Effects of DNA Topology on Transcription from rRNA Promoters in Bacillus subtilis


Abstract
:1. Introduction
2. Materials and Methods
2.1. Media and Growth Conditions
2.2. Bacterial Strains
2.3. Determination of ATP, GTP, and ppGppconcentrations
2.4. Promoter Activity Monitored by Quantitative Primer Extension (qPE)
2.5. Promoter Activity Monitored by RT–qPCR
2.6. 3H-Incorporation in Total RNA
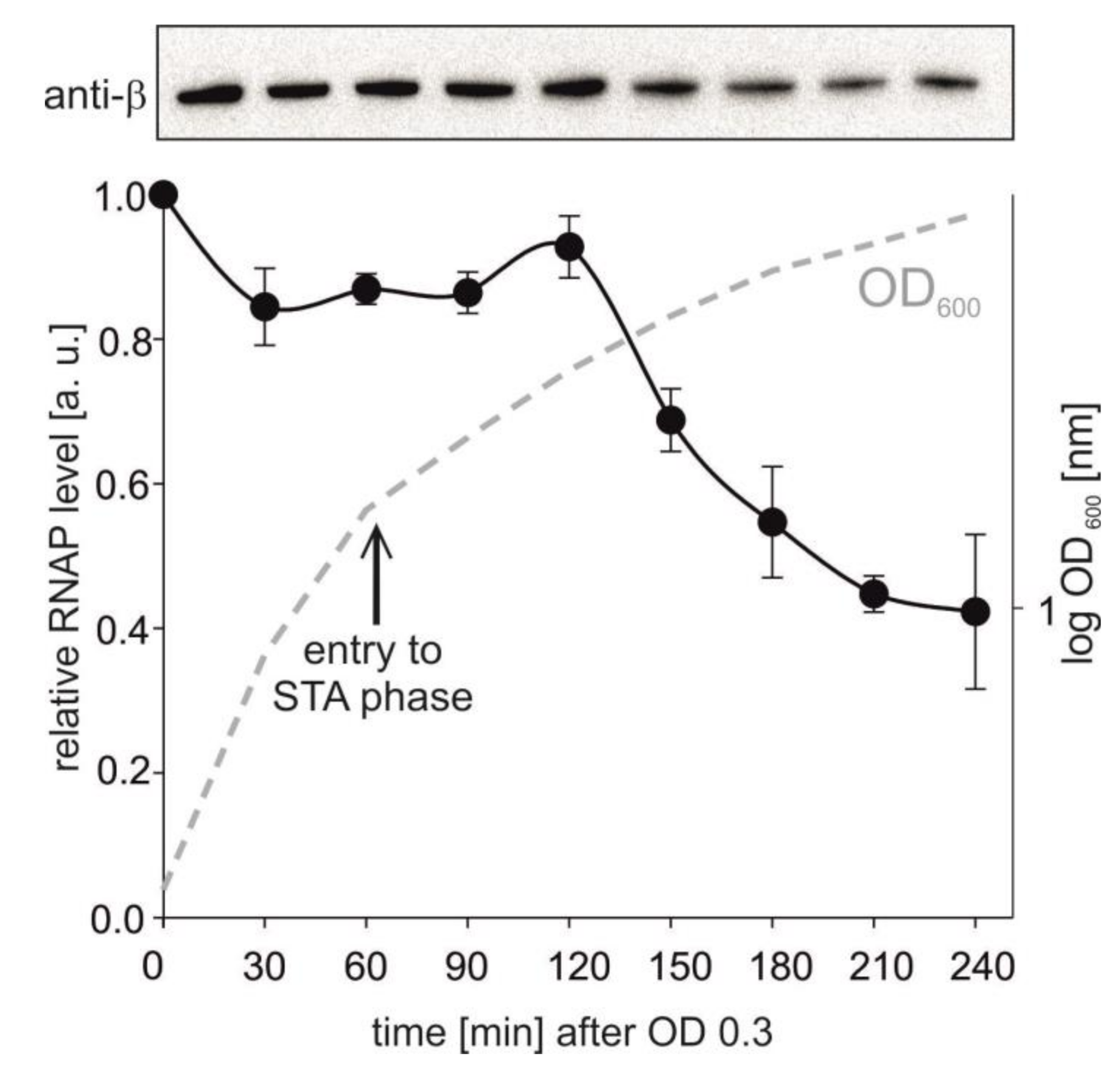
2.7. RNAP Levels in Time
2.8. Proteins and DNA for Transcription In Vitro
2.8.1. Strain Construction
2.8.2. Protein Purification
2.8.3. Promoter DNA Construction
2.8.4. List of Primers
2.9. Transcription In Vitro
3. Results
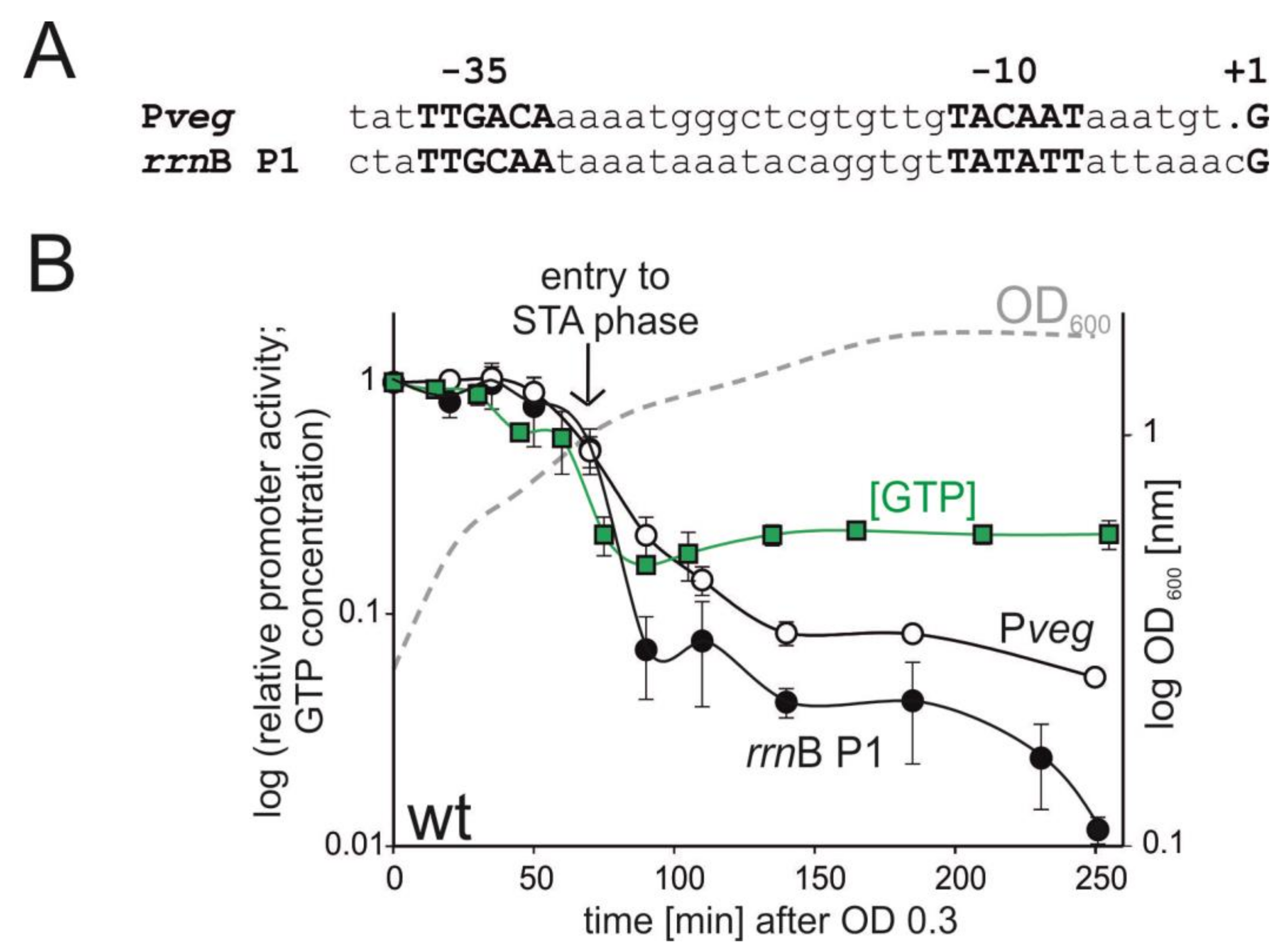
3.1. The Activity of rrnB P1 Decreases during Entry into Stationary Phase
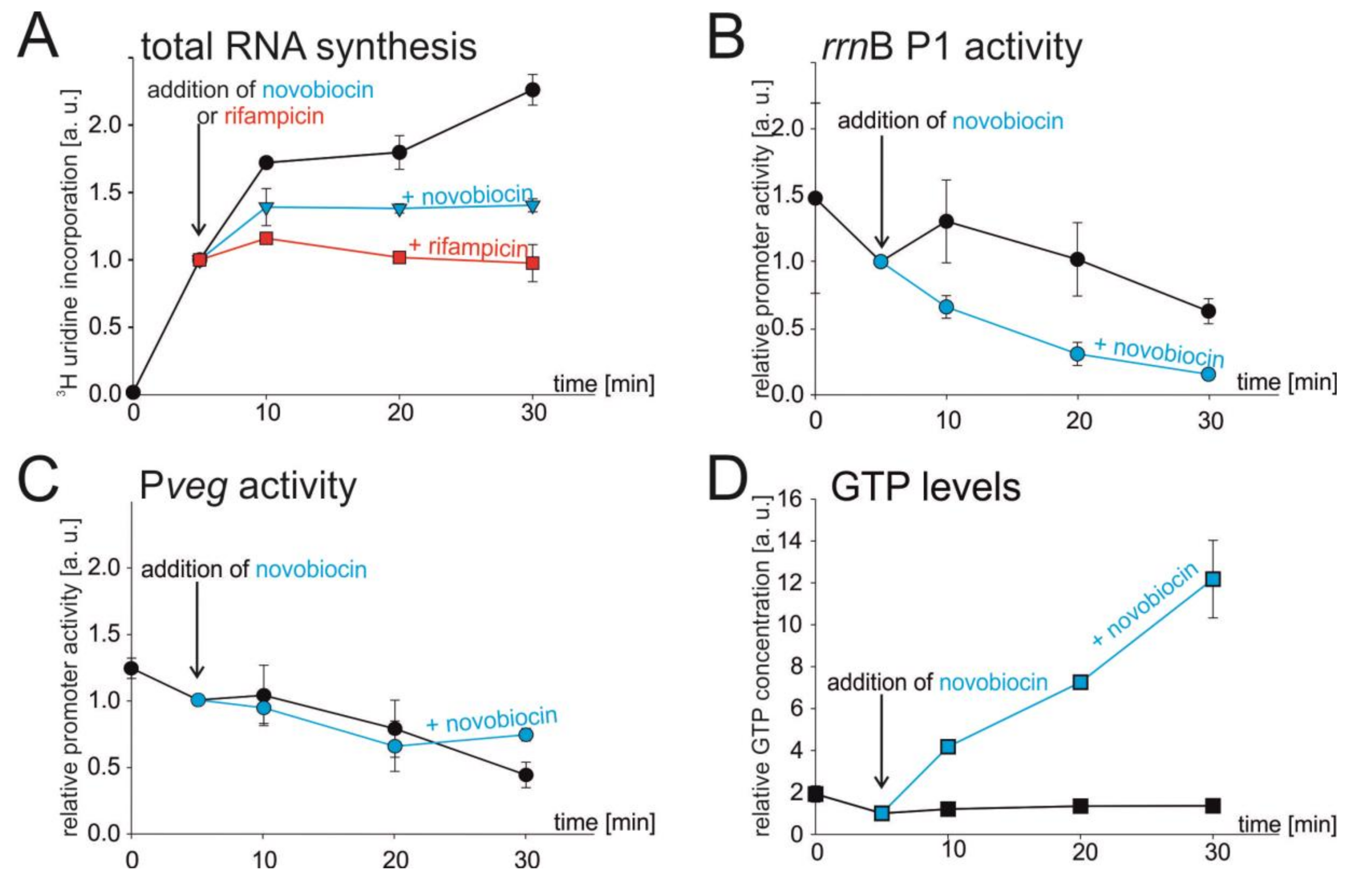
3.2. Chromosome Relaxation Inhibits Total RNA Synthesis In Vivo
3.3. Novobiocin-Induced Relaxation of DNA Affects the Activity of rrnB P1 In Vivo
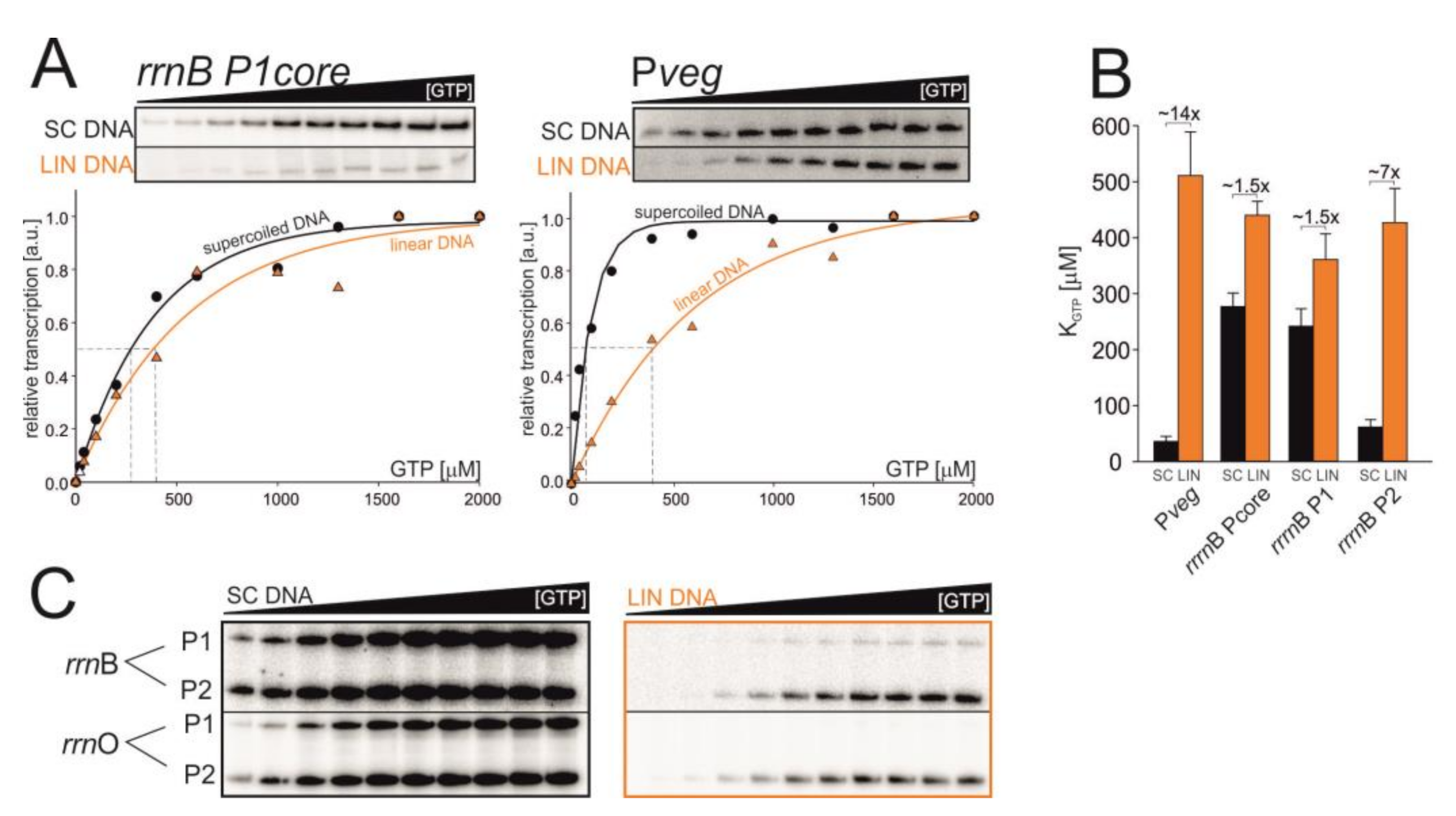
3.4. Changes in DNA Topology Affect the Affinity of RNAP for iNTP In Vitro
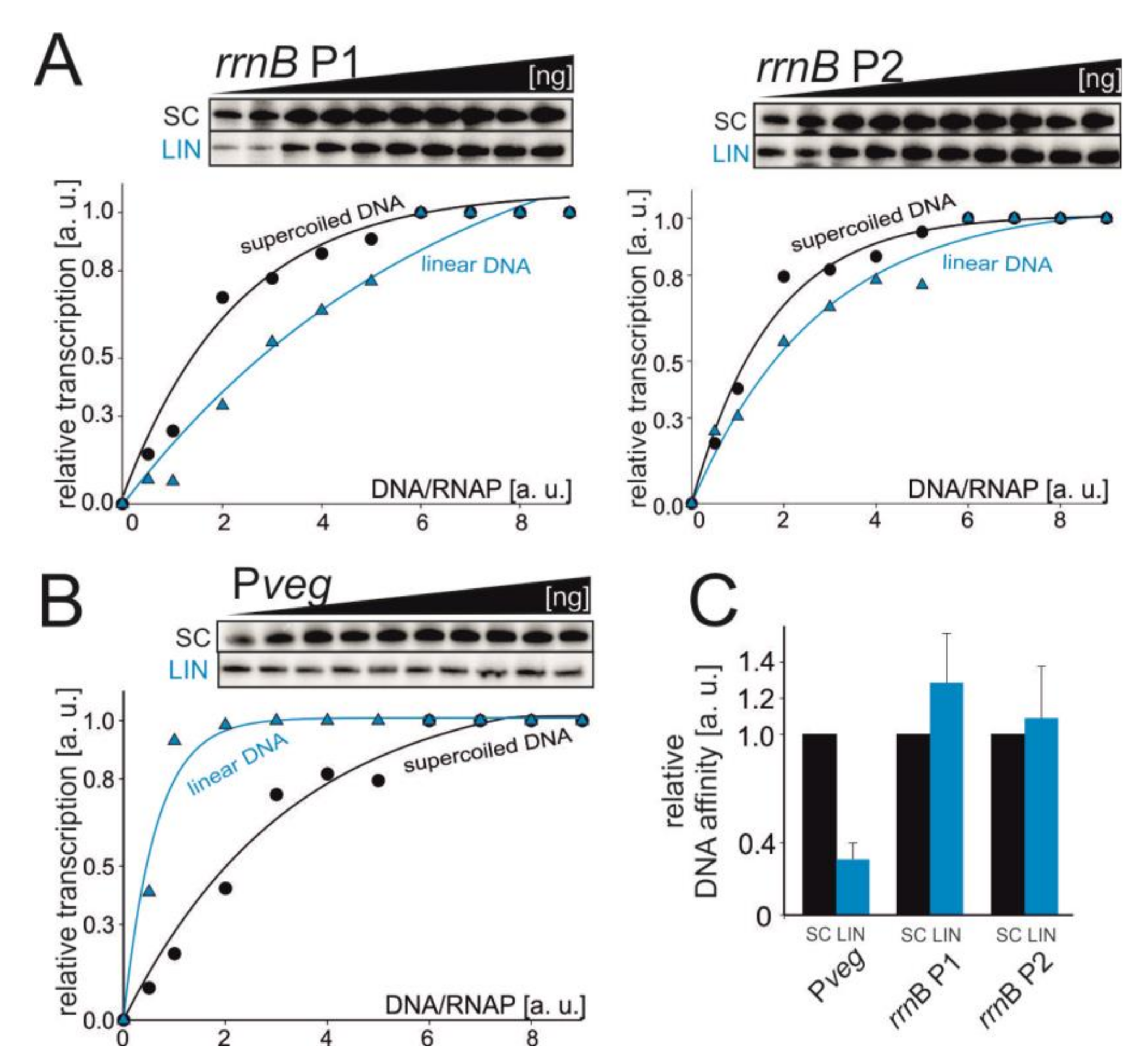
3.5. Pveg and rRNA Promoter Affinities for RNAP Change with DNA Relaxation In Vitro
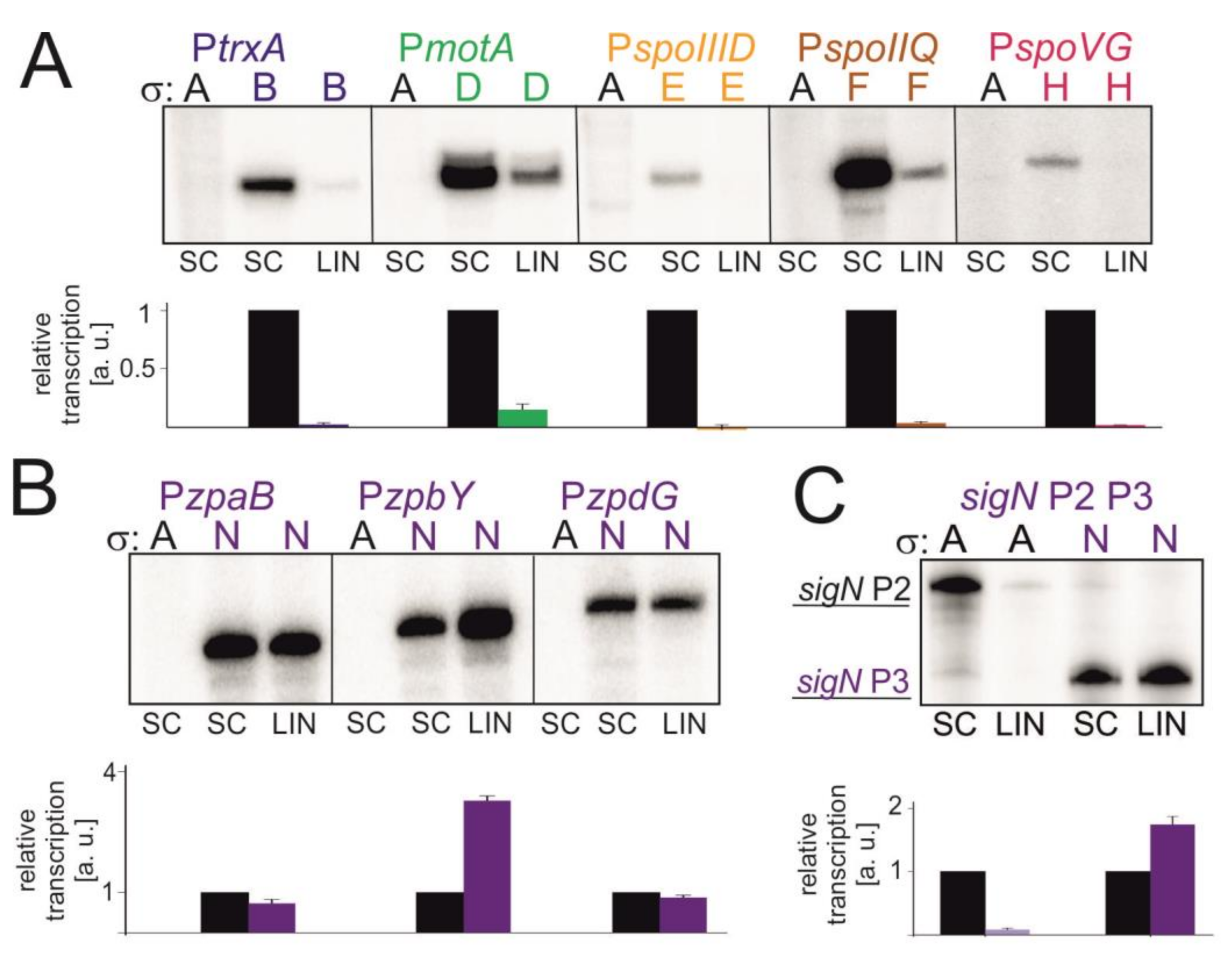
3.6. The Effect of Supercoiling on Transcription In Vitro with Alternative Sigma Factors
4. Discussion
4.1. rRNA Promoters and Pveg
4.2. Transcription with Selected Alternative σ Factors
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gourse, R.L.; Gaal, T.; Bartlett, M.S.; Appleman, J.A.; Ross, W. rRNA Transcription and growth rate–dependent regulation of ribosome synthesis in Escherichia Coli. Annu. Rev. Microbiol. 1996, 50, 645–677. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Borukhov, S. Bacterial RNA Polymerase-DNA interaction-the driving force of gene expression and the target for drug action. Front. Mol. Biosci. 2016, 3, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helmann, J.D. Where to begin? Sigma factors and the selectivity of transcription initiation in bacteria. Mol. Microbiol. 2019, 112, 335–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, C.A.; Chan, C.; Dombroski, A.; Gruber, T.; Sharp, M.; Tupy, J.; Young, B. The functional and regulatory roles of sigma factors in transcription. Cold Spring Harb. Symp. Quant. Biol. 1998, 63, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Paget, M.S. Bacterial sigma factors and anti-sigma factors: Structure, function and distribution. Biomolecules 2015, 5, 1245–1265. [Google Scholar] [CrossRef] [PubMed]
- Feklistov, A.; Darst, S.A. Structural basis for promoter -10 element recognition by the bacterial RNA polymerase σ subunit. Cell 2011, 147, 1257–1269. [Google Scholar] [CrossRef] [Green Version]
