
Diversity and Hydrocarbon-Degrading Potential of Deep-Sea Microbial Community from the Mid-Atlantic Ridge, South of the Azores (North Atlantic Ocean)

 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Material and Methods
2.1. Sampling
2.2. Environmental DNA Extraction, Amplification and Sequencing
2.3. Bioinformatic Analysis
2.4. Enrichment of Hydrocarbons-Degrading Bacteria from Deep-Sea Sediments
2.5. Microcosm Bioremediation Experiment
2.6. Abundance of Hydrocarbon-Degrading Bacteria
2.7. Determination of Total Petroleum Hydrocarbons (TPHs)
2.8. Isolation and Identification of Bacterial Strains of Microcosms’ Experiment
2.9. Statistical Analysis and Data Visualization
3. Results
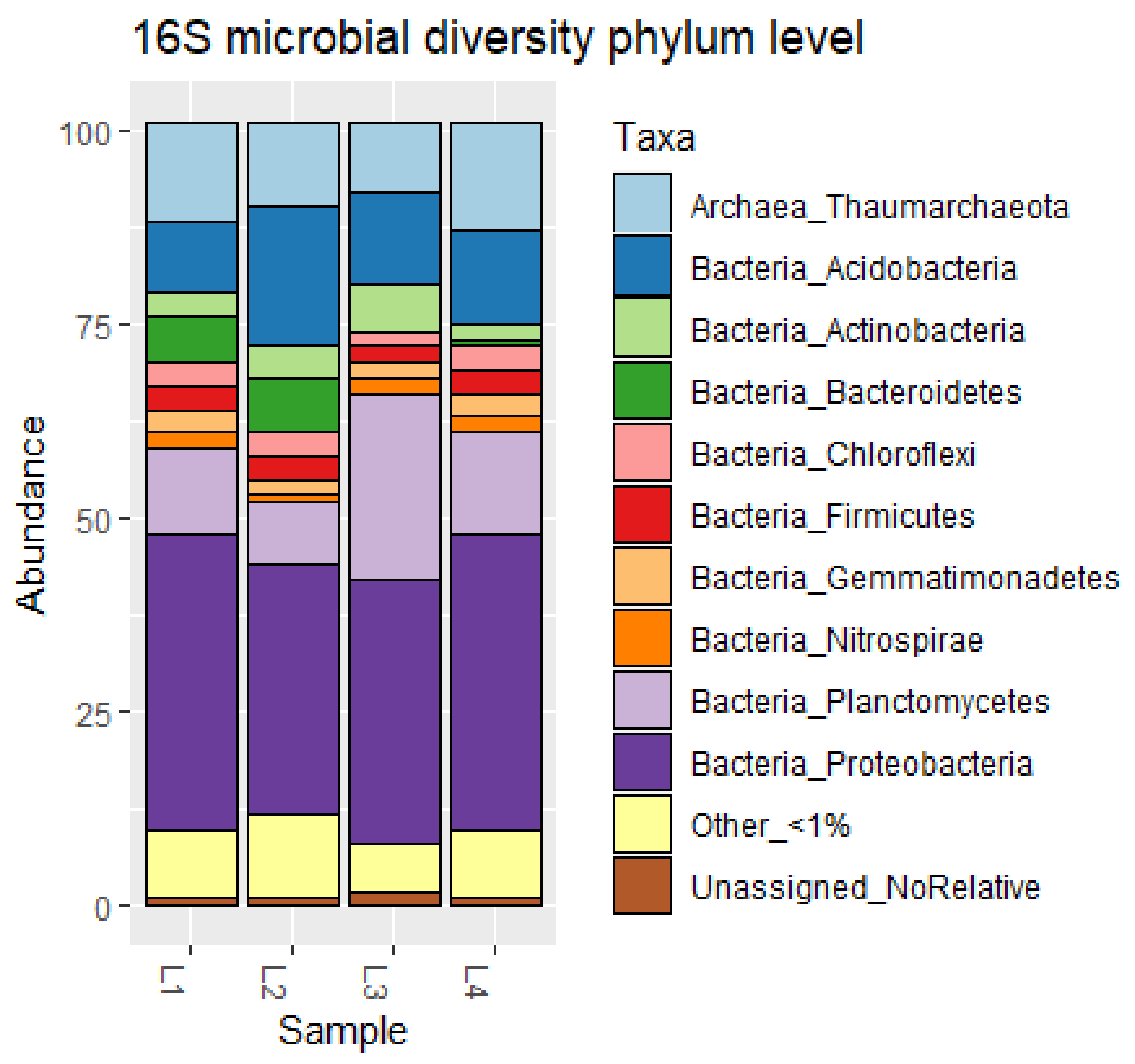
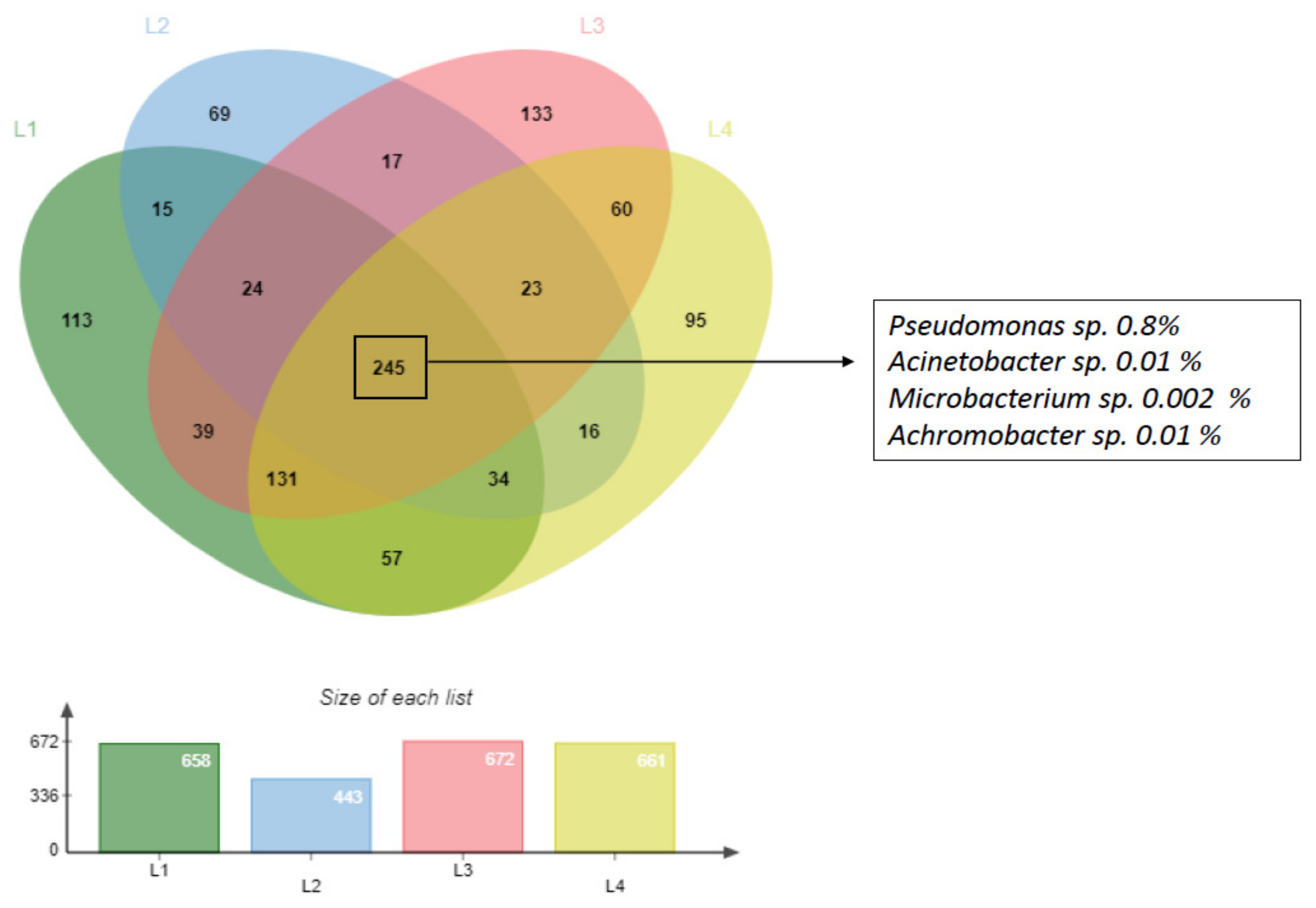
3.1. Characterization of Deep Sediment Prokaryotic Community
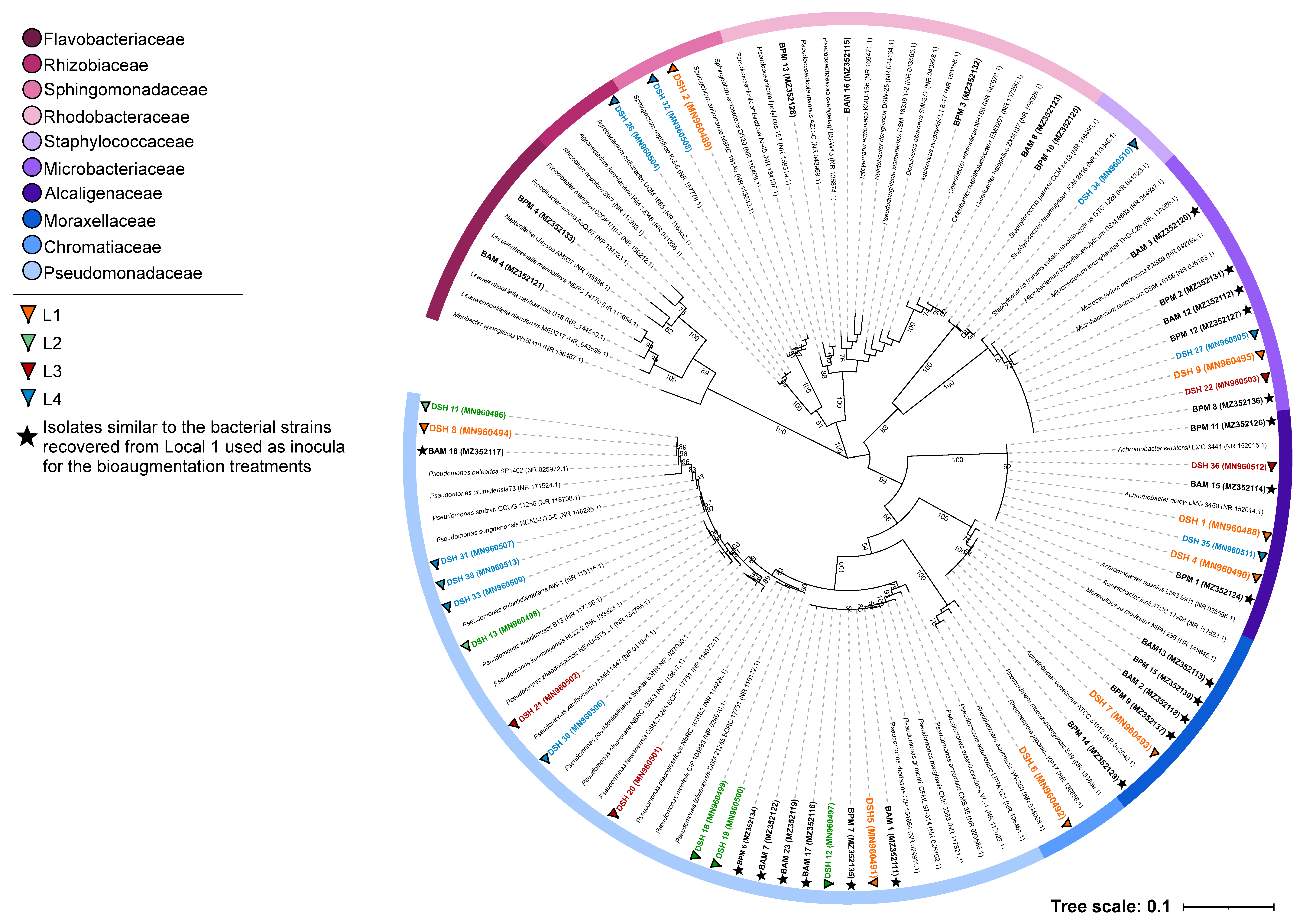
3.2. Taxonomic Identification of the Isolated Strains
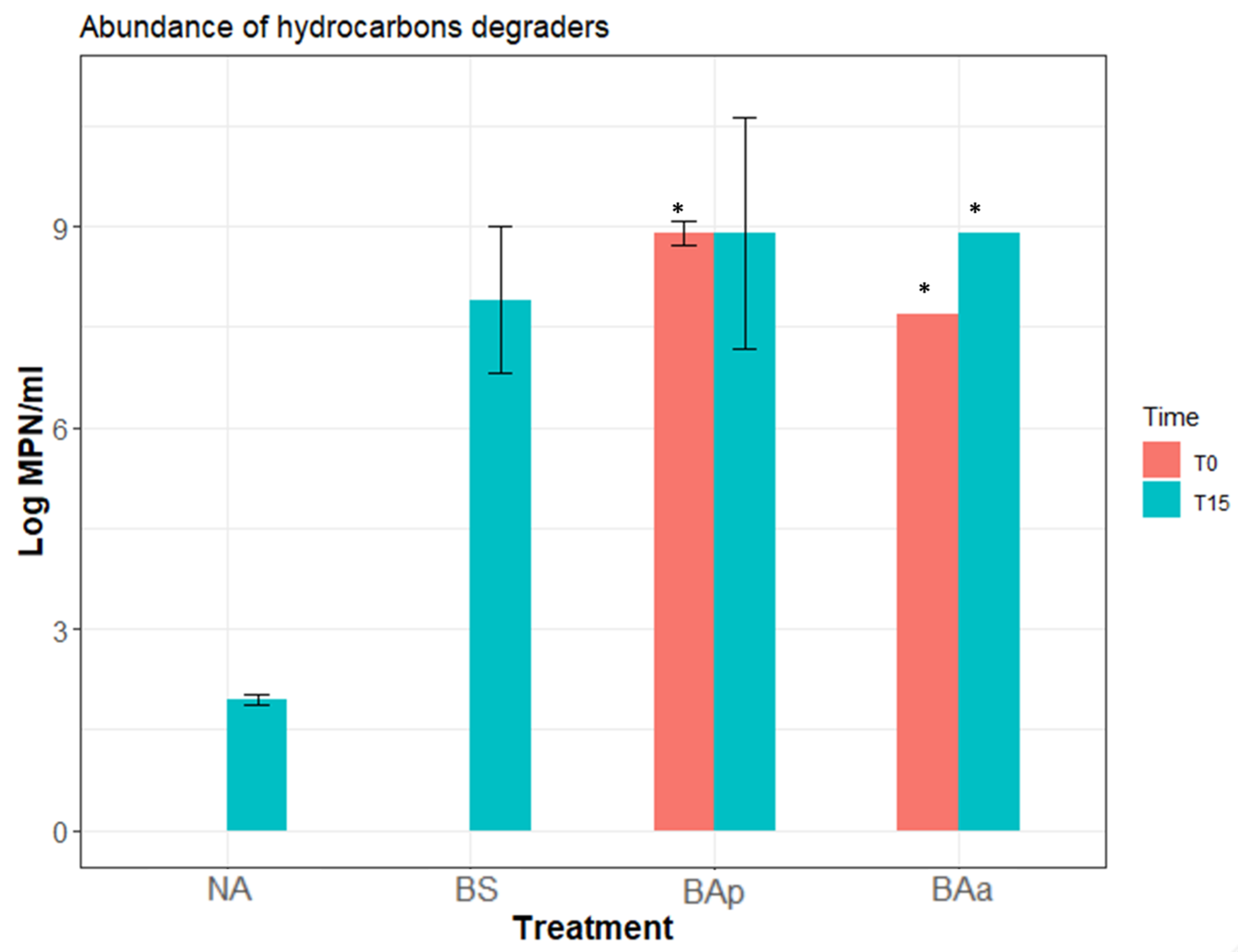
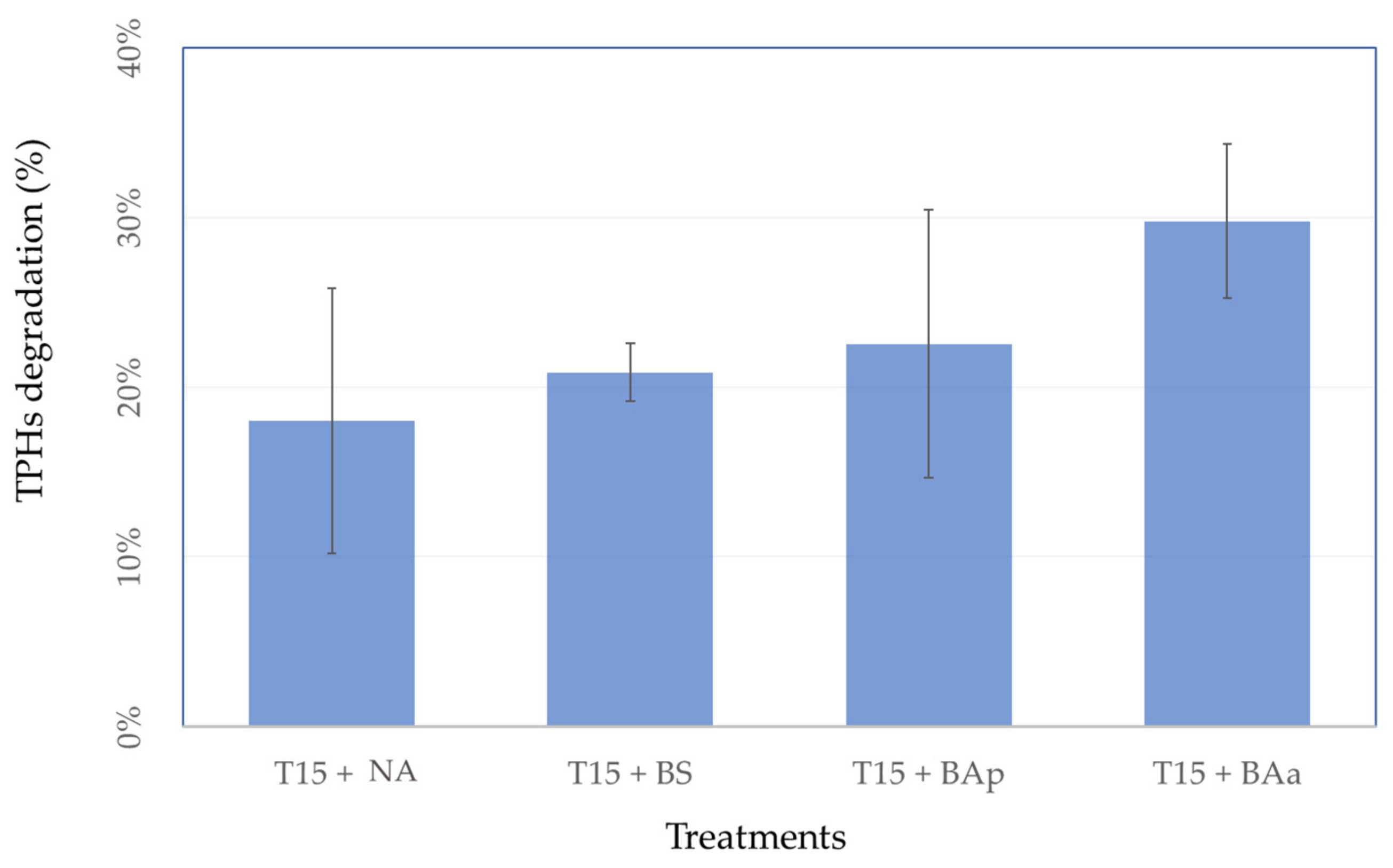
3.3. Microcosm Bioremediation Experiment
3.4. Recover of Microorganisms from Microcosm Experiments
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Atlas, R.M.; Hazen, T.C. Oil biodegradation and bioremediation: A tale of the two worst spills in U.S. history. Environ. Sci. Technol. 2011, 45, 6709–6715. [Google Scholar] [CrossRef]
- Head, I.M.; Jones, D.M.; Röling, W.F.M. Marine microorganisms make a meal of oil. Nat. Rev. Microbiol. 2006, 4, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Cappello, S.; Genovese, M.; Denaro, R.; Santisi, S.; Volta, A.; Bonsignore, M.; Mancini, G.; Giuliano, L.; Genovese, L.; Yakimov, M.M. Quick stimulation of Alcanivorax sp. By bioemulsificant EPS2003 on microcosm oil spill simulation. Braz. J. Microbiol. 2014, 45, 1317–1323. [Google Scholar] [CrossRef] [PubMed]
- Santisi, S.; Catalfamo, M.; Bonsignore, M.; Gentile, G.; Di Salvo, E.; Genovese, M.; Mahjoubi, M.; Cherif, A.; Mancini, G.; Hassanshahian, M.; et al. Biodegradation ability of two selected microbial autochthonous consortia from a chronically polluted marine coastal area (Priolo Gargallo, Italy). J. Appl. Microbiol. 2019, 127, 618–629. [Google Scholar] [CrossRef]
