Human Herpesvirus 8 and Host-Cell Interaction: Long-Lasting Physiological Modifications, Inflammation and Related Chronic Diseases



Abstract
:1. Introduction
2. Virology and Molecular Biology of HHV8
2.1. Lytic HHV8 Infection
2.2. Latent HHV8 Infection
3. HHV8 Infection, Physiological Alterations and Pathogenesis
3.1. Endothelial Cells: Glycolysis, Warburg Effect and Oncogenesis
3.2. HHV8 and Cyclooxygenase: Induction and Suppression of Immune Reaction
3.3. Immune Response and Cell Metabolism
3.4. HHV8 Prevalence in Chronic Diseases
3.5. HHV8-Infection and Oxidative Stress
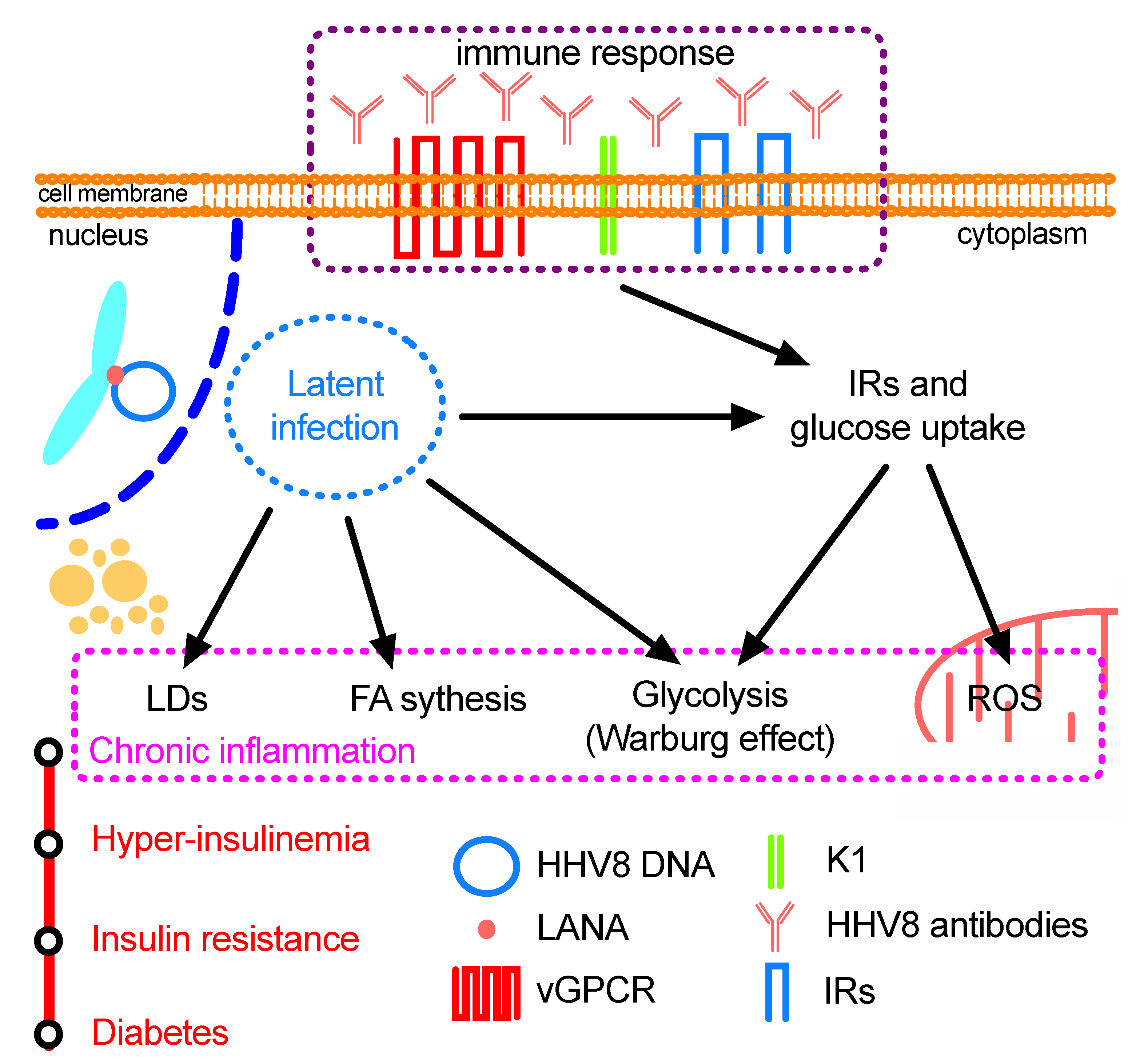
4. Concluding Remarks and Future Scenarios
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chang, Y.; Cesarman, E.; Pessin, M.S.; Lee, F.; Culpepper, J.; Knowles, D.M.; Moore, P.S. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 1994, 266, 1865–1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, K.W.; Damania, B. Kaposi sarcoma-associated herpesvirus (KSHV): Molecular biology and oncogenesis. Cancer Lett. 2010, 289, 140–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cousins, E.; Nicholas, J. Molecular biology of human herpesvirus 8: Novel functions and virus-host interactions implicated in viral pathogenesis and replication. Recent Results Cancer Res. 2014, 193, 227–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vastag, L.; Koyuncu, E.; Grady, S.L.; Shenk, T.E.; Rabinowitz, J.D. Divergent effects of human cytomegalovirus and herpes simplex virus-1 on cellular metabolism. PLoS Pathog. 2011, 7, e1002124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cesarman, E.; Damania, B.; Krown, S.E.; Martin, J.; Bower, M.; Whitby, D. Kaposi sarcoma. Nat. Rev. Dis. Primers 2019, 5, 9. [Google Scholar] [CrossRef]
- Ganem, D. KSHV and the pathogenesis of Kaposi sarcoma: Listening to human biology and medicine. J. Clin. Investig. 2010, 120, 939–949. [Google Scholar] [CrossRef]
- Ojala, P.M.; Schulz, T.F. Manipulation of endothelial cells by KSHV: Implications for angiogenesis and aberrant vascular differentiation. Semin. Cancer Biol. 2014, 26, 69–77. [Google Scholar] [CrossRef]
- Cai, X.; Cullen, B.R. Transcriptional origin of Kaposi’s sarcoma-associated herpesvirus microRNAs. J. Virol. 2006, 80, 2234–2242. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.; Lu, S.; Zhang, Z.; Gonzalez, C.M.; Damania, B.; Cullen, B.R. Kaposi’s sarcoma-associated herpesvirus expresses an array of viral microRNAs in latently infected cells. Proc. Natl. Acad. Sci. USA 2005, 102, 5570–5575. [Google Scholar] [CrossRef] [Green Version]
- Murphy, E.; Vanicek, J.; Robins, H.; Shenk, T.; Levine, A.J. Suppression of immediate-early viral gene expression by herpesvirus-coded microRNAs: Implications for latency. Proc. Natl. Acad. Sci. USA 2008, 105, 5453–5458. [Google Scholar] [CrossRef] [Green Version]
- Pfeffer, S.; Sewer, A.; Lagos-Quintana, M.; Sheridan, R.; Sander, C.; Grasser, F.A.; van Dyk, L.F.; Ho, C.K.; Shuman, S.; Chien, M.; et al. Identification of microRNAs of the herpesvirus family. Nat. Methods 2005, 2, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Samols, M.A.; Hu, J.; Skalsky, R.L.; Renne, R. Cloning and identification of a microRNA cluster within the latency-associated region of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2005, 79, 9301–9305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samols, M.A.; Skalsky, R.L.; Maldonado, A.M.; Riva, A.; Lopez, M.C.; Baker, H.V.; Renne, R. Identification of cellular genes targeted by KSHV-encoded microRNAs. PLoS Pathog. 2007, 3, e65. [Google Scholar] [CrossRef] [PubMed]