- Mustaev, A.; Roberts, J.; Gottesman, M. Transcription elongation. Transcription 2017, 8, 150–161. [Google Scholar] [CrossRef] [Green Version]
- Schneider, D.A.; Murray, H.D.; Gourse, R.L. Measuring control of transcription initiation by changing concentrations of nucleotides and their derivatives. Methods Enzymol. 2003, 370, 606–617. [Google Scholar] [CrossRef]
- Turnbough, C.L. Regulation of bacterial gene expression by the NTP substrates of transcription initiation. Mol. Microbiol. 2008, 69, 10–14. [Google Scholar] [CrossRef]
- Murray, H.D.; Schneider, D.A.; Gourse, R.L. Control of rRNA expression by small molecules is dynamic and nonredundant. Mol. Cell 2003, 12, 125–134. [Google Scholar] [CrossRef]
- Krásný, L.; Gourse, R.L. An alternative strategy for bacterial ribosome synthesis: Bacillus subtilis rRNA transcription regulation. EMBO J. 2004, 23, 4473–4483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bittner, A.N.; Kriel, A.; Wang, J.D. Lowering GTP Level increases survival of amino acid starvation but slows growth rate for Bacillus subtilis cells lacking (p)ppGpp. J. Bacteriol. 2014, 196, 2067–2076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kriel, A.; Bittner, A.N.; Kim, S.H.; Liu, K.; Tehranchi, A.K.; Zou, W.Y.; Rendon, S.; Chen, R.; Tu, B.P.; Wang, J.D. Direct regulation of GTP homeostasis by (p)ppGpp: A critical component of viability and stress resistance. Mol. Cell 2012, 48, 231–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henkin, T.M.; Yanofsky, C. Regulation by transcription attenuation in bacteria: How RNA provides instructions for transcription termination/antitermination decisions. BioEssays 2002, 24, 700–707. [Google Scholar] [CrossRef]
- Krásný, L.; Tišerová, H.; Jonák, J.; Rejman, D.; Šanderová, H. The identity of the transcription +1 position is crucial for changes in gene expression in response to amino acid starvation in Bacillus subtilis. Mol. Microbiol. 2008, 69, 42–54. [Google Scholar] [CrossRef]
- Natori, Y.; Tagami, K.; Murakami, K.; Yoshida, S.; Tanigawa, O.; Moh, Y.; Masuda, K.; Wada, T.; Suzuki, S.; Nanamiya, H.; et al. Transcription activity of individual rrn operons in Bacillus subtilis mutants deficient in (p)ppGpp synthetase genes, relA, yjbM, and ywaC. J. Bacteriol. 2009, 191, 4555–4561. [Google Scholar] [CrossRef] [Green Version]
- Kästle, B.; Geiger, T.; Gratani, F.L.; Reisinger, R.; Goerke, C.; Borisova, M.; Mayer, C.; Wolz, C. rRNA regulation during growth and under stringent conditions in S taphylococcus aureus. Environ. Microbiol. 2015, 17, 4394–4405. [Google Scholar] [CrossRef]
- Zechiedrich, E.L.; Khodursky, A.B.; Bachellier, S.; Schneider, R.; Chen, D.; Lilley, D.M.J.; Cozzarelli, N.R. Roles of topoisomerases in maintaining steady-state DNA supercoiling in Escherichia coli. J. Biol. Chem. 2000, 275, 8103–8113. [Google Scholar] [CrossRef] [Green Version]
