, 399–422. [Google Scholar] [CrossRef] [Green Version]
- Kałucka, I.L.; Jagodziński, A.M. Ectomycorrhizal Fungi: A Major Player in Early Succession. In Mycorrhiza-Function, Diversity, State of the Art; Springer: Cham, Switzerland, 2017; pp. 187–229. [Google Scholar]
- Kennedy, P.G.; Bruns, T.D. Priority effects determine the outcome of ectomycorrhizal competition between two Rhizopogon species colonizing Pinus muricata seedlings. New Phytol. 2005, 166, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, B.A.; Jones, M.D. Early-successional ectomycorrhizal fungi effectively support extracellular enzyme activities and seedling nitrogen accumulation in mature forests. Mycorrhiza 2017, 27, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, C.W.; Kennedy, P.G. Revisiting the ‘Gadgil effect’: Do interguild fungal interactions control carbon cycling in forest soils? New Phytol. 2016, 209, 1382–1394. [Google Scholar] [CrossRef]
- Sterkenburg, E.; Clemmensen, K.E.; Ekblad, A.; Finlay, R.D.; Lindahl, B.D. Contrasting effects of ectomycorrhizal fungi on early and late stage decomposition in a boreal forest. ISME J. 2018, 12, 2187–2197. [Google Scholar] [CrossRef] [Green Version]
- Schröter, K.; Wemheuer, B.; Pena, R.; Schöning, I.; Ehbrecht, M.; Schall, P.; Ammer, C.; Daniel, R.; Polle, A. Assembly processes of trophic guilds in the root mycobiome of temperate forests. Mol. Ecol. 2019, 28, 348–364. [Google Scholar] [CrossRef]
- Koskella, B.; Meaden, S.; Crowther, W.J.; Leimu, R.; Metcalf, C.J.E. A signature of tree health? Shifts in the microbiome and the ecological drivers of horse chestnut bleeding canker disease. New Phytol. 2017, 215, 737–746. [Google Scholar] [CrossRef] [Green Version]
- Feau, N.; Hamelin, R.C. Say hello to my little friends: How microbiota can modulate tree health. New Phytol. 2017, 215, 508–510. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Shared OTUs | Number of Rhizosphere OTUs | Number of Root OTUs | |
|---|---|---|---|
| Total | 2079 (54.61%) | 1457 (38.27%) | 271 (7.12%) |
| Ascomycota | 1014 (26.63%) | 673 (17.68%) | 101 (2.65%) |
| Saprotroph | 373 (9.80%) | 207 (5.44%) | 63 (1.65%) |
| Symbiotroph | 136 (3.57%) | 74 (1.94%) | 12 (0.32%) |
| Pathotroph | 58 (1.52%) | 60 (1.58%) | 2 (0.05%) |
| Unknown | 447 (11.74%) | 332 (8.72%) | 24 (0.63%) |
| Basidiomycota | 665 (17.47%) | 454 (11.93%) | 139 (3.65%) |
| Saprotroph | 153 (4.02%) | 114 (2.99%) | 52 (1.37%) |
| Symbiotroph | 146 (3.84%) | 100 (2.63%) | 48 (1.26%) |
| Pathotroph | 24 (0.63%) | 19 (0.50%) | 2 (0.05%) |
| Unknown | 342 (8.98%) | 221 (5.81%) | 37 (0.97%) |
| Other phyla | 400 (10.51%) | 330 (8.66%) | 31 (0.82%) |
| Saprotroph | 118 (3.10%) | 54 (1.42%) | 1 (0.03%) |
| Symbiotroph | 6 (0.16%) | 8 (0.21%) | 0 (0.00%) |
| Pathotroph | 4 (0.11%) | 7 (0.18%) | 0 (0.00%) |
| Unknown | 272 (7.14%) | 261 (6.85%) | 30 (0.79%) |
| Rhizosphere | Roots | |||||||
|---|---|---|---|---|---|---|---|---|
| Beech Progeny | Sampling Year | Beech Progeny | Sampling Year | |||||
| F | p | F | p | F | p | F | p | |
| Richness total fungi | 1.904 | 0.169 | 0.075 | 0.786 | 2.217 | 0.129 | 6.304 | 0.019 |
| Richness saprotrophs | 1.815 | 0.183 | 0.105 | 0.749 | 2.020 | 0.153 | 11.443 | 0.002 |
| Richness symbiotrophs | 0.361 | 0.701 | 0.216 | 0.646 | 2.081 | 0.145 | 0.948 | 0.339 |
| Richness pathotrophs | 0.803 | 0.459 | 0.861 | 0.362 | 2.846 | 0.076 | 20.421 | <0.001 |
| Rhizosphere | Roots | |||||||
|---|---|---|---|---|---|---|---|---|
| Beech progeny | Sampling year | Beech progeny | Sampling year | |||||
| R² | p | R² | p | R² | p | R² | p | |
| Total fungi | 0.030 | 1.000 | 0.081 | 0.014 | 0.053 | 0.688 | 0.160 | 0.001 |
| Saprotrophs | 0.029 | 1.000 | 0.090 | 0.005 | 0.053 | 0.618 | 0.156 | 0.001 |
| Symbiotrophs | 0.034 | 0.099 | 0.047 | 0.227 | 0.053 | 0.702 | 0.126 | 0.001 |
| Pathotrophs | 0.033 | 0.994 | 0.084 | 0.003 | 0.050 | 0.677 | 0.160 | 0.001 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goldmann, K.; Ammerschubert, S.; Pena, R.; Polle, A.; Wu, B.-W.; Wubet, T.; Buscot, F. Early Stage Root-Associated Fungi Show a High Temporal Turnover, but Are Independent of Beech Progeny. Microorganisms 2020, 8, 210. https://doi.org/10.3390/microorganisms8020210
Goldmann K, Ammerschubert S, Pena R, Polle A, Wu B-W, Wubet T, Buscot F. Early Stage Root-Associated Fungi Show a High Temporal Turnover, but Are Independent of Beech Progeny. Microorganisms. 2020; 8(2):210. https://doi.org/10.3390/microorganisms8020210
Chicago/Turabian StyleGoldmann, Kezia, Silke Ammerschubert, Rodica Pena, Andrea Polle, Bin-Wei Wu, Tesfaye Wubet, and François Buscot. 2020. "Early Stage Root-Associated Fungi Show a High Temporal Turnover, but Are Independent of Beech Progeny" Microorganisms 8, no. 2: 210. https://doi.org/10.3390/microorganisms8020210
APA StyleGoldmann, K., Ammerschubert, S., Pena, R., Polle, A., Wu, B. -W., Wubet, T., & Buscot, F. (2020). Early Stage Root-Associated Fungi Show a High Temporal Turnover, but Are Independent of Beech Progeny. Microorganisms, 8(2), 210. https://doi.org/10.3390/microorganisms8020210