- Head, I.M.; Jones, D.M.; Larter, S.R. Biological activity in the deep subsurface and the origin of heavy oil. Nature 2003, 426, 344–352. [Google Scholar] [CrossRef]
- Yap, H.S.; Zakaria, N.N.; Zulkharnain, A.; Sabri, S.; Gomez-Fuentes, C.; Ahmad, S.A. Bibliometric analysis of hydrocarbon bioremediation in cold regions and a review on enhanced soil bioremediation. Biology 2021, 10, 354. [Google Scholar] [CrossRef]
- Almeida, C.M.R.; Reis, I.; Couto, M.N.; Bordalo, A.A.; Mucha, A.P. Potential of the microbial community present in an unimpacted beach sediment to remediate petroleum hydrocarbons. Environ. Sci. Pollut. Res. 2013, 20, 3176–3184. [Google Scholar] [CrossRef]
- Omokhagbor Adams, G.; Tawari Fufeyin, P.; Eruke Okoro, S.; Ehinomen, I. Bioremediation, Biostimulation and Bioaugmention: A Review. Int. J. Environ. Bioremediat. Biodegrad. 2015, 3, 28–39. [Google Scholar] [CrossRef]
- Hosokawa, R.; Nagai, M.; Morikawa, M.; Okuyama, H. Autochthonous bioaugmentation and its possible application to oil spills. World J. Microbiol. Biotechnol. 2009, 25, 1519–1528. [Google Scholar] [CrossRef]
- Dell’Anno, F.; Rastelli, E.; Tangherlini, M.; Corinaldesi, C.; Sansone, C.; Brunet, C.; Balzano, S.; Ianora, A.; Musco, L.; Montereali, M.R.; et al. Highly Contaminated Marine Sediments Can Host Rare Bacterial Taxa Potentially Useful for Bioremediation. Front. Microbiol. 2021, 12, 1–15. [Google Scholar] [CrossRef]
- Dueholm, M.S.; Marques, I.G.; Karst, S.M.; D’Imperio, S.; Tale, V.P.; Lewis, D.; Nielsen, P.H.; Nielsen, J.L. Survival and activity of individual bioaugmentation strains. Bioresour. Technol. 2015, 186, 192–199. [Google Scholar] [CrossRef]
- Tyagi, M.; da Fonseca, M.M.R.; de Carvalho, C.C.C.R. Bioaugmentation and biostimulation strategies to improve the effectiveness of bioremediation processes. Biodegradation 2011, 22, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Rocchetti, L.; Beolchini, F.; Hallberg, K.B.; Johnson, D.B.; Dell’Anno, A. Effects of prokaryotic diversity changes on hydrocarbon degradation rates and metal partitioning during bioremediation of contaminated anoxic marine sediments. Mar. Pollut. Bull. 2012, 64, 1688–1698. [Google Scholar] [CrossRef]
