The Pattern and Function of DNA Methylation in Fungal Plant Pathogens



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
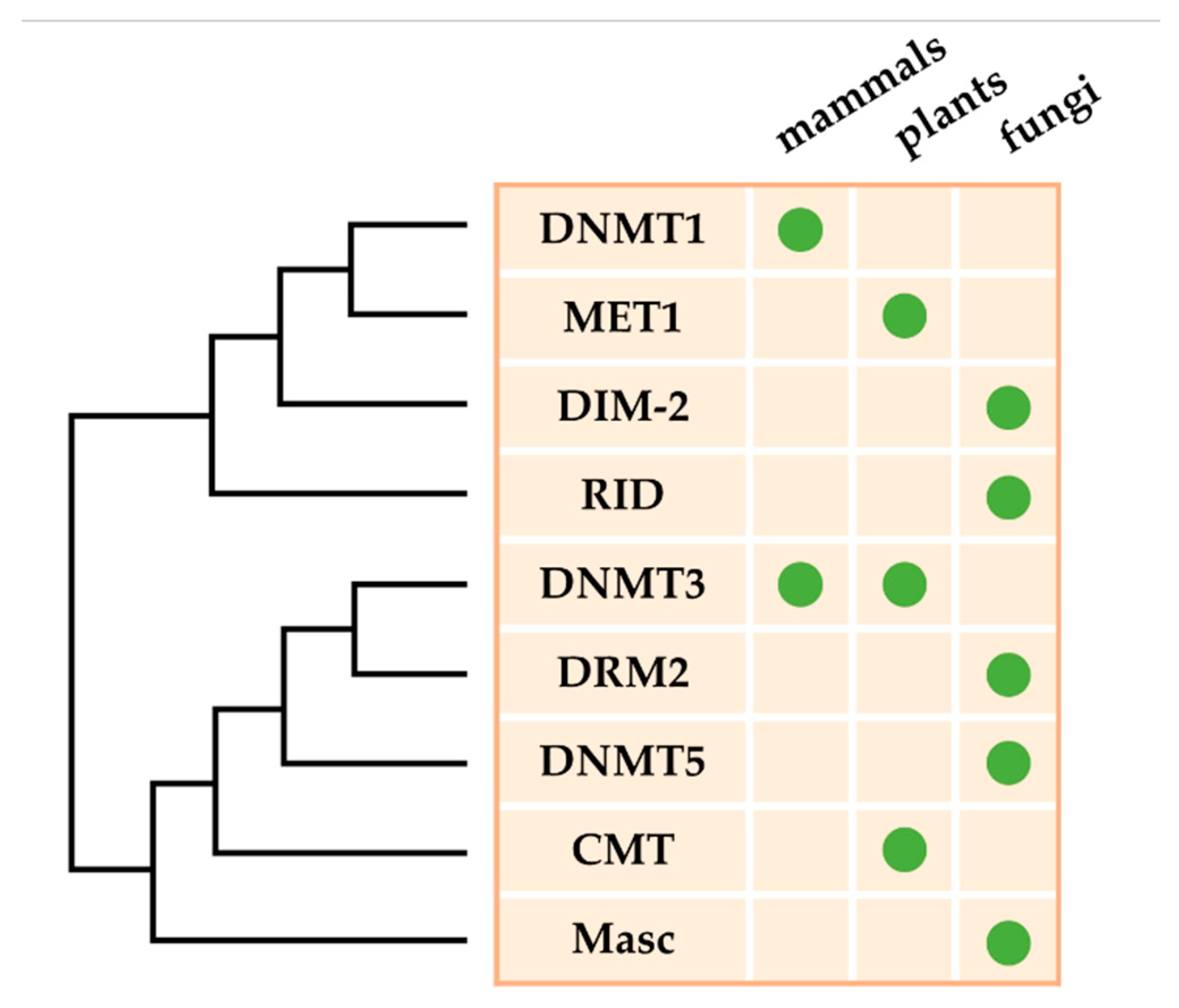
2. DNA Methyltransferases in Fungal Plant Pathogens
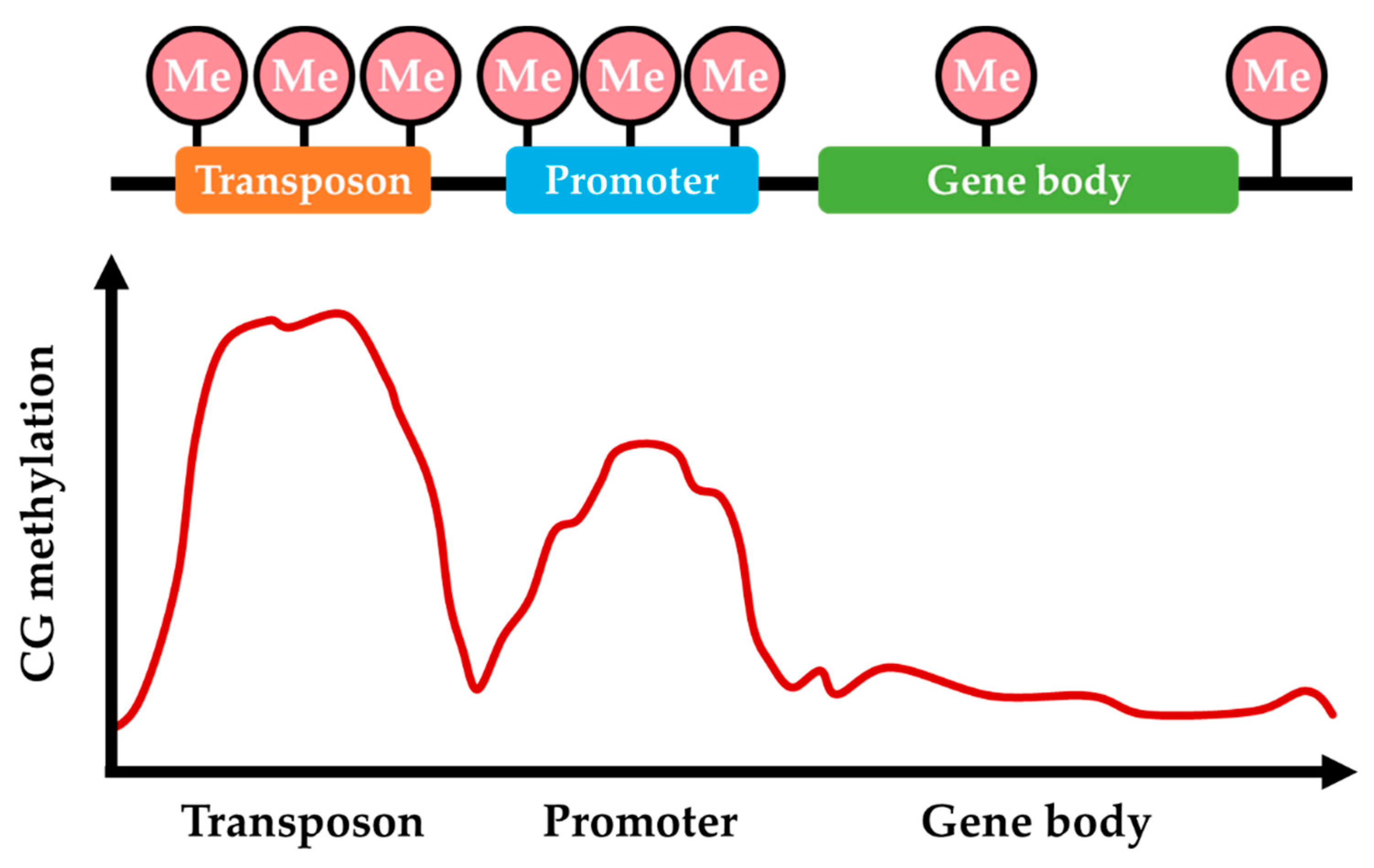
3. Patterns of DNA Methylation in Fungal Plant Pathogens
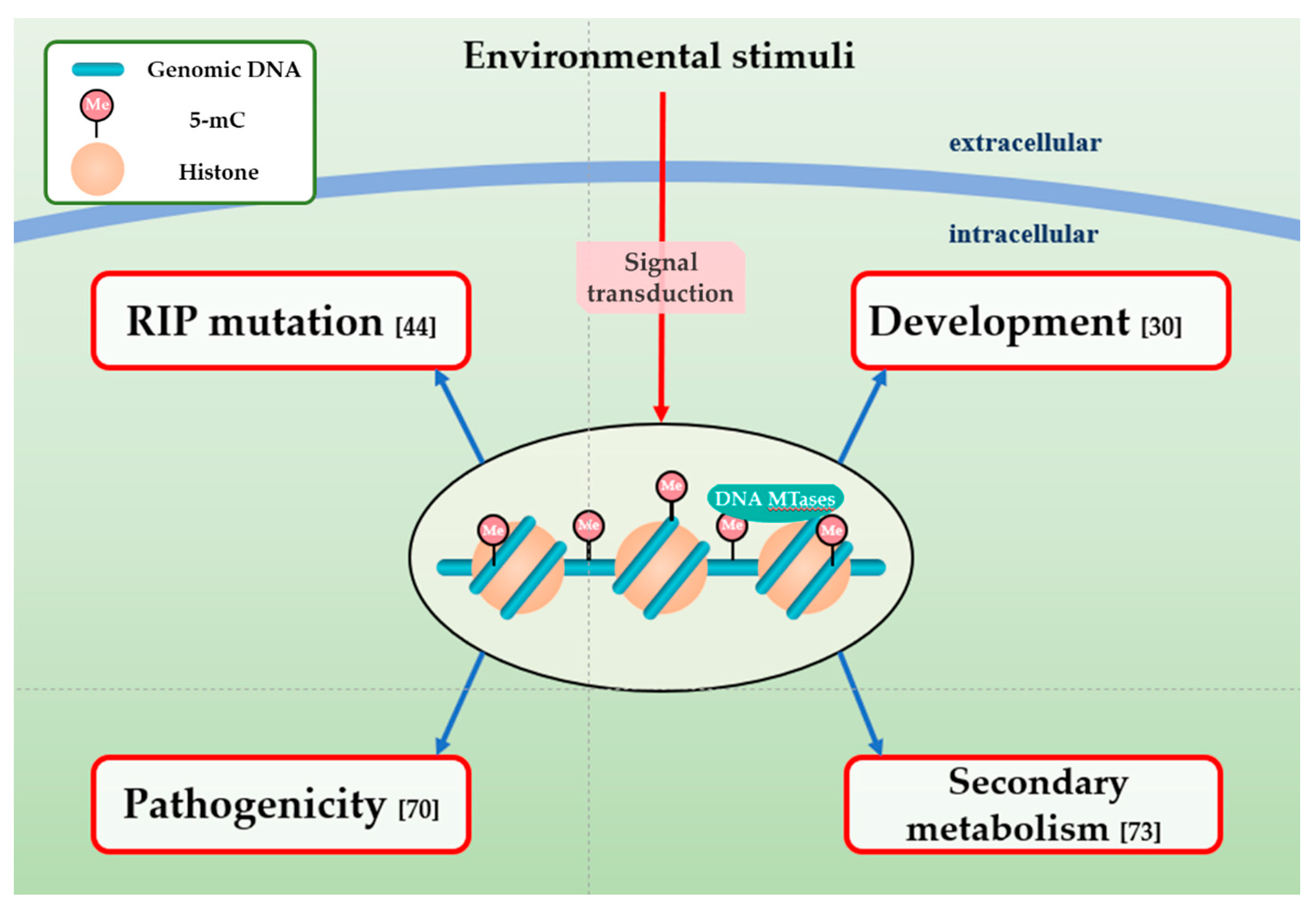
4. The Function of DNA Methylation in Fungal Plant Pathogens
4.1. DNA Methylation and RIP Mutation
4.2. Impact of DNA Methylation on the Development of Fungal Plant Pathogens
4.3. Effect of DNA Methylation on Fungal Pathogenicity
4.4. Association between DNA Methylation and Secondary Metabolism
5. DNA Methylation and Histone Methylation in Fungi
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Li, B.Q.; Zong, Y.Y.; Du, Z.L.; Shang, Y.J.; Chen, Y.; Zhang, Z.Q.; Qin, G.Z.; Zhao, W.M.; Tian, S.P. Genomic characterization reveals insights into patulin biosynthesis and pathogenicity in Penicillium species. Mol. Plant Microbe Interact. 2015, 28, 635–647. [Google Scholar] [CrossRef] [Green Version]
- Dean, R.; Kan, J.A.V.; Pretorius, Z.A.; Hammond-Kosack, K.E.; Pietro, A.D.; Spanu, P.D.; Rudd, J.J.; Dickman, M.; Kahmann, R.; Ellis, J.; et al. The top 10 fungal pathogens in molecular plant pathology. Mol. Plant Pathol. 2012, 13, 414–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malcolm, G.M.; Kuldau, G.A.; Gugino, B.K.; Jiménez-Gasco, M.D.M. Hidden host plant associations of soilborne fungal pathogens: An ecological perspective. Phytopathology 2013, 103, 538–544. [Google Scholar] [CrossRef] [Green Version]
- He, C.; Zhang, Z.Q.; Li, B.Q.; Xu, Y.; Tian, S.P. Effect of natamycin on Botrytis cinerea and Penicillium expansum-postharvest pathogens of grape berries and jujube fruit. Postharvest Biol. Technol. 2019, 151, 134–141. [Google Scholar] [CrossRef]
- Nunes, C.A. Biological control of postharvest diseases of fruit. Eur. J. Plant Pathol. 2012, 133, 181–196. [Google Scholar] [CrossRef]
- Howlett, B.J.; Lowe, R.G.T.; Marcroft, S.J.; van de Wouw, A.P. Evolution of virulence in fungal plant pathogens: Exploiting fungal genomics to control plant disease. Mycologia 2015, 107, 441–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, S.P.; Torres, R.; Ballester, A.R.; Li, B.Q.; Vilanova, L.; González-Candelas, L. Molecular aspects in pathogen-fruit interactions: Virulence and resistance. Postharvest Biol. Technol. 2016, 22, 11–21. [Google Scholar] [CrossRef] [Green Version]
- Doehlemann, G.; Ökmen, B.; Zhu, W.; Sharon, A. Plant pathogenic fungi. Microbiol. Spectr. 2017, 5. [Google Scholar] [CrossRef]
- Zhang, Z.Q.; Qin, G.Z.; Li, B.Q.; Tian, S.P. Knocking out Bcsas1 in Botrytis cinerea impacts growth, development, and secretion of extracellular proteins, which decreases virulence. Mol. Plant Microbe Interact. 2014, 27, 590–600. [Google Scholar] [CrossRef]
- Zhang, W.; Zhao, F.; Jiang, L.H.; Chen, C.; Wu, L.T.; Liu, Z.B. Different pathogen defense strategies in Arabidopsis: More than pathogen recognition. Cell 2018, 7, 252. [Google Scholar] [CrossRef] [Green Version]