- Higgins, C.F.; Dorman, C.J.; Stirling, D.A.; Waddell, L.; Booth, I.R.; May, G.; Bremer, E. A physiological role for DNA supercoiling in the osmotic regulation of gene expression in S. typhimurium and E. coli. Cell 1988, 52, 569–584. [Google Scholar] [CrossRef]
- Richardson, S.M.; Higgins, C.F.; Lilley, D.M. The genetic control of DNA supercoiling in Salmonella typhimurium. EMBO J. 1984, 3, 1745–1752. [Google Scholar] [CrossRef]
- McClure, W.R. Mechanism and control of transcription initiation in prokaryotes. Annu. Rev. Biochem. 1985, 54, 171–204. [Google Scholar] [CrossRef] [PubMed]
- Schnetz, K.; Wang, J.C. Silencing of the Escherichia Coli bgl promoter: Effects of template supercoiling and cell extracts on promoter activity in vitro. Nucleic Acids Res. 1996, 24, 2422–2428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sioud, M.; Boudabous, A.; Cekaite, L. Transcriptional responses of Bacillus subtillis and thuringiensis to antibiotics and anti-tumour drugs. Int. J. Mol. Med. 2009, 23, 33–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myagmarjav, B.-E.; Konkol, M.A.; Ramsey, J.; Mukhopadhyay, S.; Kearns, D.B. ZpdN, a Plasmid-encoded sigma factor homolog, induces pBS32-dependent cell death in Bacillus subtilis. J. Bacteriol. 2016, 198, 2975–2984. [Google Scholar] [CrossRef] [Green Version]
- Burton, A.T.; DeLoughery, A.; Li, G.W.; Kearns, D.B. Transcriptional regulation and mechanism of sigN (ZpdN), a pBS32-encoded sigma factor in bacillus subtilis. MBio 2019, 10, e01899-19. [Google Scholar] [CrossRef] [Green Version]
- Qi, Y.; Hulett, F.M. PhoP~P and RNA polymerase sigma(A) holoenzyme are sufficient for transcription of Pho regulon promoters in Bacillus subtilis: PhoP~P activator sites within the coding region stimulate transcription in vitro. Mol. Microbiol. 1998, 28, 1187–1197. [Google Scholar] [CrossRef]
- Chang, B.-Y.; Doi, R.H. Overproduction, Purification, and Characterization of Bacillus subtilis RNA Polymerase SigA Factor. J. Bacteriol. 1990, 172, 3257–3263. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-F.; Helmann, J.D. The Bacillus subtilis Flagellar Regulatory Protein SigmaD: Overproduction, Domain Analysis and DNA-binding Properties. J. Mol. Biol. 1995, 249, 743–753. [Google Scholar] [CrossRef]
- Paul, B.J.; Ross, W.; Gaal, T.; Gourse, R.L. rRNA Transcription in Escherichia coli. Annu. Rev. Genet. 2004, 38, 749–770. [Google Scholar] [CrossRef]
- Panova, N.; Zborníková, E.; Šimák, O.; Pohl, R.; Kolář, M.; Bogdanová, K.; Večeřová, R.; Seydlová, G.; Fišer, R.; Hadravová, R.; et al. Insights into the mechanism of action of bactericidal Lipophosphonoxins. PLoS ONE 2015, 10, e0145918. [Google Scholar] [CrossRef] [Green Version]