- Dell’Anno, A.; Beolchini, F.; Rocchetti, L.; Luna, G.M.; Danovaro, R. High bacterial biodiversity increases degradation performance of hydrocarbons during bioremediation of contaminated harbor marine sediments. Environ. Pollut. 2012, 167, 85–92. [Google Scholar] [CrossRef]
- Sogin, M.L.; Morrison, H.G.; Huber, J.A.; Welch, D.M.; Huse, S.M.; Neal, P.R.; Arrieta, J.M.; Herndl, G.J. Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc. Natl. Acad. Sci. USA 2006, 103, 12115–12120. [Google Scholar] [CrossRef] [PubMed]
- Louvado, A.; Gomes, N.C.M.; Simões, M.M.Q.; Almeida, A.; Cleary, D.F.R.; Cunha, A. Polycyclic aromatic hydrocarbons in deep sea sediments: Microbe-pollutant interactions in a remote environment. Sci. Total Environ. 2015, 526, 312–328. [Google Scholar] [CrossRef]
- Kostka, J.E.; Prakash, O.; Overholt, W.A.; Green, S.J.; Freyer, G.; Canion, A.; Delgardio, J.; Norton, N.; Hazen, T.C.; Huettel, M. Hydrocarbon-degrading bacteria and the bacterial community response in Gulf of Mexico beach sands impacted by the deepwater horizon oil spill. Appl. Environ. Microbiol. 2011, 77, 7962–7974. [Google Scholar] [CrossRef] [PubMed]
- Hazen, T.C.; Prince, R.C.; Mahmoudi, N. Marine Oil Biodegradation. Environ. Sci. Technol. 2016, 50, 2121–2129. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, B.B.; Boetius, A. Feast and famine—Microbial life in the deep-sea bed. Nat. Rev. Microbiol. 2007, 5, 770–781. [Google Scholar] [CrossRef]
- Zinger, L.; Amaral-Zettler, L.A.; Fuhrman, J.A.; Horner-Devine, M.C.; Huse, S.M.; Welch, D.B.M.; Martiny, J.B.H.; Sogin, M.; Boetius, A.; Ramette, A. Global patterns of bacterial beta-diversity in seafloor and seawater ecosystems. PLoS ONE 2011, 6, e24570. [Google Scholar] [CrossRef]
- Bell, S.; Gutierrez, T. Microbial Degradation of Hydrocarbons in the Marine Environment, with a Focus on the Microbial Response to the Deepwater Horizon Oil Spill. EC Microbiol. 2019, 8, 823–831. [Google Scholar]
- Joye, S.B.; Kleindienst, S.; Gilbert, J.A.; Handley, K.M.; Weisenhorn, P.; Overholt, W.A.; Kostka, J.E. Responses of microbial communities to hydrocarbon exposures. Oceanography 2016, 29, 136–149. [Google Scholar] [CrossRef]