- Renne, R.; Zhong, W.; Herndier, B.; McGrath, M.; Abbey, N.; Kedes, D.; Ganem, D. Lytic growth of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat. Med. 1996, 2, 342–346. [Google Scholar] [CrossRef]
- Gao, S.J.; Deng, J.H.; Zhou, F.C. Productive lytic replication of a recombinant Kaposi’s sarcoma-associated herpesvirus in efficient primary infection of primary human endothelial cells. J. Virol. 2003, 77, 9738–9749. [Google Scholar] [CrossRef] [Green Version]
- Moore, P.S.; Chang, Y. Kaposi’s sarcoma (KS), KS-associated herpesvirus, and the criteria for causality in the age of molecular biology. Am. J. Epidemiol. 1998, 147, 217–221. [Google Scholar] [CrossRef]
- Delgado, T.; Carroll, P.A.; Punjabi, A.S.; Margineantu, D.; Hockenbery, D.M.; Lagunoff, M. Induction of the Warburg effect by Kaposi’s sarcoma herpesvirus is required for the maintenance of latently infected endothelial cells. Proc. Natl. Acad. Sci. USA 2010, 107, 10696–10701. [Google Scholar] [CrossRef] [Green Version]
- Rose, P.P.; Bogyo, M.; Moses, A.V.; Fruh, K. Insulin-like growth factor II receptor-mediated intracellular retention of cathepsin B is essential for transformation of endothelial cells by Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2007, 81, 8050–8062. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Damania, B. Kaposi’s sarcoma-associated herpesvirus confers a survival advantage to endothelial cells. Cancer Res. 2008, 68, 4640–4648. [Google Scholar] [CrossRef] [Green Version]
- Ablashi, D.V.; Chatlynne, L.G.; Whitman, J.E., Jr.; Cesarman, E. Spectrum of Kaposi’s sarcoma-associated herpesvirus, or human herpesvirus 8, diseases. Clin. Microbiol. Rev. 2002, 15, 439–464. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Thompson, S.; Schultz, D.C.; Zhu, W.; Jiang, H.; Luo, C.; Lieberman, P.M. Discovery of selective inhibitors against EBNA1 via high throughput in silico virtual screening. PLoS ONE 2010, 5, e10126. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.G.; Sharma-Walia, N.; Chandran, B. Targeting KSHV/HHV-8 latency with COX-2 selective inhibitor nimesulide: A potential chemotherapeutic modality for primary effusion lymphoma. PLoS ONE 2011, 6, e24379. [Google Scholar] [CrossRef]
- Angius, F.; Piras, E.; Uda, S.; Madeddu, C.; Serpe, R.; Bigi, R.; Chen, W.; Dittmer, D.P.; Pompei, R.; Ingianni, A. Antimicrobial sulfonamides clear latent Kaposi sarcoma herpesvirus infection and impair MDM2-p53 complex formation. J. Antibiot. 2017, 70, 962–966. [Google Scholar] [CrossRef] [PubMed]
- Angius, F.; Piras, E.; Spolitu, S.; Marras, L.; Armas, S.F.; Ingianni, A.; Contini, P.; Pompei, R. Anti-human herpesvirus 8 antibodies affect both insulin and glucose uptake by virus-infected human endothelial cells. J. Infect. Dev. Ctries. 2018, 12, 485–491. [Google Scholar] [CrossRef] [Green Version]
- Delgado, T.; Sanchez, E.L.; Camarda, R.; Lagunoff, M. Global metabolic profiling of infection by an oncogenic virus: KSHV induces and requires lipogenesis for survival of latent infection. PLoS Pathog. 2012, 8, e1002866. [Google Scholar] [CrossRef] [Green Version]
- Fontaine, K.A.; Sanchez, E.L.; Camarda, R.; Lagunoff, M. Dengue virus induces and requires glycolysis for optimal replication. J. Virol. 2015, 89, 2358–2366. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, E.L.; Lagunoff, M. Viral activation of cellular metabolism. Virology 2015, 479–480, 609–618. [Google Scholar] [CrossRef] [Green Version]