- Lai, T.T.; Li, B.Q.; Qin, G.Z.; Tian, S.P. Oxidative damage involves in the inhibitory effect of nitric oxide on spore germination of Penicillium expansum. Curr. Microbiol. 2011, 62, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Li, B.Q.; Wang, W.H.; Zong, Y.Y.; Qin, G.Z.; Tian, S.P. Exploring pathogenic mechanisms of Botrytis cinerea under different ambient pH based on comparative proteomic analysis of secretome. J. Proteome Res. 2012, 11, 4249–4260. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.Q.; Li, H.; Qin, G.Z.; He, C.; Li, B.Q.; Tian, S.P. The MADS-Box transcription factor Bcmads1 is required for growth, sclerotia production and pathogenicity of Botrytis cinerea. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, Z.Q.; He, C.; Qin, G.Z.; Tian, S.P. Comparative proteomics reveals the potential targets of BcNoxR, a putative regulatory subunit of NADPH oxidase of Botrytis cinerea. Mol. Plant Microbe Interact. 2016, 29, 990–1003. [Google Scholar] [CrossRef] [Green Version]
- Presti, L.L.; Lanver, D.; Schweizer, G.; Tanaka, S.; Liang, L.; Tollot, M.; Zuccaro, A.; Reissmann, S.; Kahmann, R. Fungal effectors and plant susceptibility. Annu. Rev. Plant Biol. 2015, 66, 513–545. [Google Scholar] [CrossRef]
- Presti, L.L.; Kahmann, R. How filamentous plant pathogen effectors are translocated to host cells. Curr. Opin. Plant Biol. 2017, 38, 19–24. [Google Scholar] [CrossRef]
- Li, B.Q.; Lai, T.F.; Qin, G.Z.; Tian, S.P. Ambient pH stress inhibits spore germination of Penicillium expansum by impairing protein synthesis and folding: A proteomic-based study. J. Proteome Res. 2010, 9, 298–307. [Google Scholar] [CrossRef]
- Eshel, D.; Miyara, I.; Ailinng, T.; Dinoor, A.; Prusky, D. pH regulates endoglucanase expression and virulence of Alternaria alternata in persimmon fruits. Mol. Plant Microbe Interact. 2002, 15, 774–779. [Google Scholar] [CrossRef] [Green Version]
- Drori, N.; Kramer-Haimovich, H.; Rollins, J.; Dinoor, A.; Okon, Y.; Pines, O.; Prusky, D. External pH and nitrogen source affect secretion of pectate lyase by Colletotrichum gloeosporioides. Appl. Environ. Microbiol. 2003, 69, 3258–3262. [Google Scholar] [CrossRef] [Green Version]
- Caracuel, Z.; Roncero, M.I.G.; Espeso, E.A.; Gonzales-Verdejo, C.I.; Garcia-Maceira, F.I.; Di Pietro, A. The pH signaling transcription factor PacC controls virulence in the plant pathogen Fusarium oxysporum. Mol. Microbiol. 2003, 48, 765–779. [Google Scholar] [CrossRef]
- Chen, Y.; Li, B.Q.; Xu, X.D.; Zhang, Z.Q.; Tian, S.P. The pH-responsive PacC transcription factor plays pivotal roles in virulence and patulin biosynthesis in Penicillium expansum. Environ. Microbiol. 2018, 20, 4063–4078. [Google Scholar] [CrossRef] [PubMed]
- Rollins, J.A.; Dickman, M.B. pH signaling in Sclerotinia sclerotiorum: Identification of pacC/RIM1 homolog. Appl. Environ. Microbiol. 2001, 67, 75–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Ji, D.C.; Chen, T.; Li, B.Q.; Zhang, Z.Q.; Qin, G.Z.; Tian, S.P. Production, signaling and scavenging mechanisms for reactive oxygen species in fruit-pathogen interactions. Int. J. Mol. Sci. 2019, 20, e2994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, S.P.; Qin, G.Z.; Li, B.Q. Reactive oxygen species involved in regulating fruit senescence and fungal pathogenicity. Plant Mol. Biol. 2013, 82, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, J.; Simon, A.; Cohrs, K.C.; Viaud, M.; Tudzynski, P. The transcription factor BcLTF1 regulates virulence and light responses in the necrotrophic plant pathogen Botrytis cinerea. PLoS Genet. 2014, 10, e1004040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, B.; Li, B.Q.; Qin, G.Z.; Tian, S.P. Functions of small GTPase Rho3 in regulating growth, conidiation and virulence of Botrytis cinerea. Fungal Genet. Biol. 2015, 75, 46–55. [Google Scholar] [CrossRef]