- Cui, Z.; Lai, Q.; Dong, C.; Shao, Z. Biodiversity of polycyclic aromatic hydrocarbon-degrading bacteria from deep sea sediments of the Middle Atlantic Ridge. Environ. Microbiol. 2008, 10, 2138–2149. [Google Scholar] [CrossRef]
- Weiman, S.; Joye, S.B.; Kostka, J.E.; Halanych, K.M.; Colwell, R.R. Gomri insights into microbial genomics and hydrocarbon bioremediation response in marine ecosystems. Oceanography 2021, 34, 124–135. [Google Scholar] [CrossRef]
- Ma, M.; Gao, W.; Li, Q.; Han, B.; Zhu, A.; Yang, H.; Zheng, L. Biodiversity and oil degradation capacity of oil-degrading bacteria isolated from deep-sea hydrothermal sediments of the South Mid-Atlantic Ridge. Mar. Pollut. Bull. 2021, 171, 112770. [Google Scholar] [CrossRef] [PubMed]
- Parada, A.E.; Needham, D.M.; Fuhrman, J.A. Every base matters: Assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 2016, 18, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
- Comeau, A.M.; Douglas, G.M.; Langille, M.G.I. Microbiome Helper: A Custom and Streamlined Workflow for Microbiome Research. mSystems 2017, 2, e00127-16. [Google Scholar] [CrossRef] [PubMed]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef]
- Schubert, M.; Lindgreen, S.; Orlando, L. AdapterRemoval v2: Rapid adapter trimming, identification, and read merging. BMC Res. Notes 2016, 9, 1–7. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef]
- Pruesse, E.; Peplies, J.; Glöckner, F.O. SINA: Accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 2012, 28, 1823–1829. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, 590–596. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 1–9. [Google Scholar] [CrossRef]
- Bragança, I.; Mucha, A.P.; Tomasino, M.P.; Santos, F.; Lemos, P.C.; Delerue-Matos, C.; Domingues, V.F. Deltamethrin impact in a cabbage planted soil: Degradation and effect on microbial community structure. Chemosphere 2019, 220, 1179–1186. [Google Scholar] [CrossRef]