- Thai, M.; Graham, N.A.; Braas, D.; Nehil, M.; Komisopoulou, E.; Kurdistani, S.K.; McCormick, F.; Graeber, T.G.; Christofk, H.R. Adenovirus E4ORF1-induced MYC activation promotes host cell anabolic glucose metabolism and virus replication. Cell Metab. 2014, 19, 694–701. [Google Scholar] [CrossRef] [Green Version]
- Bilz, N.C.; Jahn, K.; Lorenz, M.; Ludtke, A.; Hubschen, J.M.; Geyer, H.; Mankertz, A.; Hubner, D.; Liebert, U.G.; Claus, C. Rubella viruses shift cellular bioenergetics to a more oxidative and glycolytic phenotype with a strain-specific requirement for glutamine. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Burysek, L.; Yeow, W.S.; Lubyova, B.; Kellum, M.; Schafer, S.L.; Huang, Y.Q.; Pitha, P.M. Functional analysis of human herpesvirus 8-encoded viral interferon regulatory factor 1 and its association with cellular interferon regulatory factors and p300. J. Virol. 1999, 73, 7334–7342. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.J.; Boshoff, C.; Jayachandra, S.; Weiss, R.A.; Chang, Y.; Moore, P.S. KSHV ORF K9 (vIRF) is an oncogene which inhibits the interferon signaling pathway. Oncogene 1997, 15, 1979–1985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Lee, H.; Guo, J.; Neipel, F.; Fleckenstein, B.; Ozato, K.; Jung, J.U. Kaposi’s sarcoma-associated herpesvirus viral interferon regulatory factor. J. Virol. 1998, 72, 5433–5440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pozharskaya, V.P.; Weakland, L.L.; Zimring, J.C.; Krug, L.T.; Unger, E.R.; Neisch, A.; Joshi, H.; Inoue, N.; Offermann, M.K. Short duration of elevated vIRF-1 expression during lytic replication of human herpesvirus 8 limits its ability to block antiviral responses induced by alpha interferon in BCBL-1 cells. J. Virol. 2004, 78, 6621–6635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, J.J.; Bohenzky, R.A.; Chien, M.C.; Chen, J.; Yan, M.; Maddalena, D.; Parry, J.P.; Peruzzi, D.; Edelman, I.S.; Chang, Y.; et al. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl. Acad. Sci. USA 1996, 93, 14862–14867. [Google Scholar] [CrossRef] [Green Version]
- Seo, T.; Park, J.; Lee, D.; Hwang, S.G.; Choe, J. Viral interferon regulatory factor 1 of Kaposi’s sarcoma-associated herpesvirus binds to p53 and represses p53-dependent transcription and apoptosis. J. Virol. 2001, 75, 6193–6198. [Google Scholar] [CrossRef] [Green Version]
- Arvanitakis, L.; Geras-Raaka, E.; Varma, A.; Gershengorn, M.C.; Cesarman, E. Human herpesvirus KSHV encodes a constitutively active G-protein-coupled receptor linked to cell proliferation. Nature 1997, 385, 347–350. [Google Scholar] [CrossRef]
- Bais, C.; van Geelen, A.; Eroles, P.; Mutlu, A.; Chiozzini, C.; Dias, S.; Silverstein, R.L.; Rafii, S.; Mesri, E.A. Kaposi’s sarcoma associated herpesvirus G protein-coupled receptor immortalizes human endothelial cells by activation of the VEGF receptor-2/KDR. Cancer Cell 2003, 3, 131–143. [Google Scholar] [CrossRef] [Green Version]
- Cesarman, E.; Nador, R.G.; Bai, F.; Bohenzky, R.A.; Russo, J.J.; Moore, P.S.; Chang, Y.; Knowles, D.M. Kaposi’s sarcoma-associated herpesvirus contains G protein-coupled receptor and cyclin D homologs which are expressed in Kaposi’s sarcoma and malignant lymphoma. J. Virol. 1996, 70, 8218–8223. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.G.; Browning, P.; Nicholas, J.; Hayward, G.S.; Tschachler, E.; Jiang, Y.W.; Sadowska, M.; Raffeld, M.; Colombini, S.; Gallo, R.C.; et al. Characterization of a chemokine receptor-related gene in human herpesvirus 8 and its expression in Kaposi’s sarcoma. Virology 1997, 228, 371–378. [Google Scholar] [CrossRef] [Green Version]