- An, B.; Li, B.Q.; Li, H.; Zhang, Z.Q.; Qin, G.Z.; Tian, S.P. Aquaporin8 regulates cellular development and ROS production, a critical component of virulence in Botrytis cinerea. New Phytol. 2016, 209, 1668–1680. [Google Scholar] [CrossRef]
- Oliveira-Garcia, E.; Valent, B. How eukaryotic filamentous pathogens evade plant recognition. Curr. Opin. Microbiol. 2015, 26, 92–101. [Google Scholar] [CrossRef] [Green Version]
- Shalaby, S.; Horwitz, B.A. Plant phenolic compounds and oxidative stress: Integrated signals in fungal-plant interactions. Curr. Genet. 2015, 61, 347–357. [Google Scholar] [CrossRef]
- Marshall, R.; Kombrink, A.; Motteram, J.; Loza-Reyes, E.; Lucas, J.; Hammond-Kosack, K.E.; Thomma, B.P.; Rudd, J.J. Analysis of two in planta expressed LysM effector homologs from the fungus Mycosphaerella graminicola reveals novel functional properties and varying contributions to virulence on wheat. Plant Physiol. 2011, 156, 756–769. [Google Scholar] [CrossRef] [Green Version]
- Oliveira-Garcia, E.; Deising, H.B. Attenuation of PAMP-triggered immunity in maize requires down-regulation of the key β-1,6-glucan synthesis genes KRE5 and KRE6 in biotrophic hyphae of Colletotrichum graminicola. Plant. J. 2016, 84, 355–375. [Google Scholar] [CrossRef]
- Dubey, A.; Jeon, J. Epigenetic regulation of development and pathogenesis in fungal plant pathogens. Mol. Plant Pathol. 2017, 18, 887–898. [Google Scholar] [CrossRef]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, M.M.; Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.D.; Le, T.; Fan, G.P. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [Green Version]
- Bartels, A.; Han, Q.; Nair, P.; Stacey, L.; Gaynier, H.; Mosley, M.; Huang, Q.Q.; Pearson, J.K.; Hsieh, T.F.; An, Y.C.; et al. Dynamic DNA methylation in plant growth and development. Int. J. Mol. Sci. 2018, 19, e2144. [Google Scholar] [CrossRef] [Green Version]
- Su, Z.X.; Han, L.; Zhao, Z.M. Conservation and divergence of DNA methylation in eukaryotes: New insights from single base-resolution DNA methylomes. Epigenetics 2011, 6, 134–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, S.; Fei, Z.; Chen, Y.R.; Zheng, Y.; Huang, M.; Vrebalov, J.; McQuinn, R.; Gapper, N.; Liu, B.; Xiang, J.; et al. Single-base resolution methylomes of tomato fruit development reveal epigenome modifications associated with ripening. Nat. Biotechnol. 2013, 31, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Dan, J.M.; Chen, T.P. Genetic studies on mammalian DNA methyltransferases. Adv. Exp. Med. Biol. 2016, 945, 123–150. [Google Scholar] [CrossRef]
- Martienssen, R.A.; Colot, V. DNA methylation and epigenetic inheritance in plants and filamentous fungi. Science 2001, 293, 1070–1074. [Google Scholar] [CrossRef] [Green Version]
- Cokus, S.J.; Feng, S.H.; Zhang, X.Y.; Chen, Z.G.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008, 452, 215–219. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.C.; Zhang, Y. Active DNA demethylation: Many roads lead to Rome. Nat. Rev. Mol. Cell Biol. 2010, 11, 607–620. [Google Scholar] [CrossRef] [Green Version]