- Imamura, D.; Zhou, R.; Feig, M.; Kroos, L. Evidence that the Bacillus subtilis SpoIIGA protein is a novel type of signal-transducing aspartic protease. J. Biol. Chem. 2008, 283, 15287–15299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaBell, T.L.; Trempy, J.E.; Haldenwang, W.G. Sporulation-specific σ factor σ29 of Bacillus subtilis is synthesized from a precursors protein, P31. Proc. Natl. Acad. Sci. USA 1987, 84, 1784–1788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, W.; Thompson, J.F.; Newlands, J.T.; Gourse, R.L.E. E. coli Fis protein activates ribosomal RNA transcription in vitro and in vivo. EMBO J. 1990, 9, 3733–3742. [Google Scholar] [CrossRef] [PubMed]
- Deneer, H.G.; Spiegelman, G.B. Bacillus subtilis rRNA promoters are growth rate regulated in Escherichia coli. J. Bacteriol. 1987, 169, 995–1002. [Google Scholar] [CrossRef] [Green Version]
- Samarrai, W.; Liu, D.X.; White, A.M.; Studamire, B.; Edelstein, J.; Srivastava, A.; Widom, R.L.; Rudner, R. Differential responses of Bacillus subtilis rRNA promoters to nutritional stress. J. Bacteriol. 2011, 193, 723–733. [Google Scholar] [CrossRef] [Green Version]
- Wellington, S.R.; Spiegelman, G.B. The kinetics of formation of complexes between Escherichia coli RNA polymerase and the rrnB P1 and P2 promoters of Bacillus subtilis. Effects of guanosine tetraphosphate on select steps of transcription initiation. J. Biol. Chem. 1993, 268, 7205–7214. [Google Scholar]
- Fukushima, T.; Ishikawa, S.; Yamamoto, H.; Ogasawara, N.; Sekiguchi, J. Transcriptional, functional and cytochemical analyses of the veg gene in Bacillus subtilis. J. Biochem. 2003, 133, 475–483. [Google Scholar] [CrossRef]
- Lei, Y.; Oshima, T.; Ogasawara, N.; Ishikawa, S. Functional analysis of the protein veg, which stimulates biofilm formation in Bacillus subtilis. J. Bacteriol 2013, 195, 1697–1705. [Google Scholar] [CrossRef] [Green Version]
- Conter, A.; Menchon, C.; Gutierrez, C. Role of DNA supercoiling and RpoS sigma factor in the osmotic and growth phase-dependent induction of the gene osmE of Escherichia coli K12. J. Mol. Biol. 1997, 273, 75–83. [Google Scholar] [CrossRef]
- Nicholson, W.L.; Setlow, P. Dramatic increase in negative superhelicity of plasmid DNA in the forespore compartment of sporulating cells of Bacillus subtilis. J. Bacteriol. 1990, 172, 7–14. [Google Scholar] [CrossRef] [Green Version]
- Alice, A.F.; Sanchez-Rivas, C. DNA supercoiling and osmoresistance in Bacillus subtilis 168. Curr. Microbiol. 1997, 35, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.J.; Singh, O.M.P.; Smith, C.V.; Skarzynski, T.; Maxwell, A.; Wonacott, A.J.; Wigley, D.B. The nature of inhibition of DNA Gyrase by the Coumarins and the Cyclothialidines revealed by X-ray crystallography. Embo J. 1996, 15, 1412–1420. [Google Scholar] [CrossRef] [PubMed]