- Montaner, S.; Sodhi, A.; Pece, S.; Mesri, E.A.; Gutkind, J.S. The Kaposi’s sarcoma-associated herpesvirus G protein-coupled receptor promotes endothelial cell survival through the activation of Akt/protein kinase B. Cancer Res. 2001, 61, 2641–2648. [Google Scholar]
- Hussein, H.A.M.; Alfhili, M.A.; Pakala, P.; Simon, S.; Hussain, J.; McCubrey, J.A.; Akula, S.M. miRNAs and their roles in KSHV pathogenesis. Virus Res. 2019, 266, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Dourmishev, L.A.; Dourmishev, A.L.; Palmeri, D.; Schwartz, R.A.; Lukac, D.M. Molecular genetics of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus-8) epidemiology and pathogenesis. Microbiol. Mol. Biol. Rev. 2003, 67, 175–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friborg, J., Jr.; Kong, W.; Hottiger, M.O.; Nabel, G.J. p53 inhibition by the LANA protein of KSHV protects against cell death. Nature 1999, 402, 889–894. [Google Scholar] [CrossRef] [PubMed]
- Djerbi, M.; Screpanti, V.; Catrina, A.I.; Bogen, B.; Biberfeld, P.; Grandien, A. The inhibitor of death receptor signaling, FLICE-inhibitory protein defines a new class of tumor progression factors. J. Exp. Med. 1999, 190, 1025–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Efklidou, S.; Bailey, R.; Field, N.; Noursadeghi, M.; Collins, M.K. vFLIP from KSHV inhibits anoikis of primary endothelial cells. J. Cell Sci. 2008, 121, 450–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fakhari, F.D.; Dittmer, D.P. Charting latency transcripts in Kaposi’s sarcoma-associated herpesvirus by whole-genome real-time quantitative PCR. J. Virol. 2002, 76, 6213–6223. [Google Scholar] [CrossRef] [Green Version]
- Sadler, R.; Wu, L.; Forghani, B.; Renne, R.; Zhong, W.; Herndier, B.; Ganem, D. A complex translational program generates multiple novel proteins from the latently expressed kaposin (K12) locus of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 1999, 73, 5722–5730. [Google Scholar] [CrossRef] [Green Version]
- Lagunoff, M.; Ganem, D. The structure and coding organization of the genomic termini of Kaposi’s sarcoma-associated herpesvirus. Virology 1997, 236, 147–154. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Guo, J.; Li, M.; Choi, J.K.; DeMaria, M.; Rosenzweig, M.; Jung, J.U. Identification of an immunoreceptor tyrosine-based activation motif of K1 transforming protein of Kaposi’s sarcoma-associated herpesvirus. Mol. Cell Biol. 1998, 18, 5219–5228. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Dittmer, D.P.; Tomlinson, C.C.; Fakhari, F.D.; Damania, B. Immortalization of primary endothelial cells by the K1 protein of Kaposi’s sarcoma-associated herpesvirus. Cancer Res. 2006, 66, 3658–3666. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wakisaka, N.; Tomlinson, C.C.; DeWire, S.M.; Krall, S.; Pagano, J.S.; Damania, B. The Kaposi’s sarcoma-associated herpesvirus (KSHV/HHV-8) K1 protein induces expression of angiogenic and invasion factors. Cancer Res. 2004, 64, 2774–2781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoki, Y.; Jaffe, E.S.; Chang, Y.; Jones, K.; Teruya-Feldstein, J.; Moore, P.S.; Tosato, G. Angiogenesis and hematopoiesis induced by Kaposi’s sarcoma-associated herpesvirus-encoded interleukin-6. Blood 1999, 93, 4034–4043. [Google Scholar] [CrossRef]
