A Systematic Approach to Bacterial Phylogeny Using Order Level Sampling and Identification of HGT Using Network Science


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Datasets and Clustering
2.2. Heatmap and Tree of Bacterial Phyla
2.3. Network of Organisms
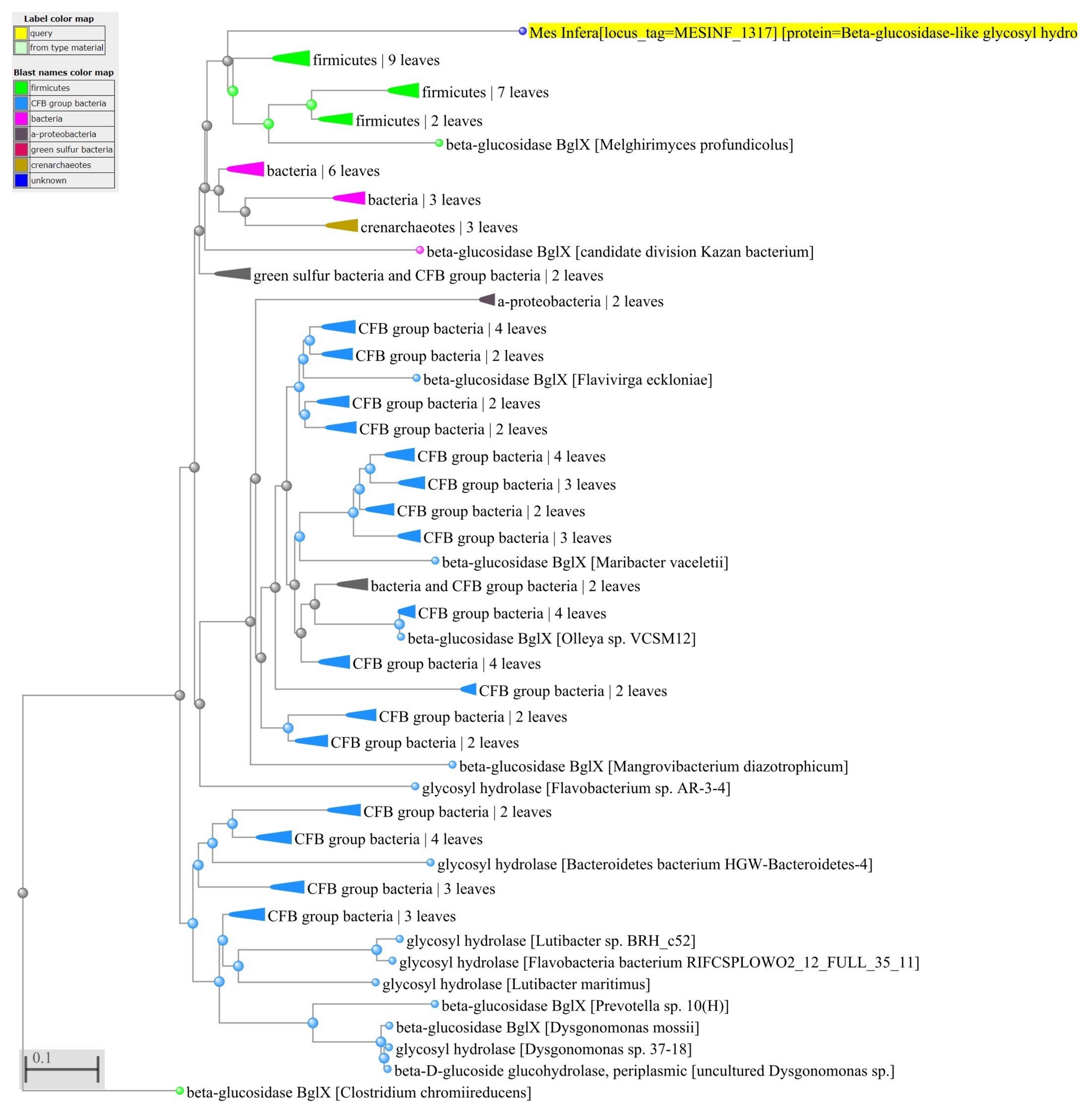
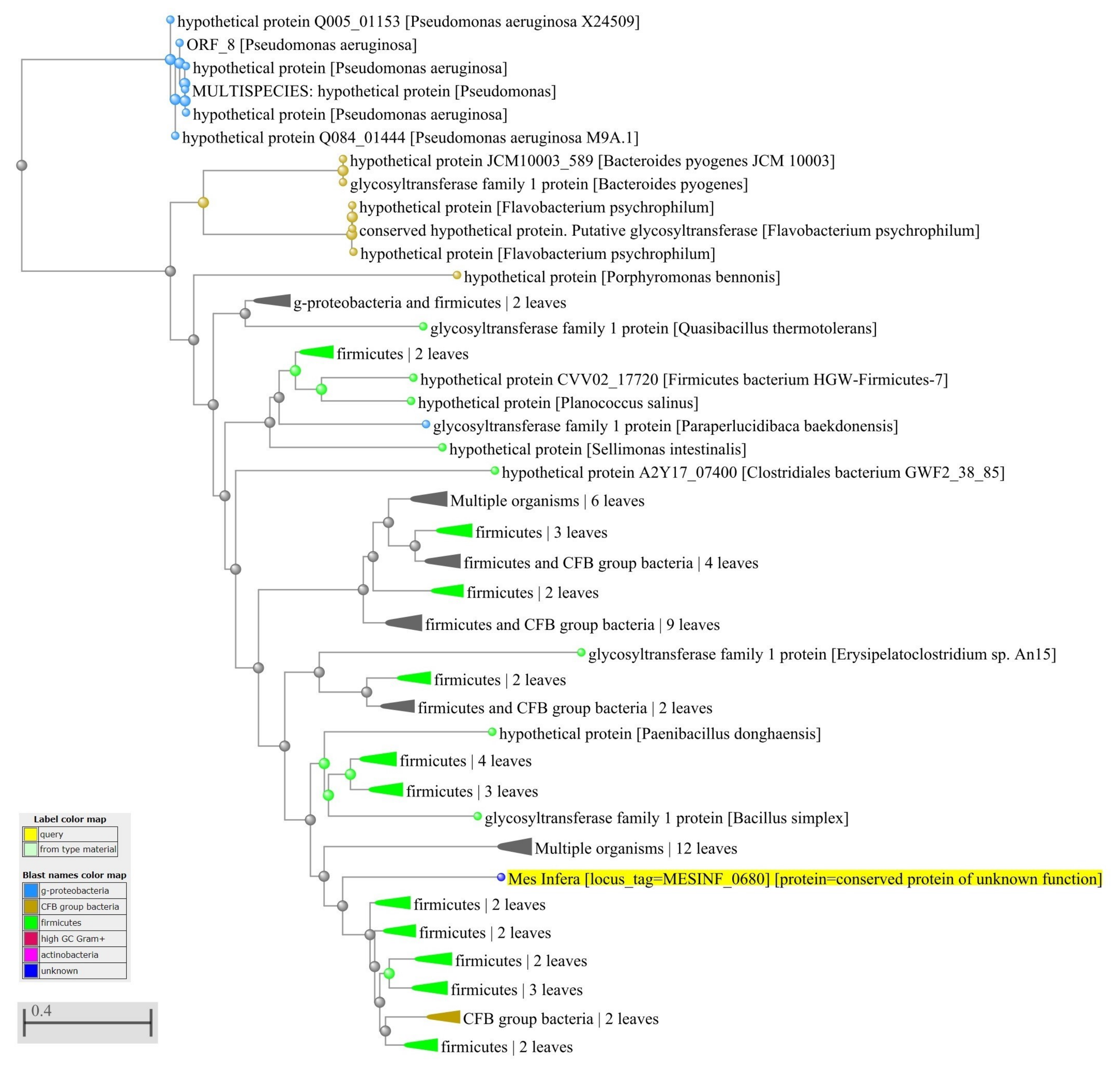
2.4. Identification of Horizontally-Transferred Genes and Protein Functions
3. Results
3.1. Clustering Protein Sequences
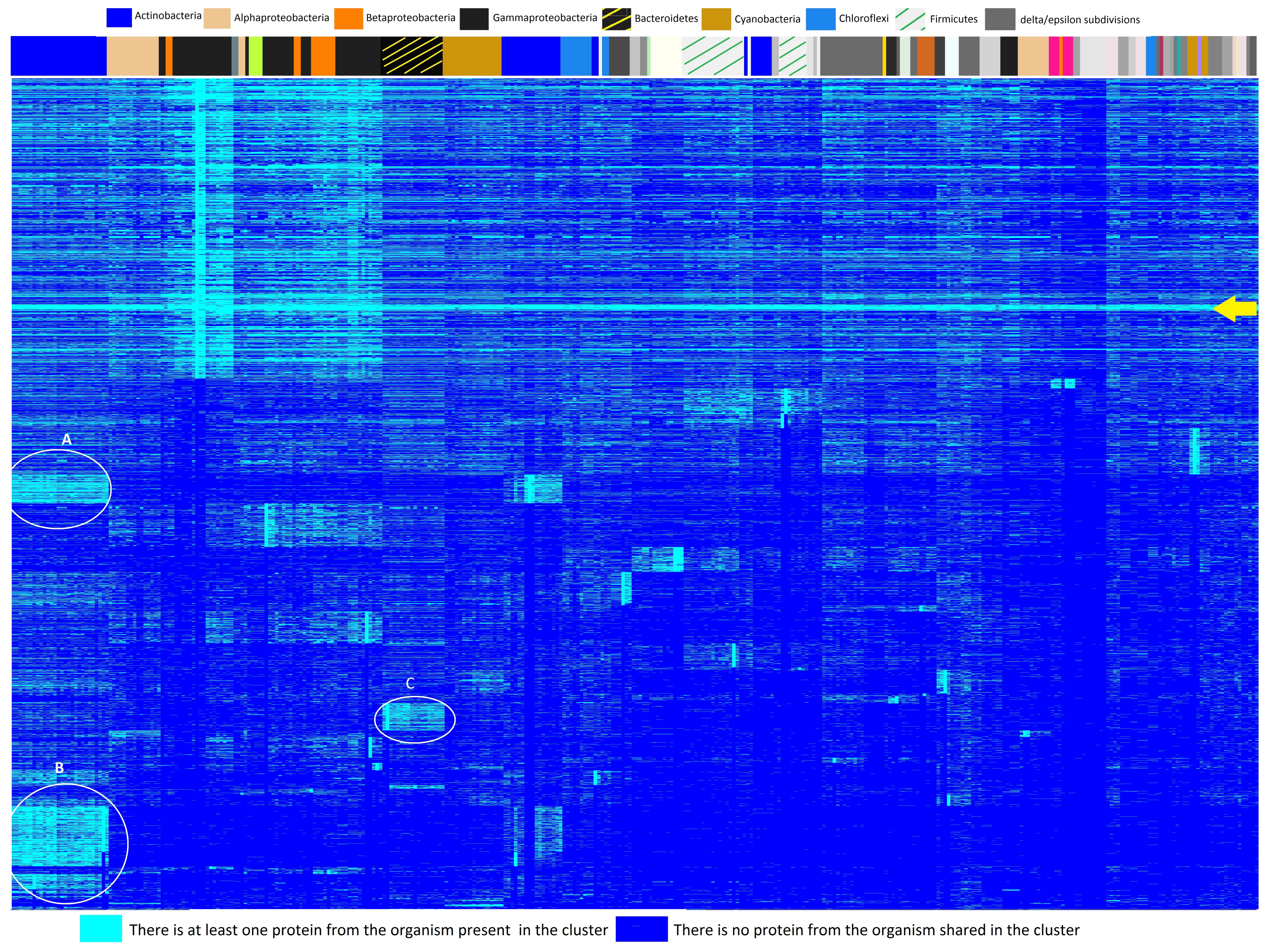
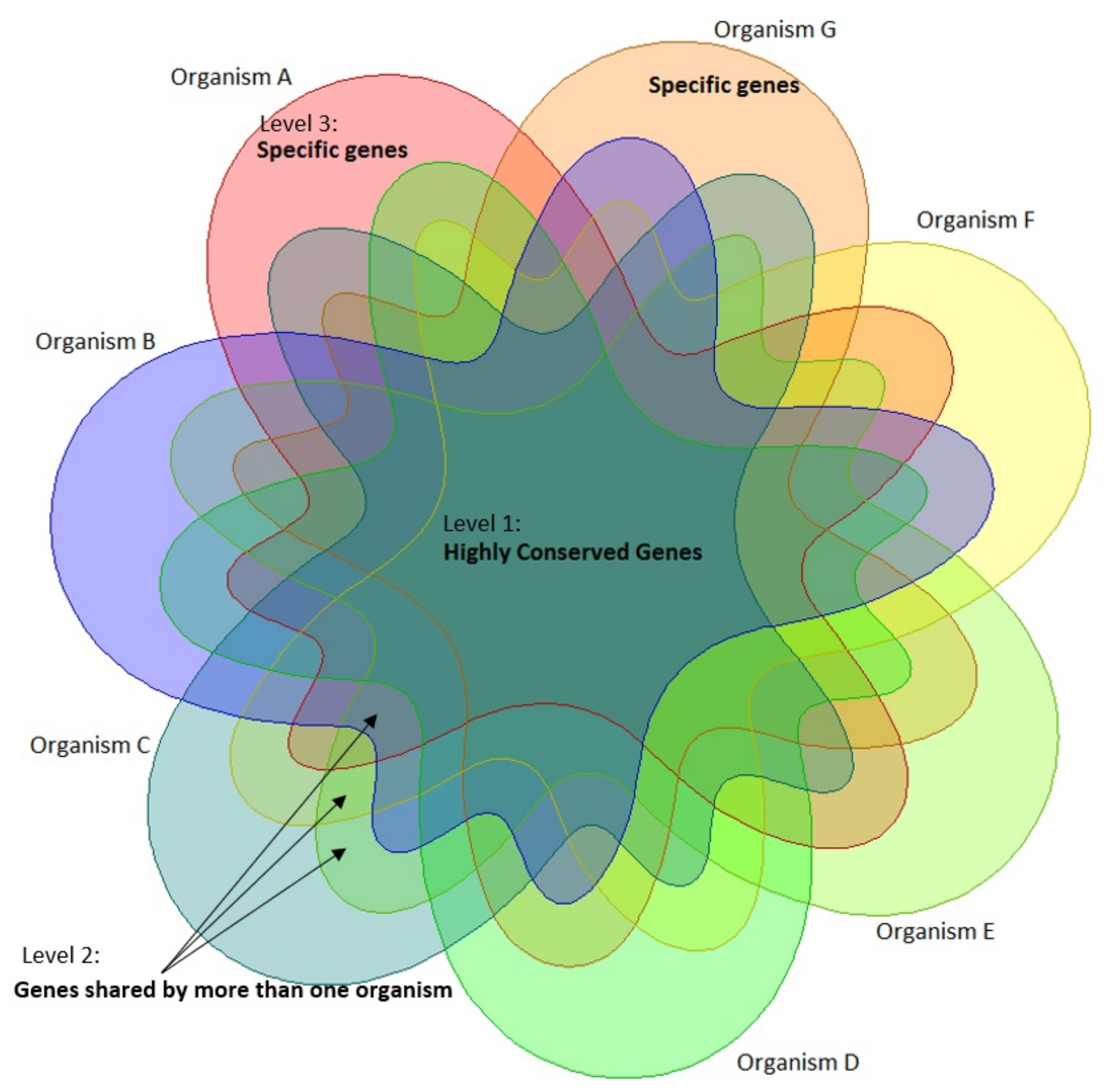
3.2. Heatmap and Essential Genes
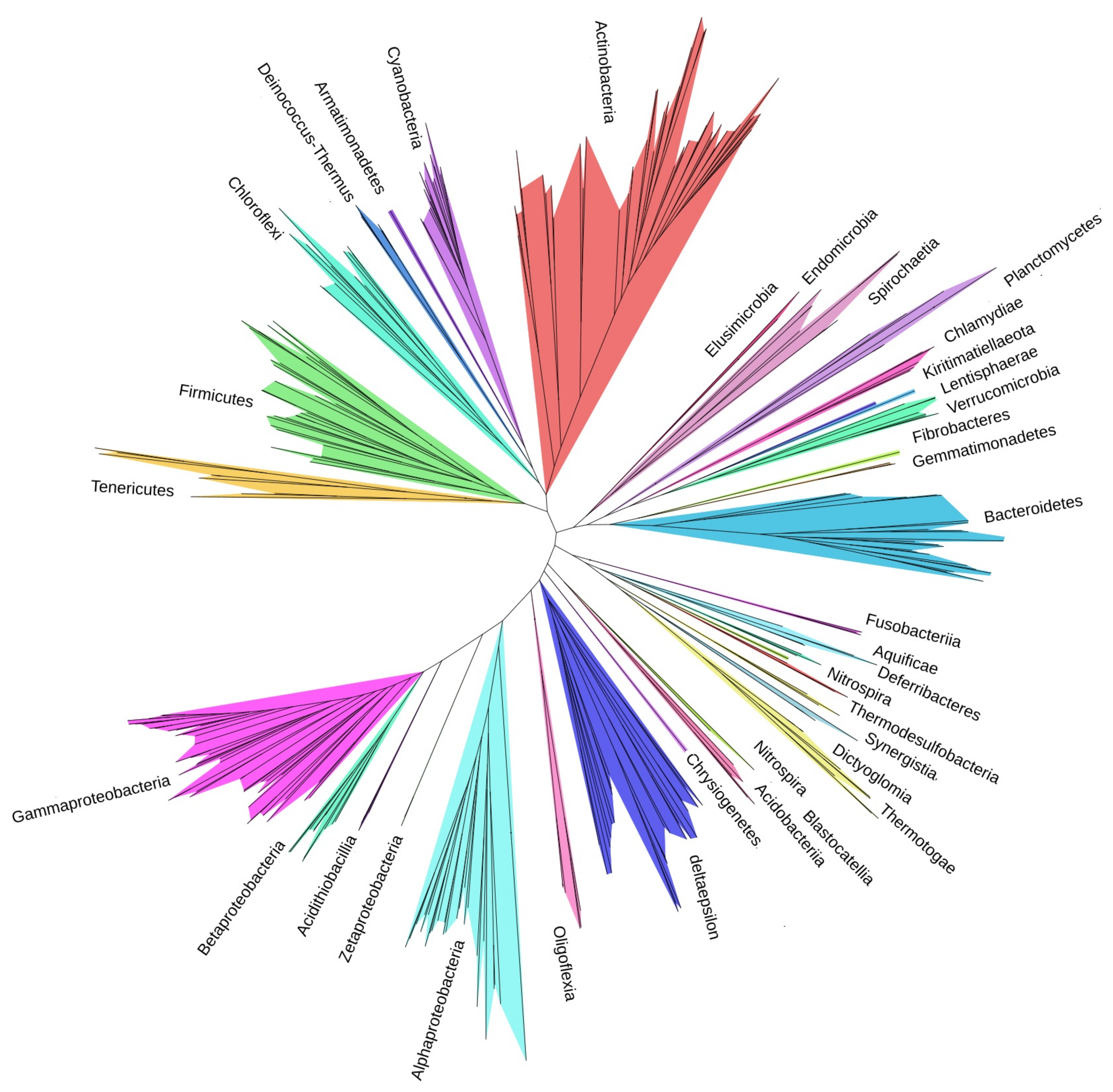