- Sugino, A.; Higginst, N.P.; Brownt, P.O.; Peeblesf, C.L.; Cozzarellitf, N.R. Energy coupling in DNA gyrase and the mechanism of action of novobiocin. Biochemistry 1978, 75, 4838–4842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gellert, M.; O’Dea, M.H.; Itoh, T.; Tomizawa, J.I. Novobiocin and coumermycin inhibit DNA supercoiling catalyzed by DNA gyrase. Proc. Natl. Acad. Sci. USA 1976, 73, 4474–4478. [Google Scholar] [CrossRef] [Green Version]
- Dennis, P.P.; Bremer, H. Modulation of chemical composition and other parameters of the cell at different exponential growth rates. EcoSal Plus 2008, 3, 1553–1569. [Google Scholar] [CrossRef]
- Sojka, L.; Kouba, T.; Barvík, I.; Šanderová, H.; Maderová, Z.; Jonák, J.; Krásný, L. Rapid changes in gene expression: DNA determinants of promoter regulation by the concentration of the transcription initiating NTP in Bacillus subtilis. Nucleic Acids Res. 2011, 39, 4598–4611. [Google Scholar] [CrossRef]
- Estrem, S.T.; Gaal, T.; Ross, W.; Gourse, R.L. Identification of an UP element consensus sequence for bacterial promoters. Proc. Natl. Acad. Sci. USA 1998, 95, 9761–9766. [Google Scholar] [CrossRef] [Green Version]
- Meng, W.; Belyaeva, T.; Savery, N.J.; Busby, S.J.W.; Ross, W.E.; Gaal, T.; Gourse, R.L.; Thomas, M.S. UP element-dependent transcription at the Escherichia coli rrnB P1 promoter: Positional requirements and role of the RNA polymerase α subunit linker. Nucleic Acids Res. 2001, 29, 4166–4178. [Google Scholar] [CrossRef]
- Rao, L.; Ross, W.; Appleman, J.A.; Gaal, T.; Leirmo, S.; Schlax, J.P.; Record, M.; Thomas, J.; Gourse, R.L. Factor independent activation of rrnB P1: An “extended” promoter with an upstream element that dramatically increases promoter strength. J. Mol. Biol. 1994, 235, 1421–1435. [Google Scholar] [CrossRef]
- Klumpp, S.; Hwa, T. Growth-rate-dependent partitioning of RNA polymerases in bacteria. Proc. Natl. Acad. Sci. USA 2008, 105, 20245–20250. [Google Scholar] [CrossRef] [Green Version]
- Liang, S.T.; Bipatnath, M.; Xu, Y.C.; Chen, S.L.; Dennis, P.; Ehrenberg, M.; Bremer, H. Activities of constitutive promoters in Escherichia coli. J. Mol. Biol. 1999, 292, 19–37. [Google Scholar] [CrossRef] [PubMed]
- Haldenwang, W.G.; Losick, R. Novel RNA polymerase σ factor from Bacillus subtilis. Proc. Natl. Acad. Sci. USA 1980, 77, 7000–7004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hecker, M.; Völker, U. General stress response of Bacillus subtilis and other bacteria. Adv. Microb. Physiol. 2001, 44, 35–91. [Google Scholar] [CrossRef] [PubMed]
- Jaehning, J.A.; Wiggs, J.L.; Chamberlin, M.J. Altered promoter selection by a novel form of Bacillus subtilis RNA polymerase. Proc. Natl. Acad. Sci. USA 1979, 76, 5470–5474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haldenwang, W.G.; Lang, N.; Losick, R. A sporulation-induced sigma-like regulatory protein from b. subtilis. Cell 1981, 23, 615–624. [Google Scholar] [CrossRef]