- Chen, D.; Nicholas, J. Structural requirements for gp80 independence of human herpesvirus 8 interleukin-6 (vIL-6) and evidence for gp80 stabilization of gp130 signaling complexes induced by vIL-6. J. Virol. 2006, 80, 9811–9821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Sandford, G.; Nicholas, J. Intracellular signaling mechanisms and activities of human herpesvirus 8 interleukin-6. J. Virol. 2009, 83, 722–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, L.; Majerciak, V.; Zheng, Z.M.; Lan, K. Towards better understanding of KSHV life cycle: From transcription and posttranscriptional regulations to pathogenesis. Virol. Sin. 2019, 34, 135–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bock, K.; Georgiadou, M.; Schoors, S.; Kuchnio, A.; Wong, B.W.; Cantelmo, A.R.; Quaegebeur, A.; Ghesquiere, B.; Cauwenberghs, S.; Eelen, G.; et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 2013, 154, 651–663. [Google Scholar] [CrossRef] [Green Version]
- Saka, H.A.; Valdivia, R. Emerging roles for lipid droplets in immunity and host-pathogen interactions. Annu. Rev. Cell Dev. Biol. 2012, 28, 411–437. [Google Scholar] [CrossRef]
- Carroll, P.A.; Kenerson, H.L.; Yeung, R.S.; Lagunoff, M. Latent Kaposi’s sarcoma-associated herpesvirus infection of endothelial cells activates hypoxia-induced factors. J. Virol. 2006, 80, 10802–10812. [Google Scholar] [CrossRef] [Green Version]
- Lagunoff, M. Activation of cellular metabolism during latent Kaposi’s Sarcoma herpesvirus infection. Curr. Opin. Virol. 2016, 19, 45–49. [Google Scholar] [CrossRef] [Green Version]
- Angius, F.; Uda, S.; Piras, E.; Spolitu, S.; Ingianni, A.; Batetta, B.; Pompei, R. Neutral lipid alterations in human herpesvirus 8-infected HUVEC cells and their possible involvement in neo-angiogenesis. BMC Microbiol. 2015, 15, 74. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, E.L.; Pulliam, T.H.; Dimaio, T.A.; Thalhofer, A.B.; Delgado, T.; Lagunoff, M. Glycolysis, glutaminolysis, and fatty acid synthesis are required for distinct stages of Kaposi’s sarcoma-associated herpesvirus lytic replication. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Dong, Y.; Atefi, M.; Liu, Y.; Elshimali, Y.; Vadgama, J.V. Lactate, a neglected factor for diabetes and cancer interaction. Mediat. Inflamm. 2016, 2016, 6456018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, P.P.; Carroll, J.M.; Carroll, P.A.; DeFilippis, V.R.; Lagunoff, M.; Moses, A.V.; Roberts, C.T., Jr.; Fruh, K. The insulin receptor is essential for virus-induced tumorigenesis of Kaposi’s sarcoma. Oncogene 2007, 26, 1995–2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guilluy, C.; Zhang, Z.; Bhende, P.M.; Sharek, L.; Wang, L.; Burridge, K.; Damania, B. Latent KSHV infection increases the vascular permeability of human endothelial cells. Blood 2011, 118, 5344–5354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingianni, A.; Piras, E.; Laconi, S.; Angius, F.; Batetta, B.; Pompei, R. Latent herpesvirus 8 infection improves both insulin and glucose uptake in primary endothelial cells. New Microbiol. 2013, 36, 257–265. [Google Scholar] [PubMed]
- Gregory, S.M.; Wang, L.; West, J.A.; Dittmer, D.P.; Damania, B. Latent Kaposi’s sarcoma-associated herpesvirus infection of monocytes downregulates expression of adaptive immune response costimulatory receptors and proinflammatory cytokines. J. Virol. 2012, 86, 3916–3923. [Google Scholar] [CrossRef] [Green Version]