- Lees-Murdock, D.J.; Walsh, C.P. DNA methylation reprogramming in the germ line. Adv. Exp. Med. Biol. 2008, 626, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.; Choi, J.; Lee, G.W.; Park, S.Y.; Huh, A.; Dean, R.A.; Lee, Y.H. Genome-Wide profiling of DNA methylation provides insights into epigenetic regulation of fungal development in a plant pathogenic fungus, Magnaporthe oryzae. Sci. Rep. 2015, 5, 8567. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Han, L. CpG islands: Algorithms and applications in methylation studies. Biochem. Biophys. Res. Commun. 2009, 382, 643–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, Y.C.; Goettel, W.; Han, Q.; Bartels, A.; Liu, Z.; Xiao, W. Dynamic changes of genome-wide DNA methylation during soybean seed development. Sci. Rep. 2017, 7, 12263. [Google Scholar] [CrossRef] [PubMed]
- Jullien, P.E.; Susaki, D.; Yelagandula, R.; Higashiyama, T.; Berger, F. DNA methylation dynamics during sexual reproduction in Arabidopsis thaliana. Curr. Biol. 2012, 22, 1825–1830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Zhu, J.D.; Hu, F.Y.; Ge, S.; Ye, M.Z.; Xiang, H.; Zhang, G.J.; Zheng, X.M.; Zhang, H.Y.; Zhang, S.L.; et al. Single-Base resolution maps of cultivated and wild rice methylomes and regulatory roles of DNA methylation in plant gene expression. BMC Genom. 2012, 13, 300. [Google Scholar] [CrossRef] [Green Version]
- Zemach, A.; McDaniel, I.E.; Silva, P.; Zilberman, D. Genome-Wide evolutionary analysis of eukaryotic DNA methylation. Science 2010, 328, 916–919. [Google Scholar] [CrossRef] [Green Version]
- Selker, E.U.; Fritz, D.Y.; Singer, M.J. Dense nonsymmetrical DNA methylation resulting from repeat-induced point mutation in Neurospora. Science 1993, 262, 1724–1728. [Google Scholar] [CrossRef] [PubMed]
- Goll, M.G.; Bestor, T.H. Eukaryotic cytosine methyltransferases. Annu. Rev. Biochem. 2005, 74, 481–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finnegan, E.J.; Dennis, E.S. Isolation and identification by sequence homology of a putative cytosine methyltransferase from Arabidopsis thaliana. Nucleic Acids Res. 1993, 21, 2383–2388. [Google Scholar] [CrossRef] [PubMed]
- Kouzminova, E.; Selker, E.U. Dim-2 encodes a DNA methyltransferase responsible for all known cytosine methylation in Neurospora. EMBO J. 2001, 20, 4309–4323. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.; Springer, N.M.; Muszynski, M.G.; Phillips, R.L.; Kaeppler, S.; Jacobsen, S.E. Conserved plant genes with similarity to mammalian de novo DNA methyltransferases. Proc. Natl. Acad. Sci. USA 2000, 97, 4979–4984. [Google Scholar] [CrossRef] [Green Version]
- Bartee, L.; Malagnac, F.; Bender, J. Arabidopsis cmt3 chromomethylase mutations block non-CG methylation and silencing of an endogenous gene. Genes Dev. 2001, 15, 1753–1758. [Google Scholar] [CrossRef] [Green Version]
- Malagnac, F.; Wendel, B.; Goyon, C.; Faugeron, G.; Zickler, D.; Rossignol, J.L.; Noyer-Weidner, M.; Vollmayr, P.; Trautner, T.A.; Walter, J. A gene essential for de novo methylation and development in Ascobolus reveals a novel type of eukaryotic DNA methyltransferase structure. Cell 1997, 91, 281–290. [Google Scholar] [CrossRef] [Green Version]