3.3. Tree of Phyla
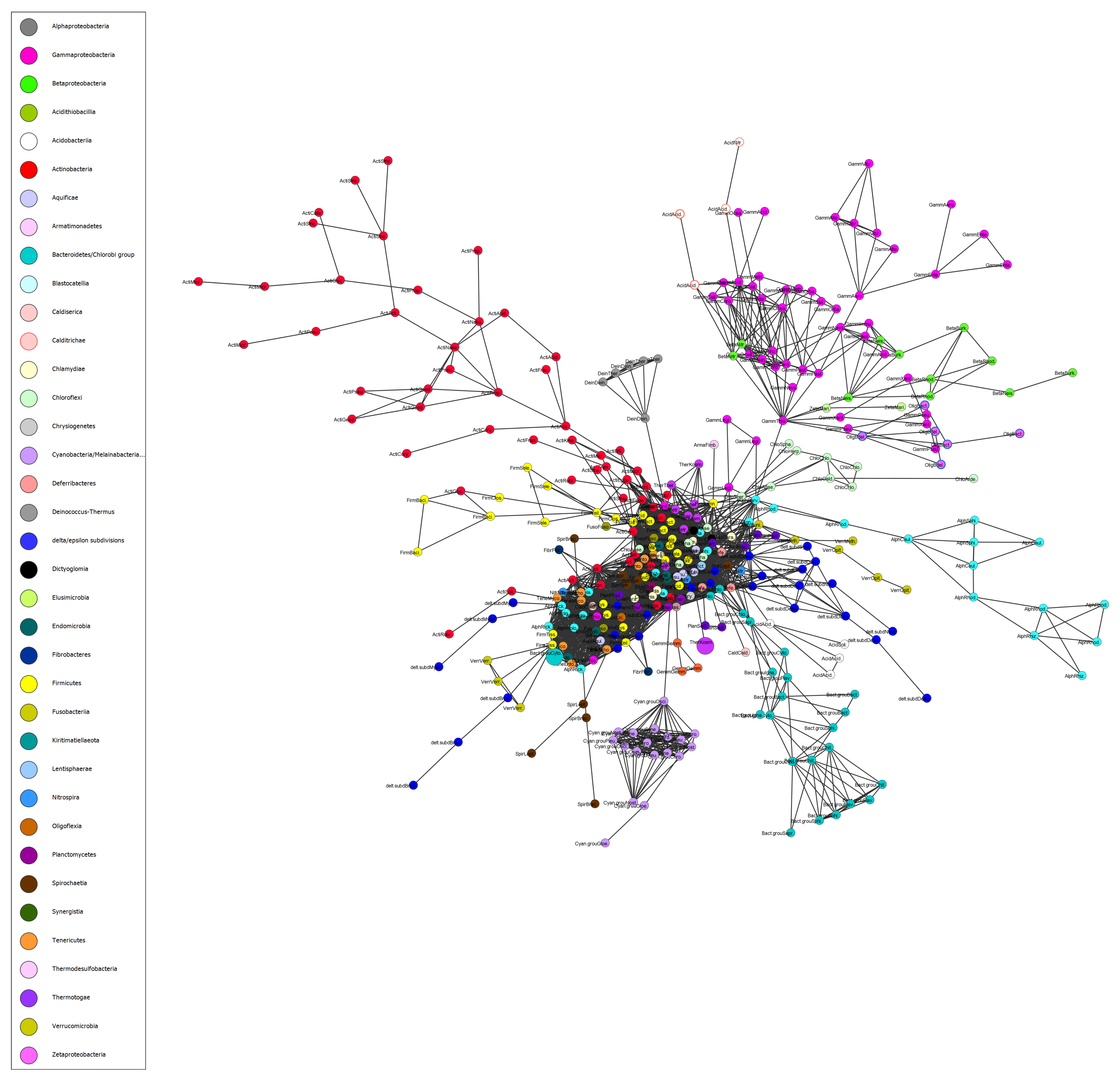
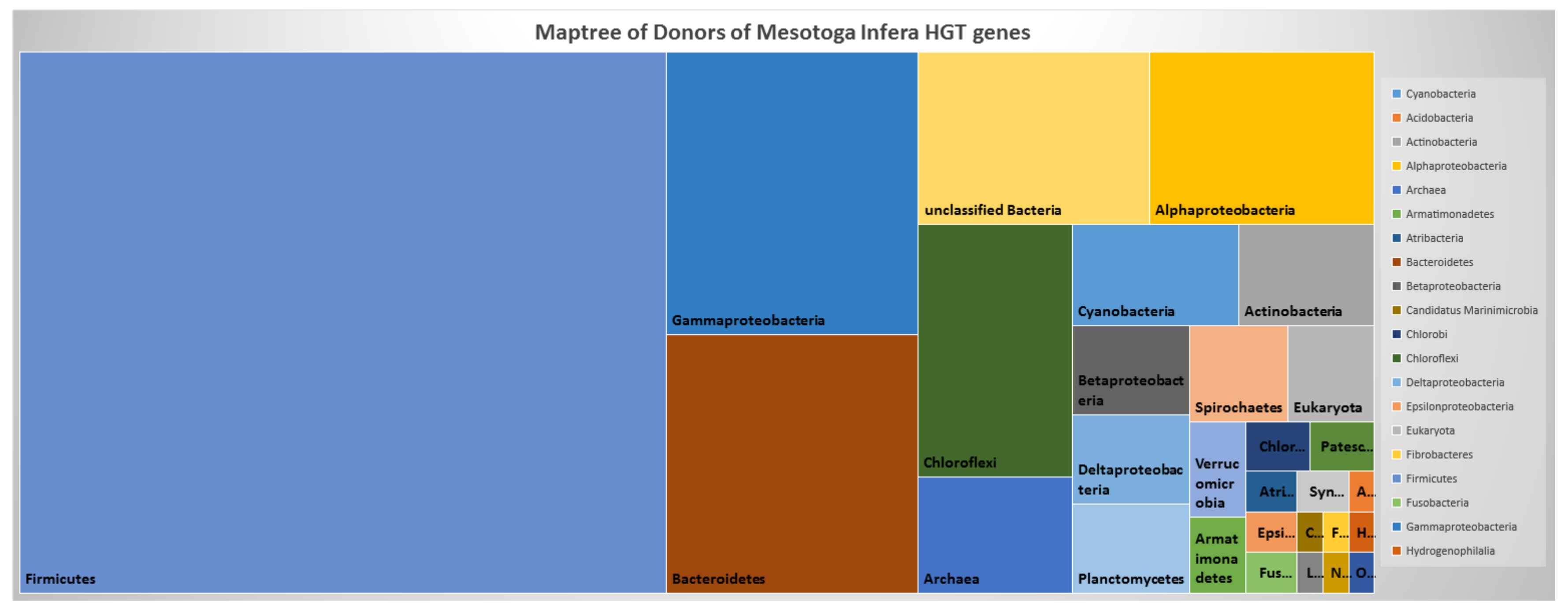
3.4. Network of Organisms and Horizontal Gene Transfer
4. Discussion
4.1. Heatmap and Essential Genes
4.2. Tree of Phyla
4.3. Network of Organisms and Horizontal Gene Transfer
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| HGT | Horizontal Gene Transfer |
References
- Lockwood, S.; Brayton, K.A.; Broschat, S.L. Comparative genomics reveals multiple pathways to mutualism for tick-borne pathogens. BMC Genom. 2016, 17, 481. [Google Scholar] [CrossRef] [Green Version]