- Partridge, S.R.; Foulger, D.; Errington, J. The role of σF in prespore-specific transcription in Bacillus subtilis. Mol. Microbiol. 1991, 5, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.C.; Moran, C.P.; Losick, R. Two RNA polymerase sigma factors from Bacillus subtilis discriminate between overlapping promoters for a developmentally regulated gene. Nature 1983, 302, 800–804. [Google Scholar] [CrossRef]
- Earl, A.M.; Losick, R.; Kolter, R. Bacillus subtilis genome diversity. J. Bacteriol. 2007, 189, 1163–1170. [Google Scholar] [CrossRef] [Green Version]
- Burby, P.E.; Simmons, L.A. A bacterial DNA repair pathway specific to a natural antibiotic. Mol. Microbiol. 2019, 111, 338–353. [Google Scholar] [CrossRef]
- Lee, Y.J.; Park, S.J.; Ciccone, S.L.M.; Kim, C.R.; Lee, S.H. An in vivo analysis of MMC-induced DNA damage and its repair. Carcinogenesis 2006, 27, 446–453. [Google Scholar] [CrossRef] [Green Version]
- Ueda, K.; Morita, J.; Komano, T. Phage inactivation and DNA strand scission activities of 7-N-(p-hydroxyphenyl)mitomycin C. J. Antibiot. 1982, 35, 1380–1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, A.; Sinai, L.; Smith, Y.; Ben-Yehuda, S. Dynamic expression of the translational machinery during Bacillus subtilis life cycle at a single cell level. PLoS ONE 2012, 7, 41921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burenina, O.Y.; Hoch, P.G.; Damm, K.; Salas, M.; Zatsepin, T.S.; Lechner, M.; Oretskaya, T.S.; Kubareva, E.A.; Hartmann, R.K. Mechanistic comparison of Bacillus subtilis 6S-1 and 6S-2 RNAs-commonalities and differences. RNA 2014, 20, 348–359. [Google Scholar] [CrossRef] [Green Version]
- Glaser, G.; Sarmientos, P.; Cashel, M. Functional interrelationship between two tandem E. coli ribosomal RNA promoters. Nature 1983, 302, 74–76. [Google Scholar] [CrossRef] [PubMed]
- Schultz, M.C.; Brill, S.J.; Ju, Q.; Sternglanz, R.; Reeder, R.H. Topoisomerases and yeast rRNA transcription: Negative supercoiling stimulates initiation and topoisomerase activity is required for elongation. Genes Dev. 1992, 6, 1332–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bird, T.; Burbulys, D.; Wu, J.; Strauch, M.; Hoch, J.; Spiegelman, G. The effect of supercoiling on the in vitro transcription of the spoIIA operon from Bacillus subtilis. Biochimie 1992, 74, 627–634. [Google Scholar] [CrossRef]
- Helmann, J.D. Compilation and analysus of Bacillus Subtilis σA-dependent promoter sequences: Evidence for extended contact between RNA polymerse and upstream promoter DNA. Nucleic Acids Res. 1995, 23, 2351–2360. [Google Scholar] [CrossRef]