- Caselli, E.; Fiorentini, S.; Amici, C.; Di Luca, D.; Caruso, A.; Santoro, M.G. Human herpesvirus 8 acute infection of endothelial cells induces monocyte chemoattractant protein 1-dependent capillary-like structure formation: Role of the IKK/NF-kappaB pathway. Blood 2007, 109, 2718–2726. [Google Scholar] [CrossRef]
- Spagnuolo, I.; Patti, A.; Sebastiani, G.; Nigi, L.; Dotta, F. The case for virus-induced type 1 diabetes. Curr. Opin. Endocrinol. Diabetes Obes. 2013, 20, 292–298. [Google Scholar] [CrossRef]
- Wan, X.; Wang, H.; Nicholas, J. Human herpesvirus 8 interleukin-6 (vIL-6) signals through gp130 but has structural and receptor-binding properties distinct from those of human IL-6. J. Virol. 1999, 73, 8268–8278. [Google Scholar] [CrossRef] [Green Version]
- Nolan, C.J.; Damm, P.; Prentki, M. Type 2 diabetes across generations: From pathophysiology to prevention and management. Lancet 2011, 378, 169–181. [Google Scholar] [CrossRef]
- Sharma-Walia, N.; Paul, A.G.; Bottero, V.; Sadagopan, S.; Veettil, M.V.; Kerur, N.; Chandran, B. Kaposi’s sarcoma associated herpes virus (KSHV) induced COX-2: A key factor in latency, inflammation, angiogenesis, cell survival and invasion. PLoS Pathog. 2010, 6, e1000777. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.T.; Qian, J.; Ang, J.; Sun, R. Vaccine prospect of Kaposi sarcoma-associated herpesvirus. Curr. Opin. Virol. 2012, 2, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Chen, D.; Xiang, Q.; Nicholas, J. Insulin-like growth factor 2 receptor expression is promoted by human herpesvirus 8-encoded interleukin-6 and contributes to viral latency and productive replication. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Bottero, V.; Chakraborty, S.; Chandran, B. Reactive oxygen species are induced by Kaposi’s sarcoma-associated herpesvirus early during primary infection of endothelial cells to promote virus entry. J. Virol. 2013, 87, 1733–1749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caselli, E.; Rizzo, R.; Ingianni, A.; Contini, P.; Pompei, R.; Di Luca, D. High prevalence of HHV8 infection and specific killer cell immunoglobulin-like receptors allotypes in Sardinian patients with type 2 diabetes mellitus. J. Med. Virol. 2014, 86, 1745–1751. [Google Scholar] [CrossRef] [PubMed]
- Gastaldi, G.; Goossens, N.; Clement, S.; Negro, F. Current level of evidence on causal association between hepatitis C virus and type 2 diabetes: A review. J. Adv. Res. 2017, 8, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Incani, A.; Marras, L.; Serreli, G.; Ingianni, A.; Pompei, R.; Deiana, M.; Angius, F. Human herpesvirus 8 infection may contribute to oxidative stress in diabetes type 2 patients. BMC Res. Notes 2020, 13, 75. [Google Scholar] [CrossRef] [Green Version]
- Pompei, R. The role of human herpesvirus 8 in diabetes mellitus type 2: State of the art and a medical hypothesis. Adv. Exp. Med. Biol. 2016, 901, 37–45. [Google Scholar] [CrossRef]
- Piras, E.; Madeddu, M.A.; Palmieri, G.; Angius, F.; Contini, P.; Pompei, R.; Ingianni, A. High prevalence of human herpesvirus 8 infection in diabetes type 2 patients and detection of a new virus subtype. Adv. Exp. Med. Biol. 2017, 973, 41–51. [Google Scholar] [CrossRef]
- Douglas, J.L.; Gustin, J.K.; Viswanathan, K.; Mansouri, M.; Moses, A.V.; Fruh, K. The great escape: Viral strategies to counter BST-2/tetherin. PLoS Pathog. 2010, 6, e1000913. [Google Scholar] [CrossRef] [Green Version]