- Perdigão, R.; Almeida, C.M.R.; Santos, F.; Carvalho, M.F.; Mucha, A.P. Optimization of an autochthonous bacterial consortium obtained from beach sediments for bioremediation of petroleum hydrocarbons. Water 2021, 13, 66. [Google Scholar] [CrossRef]
- Wrenn, B.A.; Venosa, A.D. Selective enumeration of aromatic and aliphatic hydrocarbon degrading bacteria by a most-probable-number procedure. Can. J. Microbiol. 1996, 42, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Couto, M.N.; Borges, J.R.; Guedes, P.; Almeida, R.; Monteiro, E.; Almeida, C.M.R.; Basto, M.C.P.; Vasconcelos, M.T.S.D. An improved method for the determination of petroleum hydrocarbons from soil using a simple ultrasonic extraction and fourier transform infrared spectrophotometry. Pet. Sci. Technol. 2014, 32, 426–432. [Google Scholar] [CrossRef]
- Bardou, P.; Mariette, J.; Escudié, F.; Djemiel, C.; Klopp, C. SOFTWARE Open Access jvenn: An interactive Venn diagram viewer. BMC Bioinform. 2014, 15, 293. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Rosenberg, E.; DeLong, E.F.; Lory, S.; Stackebrandt, E.; Thompson, F. The prokaryotes: Prokaryotic communities and ecophysiology. Prokaryotes Prokaryotic Communities Ecophysiol. 2012, 1–528. [Google Scholar] [CrossRef]
- Kimes, N.E.; Callaghan, A.V.; Aktas, D.F.; Smith, W.L.; Sunner, J.; Golding, B.T.; Drozdowska, M.; Hazen, T.C.; Suflita, J.M.; Morris, P.J. Metagenomic analysis and metabolite profiling of deep-sea sediments from the Gulf of Mexico following the Deepwater Horizon oil spill. Front. Microbiol. 2013, 4. [Google Scholar] [CrossRef]
- Biddle, J.F.; White, J.R.; Teske, A.P.; House, C.H. Metagenomics of the subsurface Brazos-Trinity Basin (IODP site 1320): Comparison with other sediment and pyrosequenced metagenomes. ISME J. 2011, 5, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Ribicic, D.; McFarlin, K.M.; Netzer, R.; Brakstad, O.G.; Winkler, A.; Throne-Holst, M.; Størseth, T.R. Oil type and temperature dependent biodegradation dynamics—Combining chemical and microbial community data through multivariate analysis. BMC Microbiol. 2018, 18, 83. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Lehtovirta-Morley, L.; Liu, J.; Zheng, Y.; Lin, H.; Song, D.; Todd, J.D.; Tian, J.; Zhang, X.-H. Novel insights into the Thaumarchaeota in the deepest oceans: Their metabolism and potential adaptation mechanisms. Microbiome 2020, 8, 78. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, R.; Tao, Y.; Li, G. Archaea in wastewater treatment: Current research and emerging technology. Archaea 2021, 2018. [Google Scholar] [CrossRef]