- Easley, D.; Kleinberg, J. Networks, Crowds, and Markets; Cambridge University Press: Cambridge, UK, 2010; Volume 8. [Google Scholar]
- Hiramatsu, K.; Cui, L.; Kuroda, M.; Ito, T. The emergence and evolution of methicillin-resistant Staphylococcus aureus. Trends Microbiol. 2001, 9, 486–493. [Google Scholar] [CrossRef]
- Merhej, V.; Notredame, C.; Royer-Carenzi, M.; Pontarotti, P.; Raoult, D. The rhizome of life: The sympatric Rickettsia felis paradigm demonstrates the random transfer of DNA sequences. Mol. Biol. Evol. 2011, 28, 3213–3223. [Google Scholar] [CrossRef]
- Parte, A. LPSN–list of prokaryotic names with standing in nomenclature. Nucleic Acids Res. 2014, 42, D613–D616. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.H.; Apweiler, R.; Bairoch, A.; Natale, D.A.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; et al. The Universal Protein Resource (UniProt): An expanding universe of protein information. Nucleic Acids Res. 2006, 34, D187–D191. [Google Scholar] [CrossRef]
- NCBI Resource Coordinators. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2017, 45, D12–D17. [Google Scholar] [CrossRef] [Green Version]
- Hug, L.A.; Baker, B.J.; Anantharaman, K.; Brown, C.T.; Probst, A.J.; Castelle, C.J.; Butterfield, C.N.; Hernsdorf, A.W.; Amano, Y.; Ise, K.; et al. A new view of the tree of life. Nat. Microbiol. 2016, 1, 16048. [Google Scholar] [CrossRef] [Green Version]