- Typas, A.; Hengge, R. Role of the spacer between the -35 and -10 regions in sigmaS promoter selectivity in Escherichia coli. Mol. Microbiol. 2006, 59, 1037–1051. [Google Scholar] [CrossRef]
- Bordes, P.; Conter, A.; Morales, V.; Bouvier, J.; Kolb, A.; Gutierrez, C. DNA supercoiling contributes to disconnect σS accumulation from σS-dependent transcription in Escherichia coli. Mol. Microbiol. 2003, 48, 561–571. [Google Scholar] [CrossRef]
- Kusano, S.; Ding, Q.; Fujita, N.; Ishihama, A. Promoter selectivity of Escherichia coli RNA Polymerase E and E Holoenzymes. J. Biol. Chem. 1996, 271, 1998–2004. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Original code | Construct | Description | Reference |
|---|---|---|---|---|
| B. subtilis | ||||
| LK134 | RLG7554 | rrnB P1-lacZ | MO1099 amyE::Cm rrnB P1 (−39/+1)-lacZ | [11] |
| LK135 | RLG7555 | Pveg-lacZ | MO1099 amyE::Cm Pveg (−38/−1, +1G)-lacZ | [11] |
| LK41 | RLG6943 | RM-lacZ | MO1099 amyE::Cm rrnO P2 (−77/+50)-lacZ | [11] |
| LK1723 | RLG7024 | wt RNAP | β’ with C-ter. His10x; MH5636 | [26] |
| E. coli | ||||
| LK22 | SigA | SigA; BL21(DE3) | [27] | |
| LK1207 | SigB | SigB with C-ter. His6x; BL21(DE3) | This work | |
| LK1187 | SigD | SigD; BL21(DE3) | [28] | |
| LK2580 | SigE | SigE with C-ter. His6x; BL21(DE3) | This work | |
| LK1425 | SigF | SigF with C-ter. His6x; BL21(DE3) | This work | |
| LK1208 | SigH | SigH with C-ter. His6x; BL21(DE3) | This work | |
| LK2531 | SigN | His-SUMO-SigN in pBM05; BL21(DE3) | This work | |
| LK1177 | RLG7558 | Pveg | pRLG770 with Pveg (−38/+1) +1G; DH5α | [11] |
| LK1522 | RLG7596 | rrnB P1core | pRLG770 with rrnB P1 (−39/+1); DH5α | [11] |
| LK28 | RLG6927 | rrnB P1+P2 | pRLG770 with rrnB P1+P2 (−248/+8); DH5α | [15] |
| LK17 | RLG6916 | rrnO P1+P2 | pRLG770 with rrnO P1+P2 (−314/+9); DH5α | This work |
| LK1231 | PtrxA | pRLG770 with PtrxA (−249/+11); DH5α | This work | |
| LK1233 | PmotA | pRLG770 with PmotA (−249/+11); DH5α | This work | |
| LK2594 | PspoIIID | pRLG770 with PspoIIID (−150/+10); DH5α | This work | |
| LK1495 | PspoIIQ | pRLG770 with PspoIIQ (−251/+9); DH5α | This work | |
| LK1235 | PspoVG | pRLG770 with PspoVG (−94/+11); DH5α | This work | |
| LK2672 | sigN P2+P3 | pRLG770 with sigN P2+P3 (−247/+159); DH5α | This work | |
| LK2673 | PzpaB | pRLG770 with PzpaB (−266/+175); DH5α | This work | |
| LK2608 | PzpbY | pRLG770 with PzpbY (−304/+155); DH5α | This work | |
| LK2609 | PzpdG | pRLG770 with PzpdG (−244/+170); DH5α | This work | |
| Primer No (#) | Sequence 5′→ 3′ | |
|---|---|---|
| #1001 | GGAATTCCATATGAATCTACAGAACAACAAGG | Primers for sigH cloning into pET-22b(+) |
| #1002 | CCGCTCGAGCTATTACAAACTGATTTCGCG | |
| #1004 | GGAATTCCATATGACACAACCATCAAAAAC | Primers for sigB cloning into pET-22b(+) |
| #1006 | CCGCTCGAGCATTAACTCCATCGAGGGATC | |
| #1069 | CCGGAATTCATTCCGGAGTCATTCTTACGG | Primers for PtrxA cloning into pRLG770 |