- Chao, A.; Chazdon, R.L.; Shen, T.J. A new statistical approach for assessing similarity of species composition with incidence and abundance data. Ecol. Lett. 2005, 8, 148–159. [Google Scholar] [CrossRef]
- Kim, B.R.; Shin, J.; Guevarra, R.B.; Lee, J.H.; Kim, D.W.; Seol, K.H.; Lee, J.H.; Kim, H.B.; Isaacson, R.E. Deciphering diversity indices for a better understanding of microbial communities. J. Microbiol. Biotechnol. 2017, 27, 2089–2093. [Google Scholar] [CrossRef]
- Shade, A.; Jones, S.E.; Gregory Caporaso, J.; Handelsman, J.; Knight, R.; Fierer, N.; Gilbert, J.A. Conditionally rare taxa disproportionately contribute to temporal changes in microbial diversity. MBio 2014, 5, e01371-14. [Google Scholar] [CrossRef]
- Jousset, A.; Bienhold, C.; Chatzinotas, A.; Gallien, L.; Gobet, A.; Kurm, V.; Küsel, K.; Rillig, M.C.; Rivett, D.W.; Salles, J.F.; et al. Where less may be more: How the rare biosphere pulls ecosystems strings. ISME J. 2017, 11, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Pascoal, F.; Magalhães, C.; Costa, R. The Link Between the Ecology of the Prokaryotic Rare Biosphere and Its Biotechnological Potential. Front. Microbiol. 2020, 11, 231. [Google Scholar] [CrossRef]
- Reis, I.; Almeida, C.M.R.; Magalhães, C.M.; Cochofel, J.; Guedes, P.; Basto, M.C.P.; Bordalo, A.A.; Mucha, A.P. Bioremediation potential of microorganisms from a sandy beach affected by a major oil spill. Environ. Sci. Pollut. Res. 2014, 21, 3634–3645. [Google Scholar] [CrossRef] [PubMed]
- Bento, F.M.; Camargo, F.A.O.; Okeke, B.C.; Frankenberger, W.T. Comparative bioremediation of soils contaminated with diesel oil by natural attenuation, biostimulation and bioaugmentation. Bioresour. Technol. 2005, 96, 1049–1055. [Google Scholar] [CrossRef] [PubMed]