- Schulz, F.; Eloe-Fadrosh, E.A.; Bowers, R.M.; Jarett, J.; Nielsen, T.; Ivanova, N.N.; Kyrpides, N.C.; Woyke, T. Towards a balanced view of the bacterial tree of life. Microbiome 2017, 5, 140. [Google Scholar] [CrossRef]
- Garcia-Vallvé, S.; Romeu, A.; Palau, J. Horizontal gene transfer in bacterial and archaeal complete genomes. Genome Res. 2000, 10, 1719–1725. [Google Scholar] [CrossRef]
- Daily, J. Parasail: SIMD C library for global, semi-global, and local pairwise sequence alignments. BMC Bioinf. 2016, 17, 81. [Google Scholar] [CrossRef]
- Lu, H.; Halappanavar, M.; Kalyanaraman, A. Parallel heuristics for scalable community detection. Parallel Comput. 2015, 47, 19–37. [Google Scholar] [CrossRef] [Green Version]
- Zarayeneh, N.; Kalyanaraman, A. A fast and efficient incremental approach toward dynamic community detection. In Proceedings of the 2019 IEEE/ACM International Conference on Advances in Social Networks Analysis and Mining; ASONAM ’19; ACM: New York, NY, USA, 2019; pp. 9–16. [Google Scholar] [CrossRef] [Green Version]
- Data for Networks of Organisms. Available online: https://bitbucket.org/ehdieh/network-of-organism/downloads/ (accessed on 21 February 2020).