- Freitag, M.; Williams, R.L.; Kothe, G.O.; Selker, E.U. A cytosine methyltransferase homologue is essential for repeat-induced point mutation in Neurospora crassa. Proc. Natl. Acad. Sci. USA 2002, 99, 8802–8807. [Google Scholar] [CrossRef] [Green Version]
- Bewick, A.J.; Hofmeister, B.T.; Powers, R.A.; Mondo, S.J.; Grigoriev, I.V.; James, T.Y.; Stajich, J.E.; Schmitz, R.J. Diversity of cytosine methylation across the fungal tree of life. Nat. Ecol. Evol. 2019, 3, 479–490. [Google Scholar] [CrossRef]
- Francesca, T.; Friederike, H.; Nader, A.; Sebastian, B.; Oliver, P.; Giuseppina, F.; Sonja, R.; Reinhard, L.; Georg, S.; Hermann-Josef, G.; et al. The tRNA methyltransferase Dnmt2 is required for accurate polypeptide synthesis during haematopoiesis. EMBO J. 2015, 34, 2350–2362. [Google Scholar] [CrossRef] [Green Version]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef]
- Jurkowski, T.P.; Jeltsch, A. On the evolutionary origin of eukaryotic DNA methyltransferases and Dnmt2. PLoS ONE 2011, 6, e28104. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.; Ding, Q.; Xiang, Y.; Gu, T.; Li, Y. Comparative analysis of DNA methyltransferase gene family in fungi: A focus on Basidiomycota. Front. Plant Sci. 2016, 21, 1556. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, K.; Van Vu, B.; Kadotani, N.; Tanaka, M.; Murata, T.; Shiina, K.; Chuma, I.; Tosa, Y.; Nakayashiki, H. Is the fungus Magnaporthe losing DNA methylation? Genetics 2013, 195, 845–855. [Google Scholar] [CrossRef] [Green Version]
- Nanty, L.; Carbajosa, G.; Heap, G.A.; Ratnieks, F.; van Heel, D.A.; Down, T.A.; Rakyan, V.K. Comparative methylomics reveals gene-body H3K36me3 in Drosophila predicts DNA methylation and CpG landscapes in other invertebrates. Genome Res. 2011, 21, 1841–1850. [Google Scholar] [CrossRef] [Green Version]
- Malagnac, F.; Grégoire, A.; Goyon, C.; Rossignol, J.L.; Faugeron, G. Masc2, a gene from Ascobolus encoding a protein with a DNA-methyltransferase activity In Vitro, is dispensable for In Vivo methylation. Mol. Microbiol. 1999, 31, 331–338. [Google Scholar] [CrossRef]
- Yang, K.; Liang, L.; Ran, F.; Liu, Y.; Li, Z.; Lan, H.; Gao, P.; Zhuang, Z.; Zhang, F.; Nie, X.; et al. The DmtA methyltransferase contributes to Aspergillus flavus conidiation, sclerotial production, aflatoxin biosynthesis and virulence. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.L.; Wang, T.T.; Qiao, L.T.; Zhu, J.Y.; Fan, J.R.; Zhang, T.T.; Wang, Z.X.; Li, W.Z.; Chen, A.H.; Huang, B. DNA methyltransferases contribute to the fungal development, stress tolerance and virulence of the entomopathogenic fungus Metarhizium robertsii. Appl. Microbiol. Biotechnol. 2017, 101, 4215–4226. [Google Scholar] [CrossRef]
- Lewis, Z.A.; Honda, S.; Khlafallah, T.K.; Jeffress, J.K.; Freitag, M.; Mohn, F.; Schübeler, D.; Selker, E.U. Relics of repeat-induced point mutation direct heterochromatin formation in Neurospora crassa. Genome Res. 2009, 19, 427–437. [Google Scholar] [CrossRef] [Green Version]
- Montanini, B.; Chen, P.Y.; Morselli, M.; Jaroszewicz, A.; Lopez, D.; Martin, F.; Ottonello, S.; Pellegrini, M. Non-Exhaustive DNA methylation-mediated transposon silencing in the black truffle genome, a complex fungal genome with massive repeat element content. Genome Biol. 2014, 15, 411. [Google Scholar] [CrossRef]