- Gouveia, V.; Almeida, C.M.R.; Almeida, T.; Teixeira, C.; Mucha, A.P. Indigenous microbial communities along the NW Portuguese Coast: Potential for hydrocarbons degradation and relation with sediment contamination. Mar. Pollut. Bull. 2018, 131, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Nikolopoulou, M.; Kalogerakis, N. Enhanced bioremediation of crude oil utilizing lipophilic fertilizers combined with biosurfactants and molasses. Mar. Pollut. Bull. 2008, 56, 1855–1861. [Google Scholar] [CrossRef]
- Leahy, J.G.; Colwell, R.R. Microbial degradation of hydrocarbons in the environment. Microbiol. Rev. 1990, 54, 305–315. [Google Scholar] [CrossRef]
- Potts, L.D.; Calderon, L.J.P.; Gontikaki, E.; Keith, L.; Gubry-Rangin, C.; Anderson, J.; Witte, U. Effect of spatial origin and hydrocarbon composition on bacterial consortia community structure and hydrocarbon biodegradation rates. FEMS Microbiol. Ecol. 2018, 94, 127. [Google Scholar] [CrossRef]
- Campeão, M.E.; Reis, L.; Leomil, L.; de Oliveira, L.; Otsuki, K.; Gardinali, P.; Pelz, O.; Valle, R.; Thompson, F.L.; Thompson, C.C. The deep-sea microbial community from the amazonian basin associated with oil degradation. Front. Microbiol. 2017, 8, 1–13. [Google Scholar] [CrossRef]
- Hazen, T.C.; Techtmann, S.M. Oil Biodegradation in Deep Marine Basins. Conseq. Microb. Interact. Hydrocarb. Oils Lipids Biodegrad. Bioremediat. 2019, 71–88. [Google Scholar] [CrossRef]
- Ganesh Kumar, A.; Vijayakumar, L.; Joshi, G.; Magesh Peter, D.; Dharani, G.; Kirubagaran, R. Biodegradation of complex hydrocarbons in spent engine oil by novel bacterial consortium isolated from deep sea sediment. Bioresour. Technol. 2014, 170, 556–564. [Google Scholar] [CrossRef]
- Gao, X.; Gao, W.; Cui, Z.; Han, B.; Yang, P.; Sun, C.; Zheng, L. Biodiversity and degradation potential of oil-degrading bacteria isolated from deep-sea sediments of South Mid-Atlantic Ridge. Mar. Pollut. Bull. 2015, 97, 373–380. [Google Scholar] [CrossRef]
- Chikere, C.B.; Okpokwasili, G.C.; Chikere, B.O. Monitoring of microbial hydrocarbon remediation in the soil. 3 Biotech 2011, 1, 117–138. [Google Scholar] [CrossRef] [PubMed]