- Csardi, M.G. Package ‘igraph’. Last Accessed 2013, 3, 2013. [Google Scholar]
- Murtagh, F.; Legendre, P. Ward’s hierarchical agglomerative clustering method: Which algorithms implement Ward’s criterion? J. Classif. 2014, 31, 274–295. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Briefings Bioinf. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Ogden, T.H.; Rosenberg, M.S. Multiple sequence alignment accuracy and phylogenetic inference. Syst. Biol. 2006, 55, 314–328. [Google Scholar] [CrossRef] [Green Version]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 1–7. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2014, 32, 268–274. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef]
- Madhulatha, T.S. An overview on clustering methods. arXiv 2012, arXiv:1205.1117. [Google Scholar] [CrossRef]
- Khaledian, E.; Gebremedhin, A.H.; Brayton, K.A.; Broschat, S.L. A Network Science Approach for Determining the Ancestral Phylum of Bacteria. In Proceedings of the 2018 ACM International Conference on Bioinformatics, Computational Biology and Health Informatics; ACM: New York, NY, USA, 2018; pp. 398–403. [Google Scholar]
- Csardi, G.; Nepusz, T. The igraph software package for complex network research. Int. J. Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Baur, M.; Benkert, M.; Brandes, U.; Cornelsen, S.; Gaertler, M.; Köpf, B.; Lerner, J.; Wagner, D. Visone Software for visual social network analysis. In International Symposium on Graph Drawing; Springer: Berlin, Germany, 2001; pp. 463–464. [Google Scholar]
- Nocaj, A.; Ortmann, M.; Brandes, U. Untangling the hairballs of multi-centered, small-world online social media networks. J. Graph Alg. Appl. JGAA 2015, 19, 595–618. [Google Scholar] [CrossRef] [Green Version]
- Charif, D.; Lobry, J.R. SeqinR 1.0-2: A contributed package to the R project for statistical computing devoted to biological sequences retrieval and analysis. In Structural Approaches to Sequence Evolution; Springer: Berlin, Germany, 2007; pp. 207–232. [Google Scholar]
- Boratyn, G.M.; Camacho, C.; Cooper, P.S.; Coulouris, G.; Fong, A.; Ma, N.; Madden, T.L.; Matten, W.T.; McGinnis, S.D.; Merezhuk, Y.; et al. BLAST: A more efficient report with usability improvements. Nucleic Acids Res. 2013, 41, W29–W33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Consortium, G.O. Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res. 2016, 45, 331–338. [Google Scholar]
- Hillis, D.M.; Pollock, D.D.; McGuire, J.A.; Zwickl, D.J. Is sparse taxon sampling a problem for phylogenetic inference? Syst. Biol. 2003, 52, 124–126. [Google Scholar] [CrossRef]
- Timmermans, M.J.; Barton, C.; Haran, J.; Ahrens, D.; Culverwell, C.L.; Ollikainen, A.; Dodsworth, S.; Foster, P.G.; Bocak, L.; Vogler, A.P. Family-level sampling of mitochondrial genomes in Coleoptera: Compositional heterogeneity and phylogenetics. Genome Biol. Evol. 2015, 8, 161–175. [Google Scholar] [CrossRef] [Green Version]
- Woese, C.R.; Kandler, O.; Wheelis, M.L. Towards a natural system of organisms: Proposal for the domains Archaea, Bacteria, and Eucarya. Proc. Natl. Acad. Sci. USA 1990, 87, 4576–4579. [Google Scholar] [CrossRef] [Green Version]
- Parks, D.H.; Chuvochina, M.; Waite, D.W.; Rinke, C.; Skarshewski, A.; Chaumeil, P.A.; Hugenholtz, P. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 2018, 36, 996–1004. [Google Scholar] [CrossRef]
- Whitton, B.A.; Potts, M. Introduction to the Cyanobacteria. In Ecology of Cyanobacteria II; Springer: Berlin, Germany, 2012; pp. 1–13. [Google Scholar]
- Fan, X.T.; Li, H.; Chen, Q.L.; Zhang, Y.S.; Ye, J.; Zhu, Y.G.; Su, J.Q. Fate of antibiotic resistant Pseudomonas putida and broad host range plasmid in natural soil microcosms. Front. Microbiol. 2019, 10, 194. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Z.; Fu, Y.; Guo, D.; Wu, Y.; Ajayi, O.E.; Wu, Q. Bacterial endosymbiont Cardinium cSfur genome sequence provides insights for understanding the symbiotic relationship in Sogatella furcifera host. BMC Genom. 2018, 19, 688. [Google Scholar] [CrossRef]
- Ravenhall, M.; Škunca, N.; Lassalle, F.; Dessimoz, C. Inferring horizontal gene transfer. PLoS Comput. Biol. 2015, 11, e1004095. [Google Scholar] [CrossRef] [Green Version]
- Dereeper, A.; Audic, S.; Claverie, J.M.; Blanc, G. BLAST-EXPLORER helps you building datasets for phylogenetic analysis. BMC Evol. Biol. 2010, 10, 8. [Google Scholar] [CrossRef] [Green Version]