- Ingianni, A.; Carta, F.; Reina, A.; Manai, M.; Desogus, A.; Pompei, R. Prevalence of herpesvirus 8 infection in type 2 diabetes mellitus patients. Am. J. Infect. Dis. 2007, 3, 123–127. [Google Scholar] [CrossRef]
- Sobngwi, E.; Choukem, S.P.; Agbalika, F.; Blondeau, B.; Fetita, L.S.; Lebbe, C.; Thiam, D.; Cattan, P.; Larghero, J.; Foufelle, F.; et al. Ketosis-prone type 2 diabetes mellitus and human herpesvirus 8 infection in sub-saharan africans. JAMA 2008, 299, 2770–2776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingianni, A.; Madeddu, M.A.; Carta, F.; Reina, A.; Lai, C.; Pompei, R. Epidemiology of human herpesvirus type 8 infection in cardiopathic patients. OnLine J. Biol. Sci. 2009, 9, 36–39. [Google Scholar] [CrossRef]
- Ye, F.; Zeng, Y.; Sha, J.; Jones, T.; Kuhne, K.; Wood, C.; Gao, S.J. High glucose induces reactivation of latent Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angius, F.; Madeddu, M.; Pompei, R. Commentary: High glucose induces reactivation of latent Kaposi’s sarcoma-associated herpesvirus. Front. Microbiol. 2017, 8, 1796. [Google Scholar] [CrossRef] [PubMed]
- Angius, F.; Marras, L.; Ingianni, A.; Pompei, R. Latent-persistent virus infections. Hepatitis C virus and human herpesvirus 8: Immunological response, modification of cell metabolism and association with type 2 diabetes. In Emerging and Reemerging Viral Pathogens; Academic Press: Cambridge, MA, USA, 2020; Volume 1, pp. 169–181. [Google Scholar]
- Abenavoli, L.; Masarone, M.; Peta, V.; Milic, N.; Kobyliak, N.; Rouabhia, S.; Persico, M. Insulin resistance and liver steatosis in chronic hepatitis C infection genotype. World J. Gastroenterol. 2014, 20, 15233–15240. [Google Scholar] [CrossRef]
- Chen, S.; de Craen, A.J.; Raz, Y.; Derhovanessian, E.; Vossen, A.C.; Westendorp, R.G.; Pawelec, G.; Maier, A.B. Cytomegalovirus seropositivity is associated with glucose regulation in the oldest old. Results from the leiden 85-plus study. Immun. Ageing 2012, 9, 18. [Google Scholar] [CrossRef] [Green Version]
- Lontchi-Yimagou, E.; Legoff, J.; Nguewa, J.L.; Boudou, P.; Balti, E.V.; Noubiap, J.J.; Kamwa, V.; Atogho-Tiedeu, B.; Azabji-Kenfack, M.; Djahmeni, E.N.; et al. Human herpesvirus 8 infection DNA positivity is associated with low insulin secretion: A case-control study in a sub-Saharan African population with diabetes. J. Diabetes 2018, 10, 866–873. [Google Scholar] [CrossRef]
- Falkenberg, K.D.; Rohlenova, K.; Luo, Y.L.; Carmeliet, P. The metabolic engine of endothelial cells. Nat. Metab. 2019, 1, 937–946. [Google Scholar] [CrossRef]
- Gimbrone, M.A., Jr.; Garcia-Cardena, G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [Green Version]
- Tabit, C.E.; Chung, W.B.; Hamburg, N.M.; Vita, J.A. Endothelial dysfunction in diabetes mellitus: Molecular mechanisms and clinical implications. Rev. Endocr. Metab. Disord. 2010, 11, 61–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Incani, A.; Serra, G.; Atzeri, A.; Melis, M.P.; Serreli, G.; Bandino, G.; Sedda, P.; Campus, M.; Tuberoso, C.I.; Deiana, M. Extra virgin olive oil phenolic extracts counteract the pro-oxidant effect of dietary oxidized lipids in human intestinal cells. Food Chem. Toxicol. 2016, 90, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Corkey, B.E. Banting lecture 2011: Hyperinsulinemia: Cause or consequence? Diabetes 2012, 61, 4–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erion, K.A.; Corkey, B.E. Hyperinsulinemia: A cause of obesity? Curr. Obes. Rep. 2017, 6, 178–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidali, M.; Tripodi, M.F.; Ivaldi, A.; Zampino, R.; Occhino, G.; Restivo, L.; Sutti, S.; Marrone, A.; Ruggiero, G.; Albano, E.; et al. Interplay between oxidative stress and hepatic steatosis in the progression of chronic hepatitis C. J. Hepatol. 2008, 48, 399–406. [Google Scholar] [CrossRef]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vasc. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef]