- Antequera, F.; Tamame, M.; Villanuevaz, J.R.; Santos, Q.T. DNA methylation in the fungi. J. Biol. Chem. 1984, 259, 8033–8036. [Google Scholar]
- Zeng, Z.; Wu, J.Y.; Kovalchuk, A.; Raffaello, T.; Wen, Z.L.; Liu, M.X.; Asiegbu, F.O. Genome-Wide DNA methylation and transcriptomic profiles in the lifestyle strategies and asexual development of the forest fungal pathogen Heterobasidion parviporum. Epigenetics 2019, 14, 16–40. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Z.; Sun, H.; Vainio, E.J.; Raffaello, T.; Kovalchuk, A.; Morin, E.; Duplessis, S.; Asiegbu, F.O. Intraspecific comparative genomics of isolates of the Norway spruce pathogen (Heterobasidion parviporum) and identification of its potential virulence factors. BMC Genom. 2018, 19, 220. [Google Scholar] [CrossRef] [Green Version]
- Brenet, F.; Moh, M.; Funk, P.; Feierstein, E.; Viale, A.J.; Socci, N.D.; Scandura, J.M. DNA methylation of the first exon is tightly linked to transcriptional silencing. PLoS ONE 2011, 6, e14524. [Google Scholar] [CrossRef]
- Wang, Y.L.; Wang, Z.X.; Liu, C.; Wang, S.B.; Huang, B. Genome-Wide analysis of DNA methylation in the sexual stage of the insect pathogenic fungus Cordyceps militaris. Fungal Biol. 2015, 119, 1246–1254. [Google Scholar] [CrossRef]
- So, K.K.; Ko, Y.H.; Chun, J.; Bal, J.; Jeon, J.; Kim, J.M.; Choi, J.; Lee, Y.H.; Huh, J.H.; Kim, D.H. Global DNA methylation in the chestnut blight fungus Cryphonectria parasitica and genome-wide changes in DNA methylation accompanied with sectorization. Front. Plant Sci. 2018, 9, 103. [Google Scholar] [CrossRef] [Green Version]
- Hane, J.; Williams, A.; Taranto, A.; Solomon, P.; Oliver, R.; VanDenBerg, M.; Maruthachalam, K. Repeat-Induced point mutation: A fungal-specific, endogenous mutagenesis process. Genet. Transform. Syst. Fungi 2015, 2, 55–68. [Google Scholar] [CrossRef]
- Galagan, J.E.; Selker, E.U. RIP: The evolutionary cost of genome defense. Trends Genet. 2004, 20, 417–423. [Google Scholar] [CrossRef]
- Gladyshev, E. Repeat-Induced point mutation and other genome defense mechanisms in fungi. Microbiol. Spectr. 2017, 5. [Google Scholar] [CrossRef] [Green Version]
- Nakayashiki, H.; Nishimoto, N.; Ikeda, K.; Tosa, Y.; Mayama, S. Degenerate MAGGY elements in a subgroup of Pyricularia grisea: A possible example of successful capture of a genetic invader by a fungal genome. Mol. Gen. Genet. 1999, 261, 958–966. [Google Scholar] [CrossRef]
- Ikeda, K.; Nakayashiki, H.; Kataoka, T.; Tamba, H.; Hashimoto, Y.; Tosa, Y.; Mayama, S. Repeat-Induced point mutation (RIP) in Magnaporthe grisea: Implications for its sexual cycle in the natural field context. Mol. Microbiol. 2002, 45, 1355–1364. [Google Scholar] [CrossRef]
- Cuomo, C.A.; Guldener, U.; Xu, J.R.; Trail, F.; Turgeon, B.G.; Di Pietro, A.; Walton, J.D.; Ma, L.J.; Baker, S.E.; Rep, M.; et al. The Fusarium graminearum genome reveals a link between localized polymorphism and pathogen specialization. Science 2007, 317, 1400–1402. [Google Scholar] [CrossRef] [Green Version]
- Galagan, J.E.; Calvo, S.E.; Cuomo, C.; Ma, L.J.; Wortman, J.R.; Batzoglou, S.; Lee, S.I.; Basturkmen, M.; Spevak, C.C.; Clutterbuck, J.; et al. Sequencing of Aspergillus nidulans and comparative analysis with A. fumigatus and A. oryzae. Nature 2005, 438, 1105–1115. [Google Scholar] [CrossRef]