| #1070 | CCCAAGCTTCACTGTCATGTACTTTACCATG | |
| #1075 | CCGGAATTCCTTTACACTTTTTTAAGGAGG | Primers for PmotA cloning into pRLG770 |
| #1076 | CCCAAGCTTCTAGCTTGTCTATGGTTAATATC | |
| #1079 | CCGGAATTCTTTATGACCTAATTGTGTAAC | Primers for PspoVG cloning into pRLG770 |
| #1080 | CCCAAGCTTATAAAAGCATTAGTGTATC | |
| #1309 | GGAATTCCATATGGATGTGGAGGTTAAGAAAAAC | Primers for sigF cloning into pET-22b(+) |
| #1311 | CCGCTCGAGGCCATCCGTATGATCCATTTG | |
| #1425 | CCGGAATTCCATTCCATCCGGTCTTCAGG | Primers for PspoIIQ cloning into pRLG770 |
| #1426 | CCCAAGCTTCATCACCTCAGCAACATTCTG | |
| #2973 | CAGTAACTTCCACAGTAGTTCACCAC | universal reverse primer for PE and qPCR |
| #2974 | TCTAAGCTTCTAGGATCCCC | test RNA-specific forward primer for PE and qPCR |
| #2975 | GTCGCTTTGAGAGAAGCACA | RM RNA-specific forward primer for PE and qPCR |
| #3109 | GCGAATTCCGTGTCGGTCAACATAATAAAGG | Primers for sigN P2+P3 cloning into pRLG770 |
| #3110 | GCAAGCTTCGGCAAAAATCTTTCTCTCACC | |
| #3111 | GCGAATTCGCGATGAATGAAGAGACACGG | Primers for PzpaB cloning into pRLG770 |
| #3112 | GCAAGCTTAGTCCATCTCGAAGATCTGGT | |
| #3113 | GCGAATTCGACTCCAACATTTCTATTCC | Primers for PzpbY cloning into pRLG770 |
| #3114 | GCAAGCTTGGTCTTCTTCACTTAATTCA | |
| #3117 | GCGAATTCTCAAAGATCTTCTAACTTGT | Primers for PzpdG cloning into pRLG770 |
| #3118 | GCAAGCTTGGCAGTAATCAATCAATTCT | |
| #3166 | CGGCATATGTACATAGGCGGGAGTGAAGCC | Primers for sigE active form cloning into pET-22b(+) |
| #3167 | CCGCTCGAGCACCATTTTGTTGAACTCTTTTC | |
| #3170 | GGCGAATTCGCTTATTTCATTTTACAGGAG | Primers for PspoIIID cloning into pRLG770 |
| #3171 | CCGAAGCTTTGTTAGGTTTGTAACAGTGT | |
| primer A | GGGAATTCATGGACATCAATGATATCTC | Primers for rrnO P1+P2 cloning into pRLG770 |
| primer B | GGAAGCTTTCAAAGCGACTACTTAATAG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sudzinová, P.; Kambová, M.; Ramaniuk, O.; Benda, M.; Šanderová, H.; Krásný, L. Effects of DNA Topology on Transcription from rRNA Promoters in Bacillus subtilis. Microorganisms 2021, 9, 87. https://doi.org/10.3390/microorganisms9010087
Sudzinová P, Kambová M, Ramaniuk O, Benda M, Šanderová H, Krásný L. Effects of DNA Topology on Transcription from rRNA Promoters in Bacillus subtilis. Microorganisms. 2021; 9(1):87. https://doi.org/10.3390/microorganisms9010087
Chicago/Turabian StyleSudzinová, Petra, Milada Kambová, Olga Ramaniuk, Martin Benda, Hana Šanderová, and Libor Krásný. 2021. "Effects of DNA Topology on Transcription from rRNA Promoters in Bacillus subtilis" Microorganisms 9, no. 1: 87. https://doi.org/10.3390/microorganisms9010087
APA StyleSudzinová, P., Kambová, M., Ramaniuk, O., Benda, M., Šanderová, H., & Krásný, L. (2021). Effects of DNA Topology on Transcription from rRNA Promoters in Bacillus subtilis. Microorganisms, 9(1), 87. https://doi.org/10.3390/microorganisms9010087