- Lo, A.K.; Dawson, C.W.; Young, L.S.; Lo, K.W. The role of metabolic reprogramming in gamma-herpesvirus-associated oncogenesis. Int. J. Cancer 2017, 141, 1512–1521. [Google Scholar] [CrossRef] [Green Version]
- Zecchin, A.; Kalucka, J.; Dubois, C.; Carmeliet, P. How endothelial cells adapt their metabolism to form vessels in tumors. Front. Immunol. 2017, 8, 1750. [Google Scholar] [CrossRef] [Green Version]
- Robson, R.; Kundur, A.R.; Singh, I. Oxidative stress biomarkers in type 2 diabetes mellitus for assessment of cardiovascular disease risk. Diabetes Metab. Syndr. 2018, 12, 455–462. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Feng, J.; Sun, R. Oxidative stress induces reactivation of Kaposi’s sarcoma-associated herpesvirus and death of primary effusion lymphoma cells. J. Virol. 2011, 85, 715–724. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q.; Cavallin, L.E.; Leung, H.J.; Chiozzini, C.; Goldschmidt-Clermont, P.J.; Mesri, E.A. A role for virally induced reactive oxygen species in Kaposi’s sarcoma herpesvirus tumorigenesis. Antioxid. Redox Signal 2013, 18, 80–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poli, G.; Sottero, B.; Gargiulo, S.; Leonarduzzi, G. Cholesterol oxidation products in the vascular remodeling due to atherosclerosis. Mol. Asp. Med. 2009, 30, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.D.; Corkey, B.E.; Istfan, N.W.; Apovian, C.M. Hyperinsulinemia: An early indicator of Metabolic dysfunction. J. Endocr. Soc. 2019, 3, 1727–1747. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Metabolites | HHV8 Lytic Infection | HHV8 Latent Infection |
|---|---|---|
| * Cholesterol esters | − | +++ |
| ** Fatty acids | ++ | − |
| ** Spermidine | − | ++ |
| ** 7-beta-hydroxycholesterol | − | + |
| ** Mannose-6-phosphate | − | ++ |
| * Glucose uptake | − | +++ |
| ** phospho-enol-pyruvate | − | +++ |
| ** 6-phosphogluconate | − | + |
| * Triglycerides | +++ | − |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angius, F.; Ingianni, A.; Pompei, R. Human Herpesvirus 8 and Host-Cell Interaction: Long-Lasting Physiological Modifications, Inflammation and Related Chronic Diseases. Microorganisms 2020, 8, 388. https://doi.org/10.3390/microorganisms8030388
Angius F, Ingianni A, Pompei R. Human Herpesvirus 8 and Host-Cell Interaction: Long-Lasting Physiological Modifications, Inflammation and Related Chronic Diseases. Microorganisms. 2020; 8(3):388. https://doi.org/10.3390/microorganisms8030388
Chicago/Turabian StyleAngius, Fabrizio, Angela Ingianni, and Raffaello Pompei. 2020. "Human Herpesvirus 8 and Host-Cell Interaction: Long-Lasting Physiological Modifications, Inflammation and Related Chronic Diseases" Microorganisms 8, no. 3: 388. https://doi.org/10.3390/microorganisms8030388
APA StyleAngius, F., Ingianni, A., & Pompei, R. (2020). Human Herpesvirus 8 and Host-Cell Interaction: Long-Lasting Physiological Modifications, Inflammation and Related Chronic Diseases. Microorganisms, 8(3), 388. https://doi.org/10.3390/microorganisms8030388