- Zhu, Y.J.; Xu, J.; Sun, C.; Zhou, S.; Xu, H.; Nelson, D.R.; Qian, J.; Song, J.; Luo, H.; Xiang, L.; et al. Chromosome-Level genome map provides insights into diverse defense mechanisms in the medicinal fungus Ganoderma sinense. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [Green Version]
- Jeon, J.; Lee, Y.H.; Dean, R.A. Characterization of Experimental Evolution and DNA Methylation in the Rice Blast Fungus. Functional Genomics. Ph.D. Thesis, North Carolina State University, Raleigh, NC, USA, 2012. [Google Scholar]
- Gowher, H.; Ehrlich, K.C.; Jeltsch, A. DNA from Aspergillus flavus contains 5-methylcytosine. FEMS Microbiol. Lett. 2001, 205, 151–155. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.L.; Zhuang, Z.H.; Zhang, F.; Song, F.Q.; Zhong, H.; Ran, F.L.; Yu, S.; Xu, G.P.; Lan, F.X.; Wang, S.H. Inhibition of aflatoxin metabolism and growth of Aspergillus flavus in liquid culture by a DNA methylation inhibitor. Food Addit. Contam. Part A Chem. Anal. Control. Expo. Risk Assess. 2015, 32, 554–563. [Google Scholar] [CrossRef]
- Liu, S.Y.; Lin, J.Q.; Wu, H.L.; Wang, C.C.; Huang, S.J.; Luo, Y.F.; Sun, J.H.; Zhou, J.X.; Yan, S.J.; He, J.G.; et al. Bisulfite sequencing reveals that Aspergillus flavus holds a hollow in DNA methylation. PLoS ONE 2012, 7, e30349. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.H.; Shan, W.X.; Ayliffe, M.A.; Wang, M.B. Epigenetic mechanisms: An emerging player in plant-microbe interactions. Mol. Plant Microbe Interact. 2016, 29, 187–196. [Google Scholar] [CrossRef] [Green Version]
- Pfannenstiel, B.T.; Keller, N.P. On top of biosynthetic gene clusters: How epigenetic machinery influences secondary metabolism in fungi. Biotechnol. Adv. 2019, 37, 107345. [Google Scholar] [CrossRef]
- Sun, J.; Awakawa, T.; Noguchi, H.; Abe, I. Induced production of mycotoxins in an endophytic fungus from the medicinal plant Datura stramonium. Bioorg. Med. Chem. Lett. 2012, 22, 6397–6400. [Google Scholar] [CrossRef]
- Fisch, K.M.; Gillaspy, A.F.; Gipson, M.; Henrikson, J.C.; Hoover, A.R.; Jackson, L.; Najar, F.Z.; Wägele, H.; Cichewicz, R.H. Chemical induction of silent biosynthetic pathway transcription in Aspergillus niger. J. Ind. Microbiol. Biotechnol. 2009, 36, 1199–1213. [Google Scholar] [CrossRef]
- Williams, R.B.; Henrikson, J.C.; Hoover, A.R.; Lee, A.E.; Cichewicz, R.H. Epigenetic remodeling of the fungal secondary metabolome. Org. Biomol. Chem. 2008, 6, 1895. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Sena-Filho, J.G.; Hoover, A.R.; King, J.B.; Ellis, T.K.; Powell, D.R.; Cichewicz, R.H. Chemical epigenetics alters the secondary metabolite composition of guttate excreted by an Atlantic-forestsoil-derived Penicillium citregonigrum. J. Nat. Prod. 2010, 73, 942–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asai, T.; Chung, Y.M.; Sakurai, H.; Ozeki, T.; Chang, F.R.; Yamashita, K.; Oshima, Y. Tenuipyrone, a novel skeletal polyketide from the entomopathogenic fungus, Isaria tenuipes, cultivated in the presence of epigenetic modifiers. Org. Lett. 2012, 14, 513–515. [Google Scholar] [CrossRef] [PubMed]