- Huber, R.; Hannig, M. Thermotogales. In The Prokaryotes: Volume 7: Proteobacteria: Delta, Epsilon Subclass; Springer: New York, NY, USA, 2006; pp. 899–922. [Google Scholar]
- Yamaguchi, K.; Subramanian, A.R. The plastid ribosomal proteins identification of all the proteins in the 50S subunit of an organelle ribosome (chloroplast). J. Biol. Chem. 2000, 275, 28466–28482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bieri, P.; Leibundgut, M.; Saurer, M.; Boehringer, D.; Ban, N. The complete structure of the chloroplast 70S ribosome in complex with translation factor pY. EMBO J. 2017, 36, 475–486. [Google Scholar] [CrossRef] [PubMed]
- Nowotny, V.; Nierhaus, K.H. Assembly of the 30S subunit from Escherichia coli ribosomes occurs via two assembly domains which are initiated by S4 and S7. Biochemistry 1988, 27, 7051–7055. [Google Scholar] [CrossRef]
- Olins, P.O.; Nomura, M. Translational regulation by ribosomal protein S8 in Escherichia coli: Structural homology between rRNA binding site and feedback target on mRNA. Nucleic Acids Res. 1981, 9, 1757–1764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herold, M.; Nierhaus, K. Incorporation of six additional proteins to complete the assembly map of the 50S subunit from Escherichia coli ribosomes. J. Biol. Chem. 1987, 262, 8826–8833. [Google Scholar] [PubMed]
- Shpanchenko, O.V.; Zvereva, M.I.; Dontsova, O.A.; Nierhaus, K.H.; Bogdanov, A.A. 5S rRNA sugar-phosphate backbone protection in complexes with specific ribosomal proteins. FEBS Lett. 1996, 394, 71–75. [Google Scholar] [CrossRef] [Green Version]
- Ouzounis, C.A.; Kunin, V.; Darzentas, N.; Goldovsky, L. A minimal estimate for the gene content of the last universal common ancestor—exobiology from a terrestrial perspective. Res. Microbiol. 2006, 157, 57–68. [Google Scholar] [CrossRef]
- Weiss, M.C.; Sousa, F.L.; Mrnjavac, N.; Neukirchen, S.; Roettger, M.; Nelson-Sathi, S.; Martin, W.F. The physiology and habitat of the last universal common ancestor. Nat. Microbiol. 2016, 1, 16116. [Google Scholar] [CrossRef]
- Mushegian, A.R.; Koonin, E.V. A minimal gene set for cellular life derived by comparison of complete bacterial genomes. Proc. Natl. Acad. Sci. USA 1996, 93, 10268–10273. [Google Scholar] [CrossRef] [Green Version]
- Klasson, L.; Andersson, S.G. Evolution of minimal-gene-sets in host-dependent bacteria. Trends Microbiol. 2004, 12, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Chambers, M.C.; Tabb, D.L. Proteomic parsimony through bipartite graph analysis improves accuracy and transparency. J. Proteome Res. 2007, 6, 3549–3557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkins, C.; Fuerst, J.A. Phylogenetic analysis of evolutionary relationships of the planctomycete division of the domain bacteria based on amino acid sequences of elongation factor Tu. J. Mol. Evol. 2001, 52, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Soucy, S.M.; Huang, J.; Gogarten, J.P. Horizontal gene transfer: Building the web of life. Nat. Rev. Genet. 2015, 16, 472–482. [Google Scholar] [CrossRef]
- Skippington, E.; Ragan, M.A. Phylogeny rather than ecology or lifestyle biases the construction of Escherichia coli–Shigella genetic exchange communities. Open Biol. 2012, 2, 120112. [Google Scholar] [CrossRef] [PubMed] [Green Version]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khaledian, E.; Brayton, K.A.; Broschat, S.L. A Systematic Approach to Bacterial Phylogeny Using Order Level Sampling and Identification of HGT Using Network Science. Microorganisms 2020, 8, 312. https://doi.org/10.3390/microorganisms8020312
Khaledian E, Brayton KA, Broschat SL. A Systematic Approach to Bacterial Phylogeny Using Order Level Sampling and Identification of HGT Using Network Science. Microorganisms. 2020; 8(2):312. https://doi.org/10.3390/microorganisms8020312
Chicago/Turabian StyleKhaledian, Ehdieh, Kelly A. Brayton, and Shira L. Broschat. 2020. "A Systematic Approach to Bacterial Phylogeny Using Order Level Sampling and Identification of HGT Using Network Science" Microorganisms 8, no. 2: 312. https://doi.org/10.3390/microorganisms8020312
APA StyleKhaledian, E., Brayton, K. A., & Broschat, S. L. (2020). A Systematic Approach to Bacterial Phylogeny Using Order Level Sampling and Identification of HGT Using Network Science. Microorganisms, 8(2), 312. https://doi.org/10.3390/microorganisms8020312