- Kensler, T.W.; Roebuck, B.D.; Wogan, G.N.; Groopman, J.D. Aflatoxin: A 50-year odyssey of mechanistic and translational toxicology. Toxicol. Sci. 2011, 1, 28–48. [Google Scholar] [CrossRef] [Green Version]
- Kritskiĭ, M.S.; Filippovich, S.I.; Afanas’eva, T.P.; Bachurina, G.P.; Russo, V.E. Effect of inhibitors of enzymatic DNA methylation on the formation of reproductive structures and carotenoid production in Neurospora crassa. Prikl. Biokhim. Mikrobiol. 2001, 37, 279–284. [Google Scholar] [CrossRef]
- Cedar, H.; Bergman, Y. Linking DNA methylation and histone modification: Patterns and paradigms. Nat. Rev. Genet. 2009, 10, 295–304. [Google Scholar] [CrossRef]
- Migliori, V.; Phalke, S.; Bezzi, M.; Guccione, E. Arginine/lysine-methyl/methyl switches: Biochemical role of histone arginine methylation in transcriptional regulation. Epigenomics 2012, 2, 119–137. [Google Scholar] [CrossRef]
- Li, Y.; He, Y.Z.; Li, X.; Fasoyin, O.E.; Hu, Y.L.; Liu, Y.J.; Yuan, J.; Zhuang, Z.H.; Wang, S.H. Histone methyltransferase aflrmtA gene is involved in the morphogenesis, mycotoxin biosynthesis, and pathogenicity of Aspergillus Flavus. Toxicon 2017, 127, 112–121. [Google Scholar] [CrossRef]
- Du, J.; Johnson, L.M.; Jacobsen, S.E.; Patel, D.J. DNA methylation pathways and their crosstalk with histone methylation. Nat. Rev. Mol. Cell Biol. 2015, 16, 519–532. [Google Scholar] [CrossRef] [Green Version]
- Tamaru, H.; Zhang, X.; McMillen, D.; Singh, P.B.; Nakayama, J.; Grewal, S.I.; Allis, C.D.; Cheng, X.; Selker, E.U. Trimethylated lysine 9 of histone H3 is a mark for DNA methylation in Neurospora crassa. Nat. Genet. 2003, 34, 75–79. [Google Scholar] [CrossRef]
- Lewis, Z.A.; Adhvaryu, K.K.; Honda, S.; Shiver, A.L.; Knip, M.; Sack, R.; Selker, E.U. DNA methylation and normal chromosome behavior in Neurospora depend on five components of a histone methyltransferase complex, DCDC. PLoS Genet. 2010, 6, e1001196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gessaman, J.D.; Selker, E.U. Induction of H3K9me3 and DNA methylation by tethered heterochromatin factors in Neurospora crassa. Proc. Natl. Acad. Sci. USA 2017, 114, e9598–e9607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasuga, T.; Gijzen, M. Epigenetics and the evolution of virulence. Trends Microbiol. 2013, 21, 575–582. [Google Scholar] [CrossRef]
- Gomez-Diaz, E.; Jorda, M.; Peinado, M.A.; Rivero, A. Epigenetics of host–pathogen interactions: The road ahead and the road behind. PLoS Pathog. 2012, 8, e1003007. [Google Scholar] [CrossRef] [PubMed] [Green Version]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, C.; Zhang, Z.; Li, B.; Tian, S. The Pattern and Function of DNA Methylation in Fungal Plant Pathogens. Microorganisms 2020, 8, 227. https://doi.org/10.3390/microorganisms8020227
He C, Zhang Z, Li B, Tian S. The Pattern and Function of DNA Methylation in Fungal Plant Pathogens. Microorganisms. 2020; 8(2):227. https://doi.org/10.3390/microorganisms8020227
Chicago/Turabian StyleHe, Chang, Zhanquan Zhang, Boqiang Li, and Shiping Tian. 2020. "The Pattern and Function of DNA Methylation in Fungal Plant Pathogens" Microorganisms 8, no. 2: 227. https://doi.org/10.3390/microorganisms8020227
APA StyleHe, C., Zhang, Z., Li, B., & Tian, S. (2020). The Pattern and Function of DNA Methylation in Fungal Plant Pathogens. Microorganisms, 8(2), 227. https://doi.org/10.3390/microorganisms8020227