- Yao, M.; Elling, F.J.; Jones, C.; Nomosatryo, S.; Long, C.P.; Crowe, S.A.; Antoniewicz, M.R.; Hinrichs, K.U.; Maresca, J.A. Heterotrophic bacteria from an extremely phosphate-poor lake have conditionally reduced phosphorus demand and utilize diverse sources of phosphorus. Environ. Microbiol. 2016, 18, 656–667. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; Park, S.E.; Kang, S.J.; Oh, T.K. Rheinheimera aquimaris sp. nov., isolated from seawater of the East Sea in Korea. Int. J. Syst. Evol. Microbiol. 2007, 57, 1386–1390. [Google Scholar] [CrossRef]
- Sun, S.; Dai, X.; Sun, J.; Bu, X.; Weng, C.; Li, H.; Zhu, H. A diketopiperazine factor from Rheinheimera aquimaris QSI02 exhibits anti-quorum sensing activity. Sci. Rep. 2016, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Cappello, S.; Volta, A.; Santisi, S.; Morici, C.; Mancini, G.; Quatrini, P.; Genovese, M.; Yakimov, M.M.; Torregrossa, M. Oil-degrading bacteria from a membrane bioreactor (BF-MBR) system for treatment of saline oily waste: Isolation, identification and characterization of the biotechnological potential. Int. Biodeterior. Biodegrad. 2016, 110, 235–244. [Google Scholar] [CrossRef]
- Nedashkovskaya, O.I.; Vancanneyt, M.; Dawyndt, P.; Engelbeen, K.; Vandemeulebroecke, K.; Cleenwerck, I.; Hoste, B.; Mergaert, J.; Tan, T.L.; Frolova, G.M.; et al. Reclassification of [Cytophaga] marinoflava Reichenbach 1989 as Leeuwenhoekiella marinoflava gen. nov., comb. nov. and description of Leeuwenhoekiella aequorea sp. nov. Int. J. Syst. Evol. Microbiol. 2005, 55, 1033–1038. [Google Scholar] [CrossRef] [PubMed]
- Viñas, M.; Sabaté, J.; Espuny, M.J.; Solanas, A.M. Bacterial community dynamics and polycyclic aromatic hydrocarbon degradation during bioremediation of heavily creosote-contaminated soil. Appl. Environ. Microbiol. 2005, 71, 7008–7018. [Google Scholar] [CrossRef]
- Feng, T.; Kim, K.H.; Jeong, S.E.; Kim, W.; Jeon, C.O. Aquicoccus porphyridii gen. nov., sp. nov., isolated from a small marine red alga, Porphyridium marinum. Int. J. Syst. Evol. Microbiol. 2018, 68, 283–288. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N | Lat (N) | Lon (W) | Depth (m) | Content |
|---|---|---|---|---|
| L1 | 33.9175 | −37.5054 | −1067 | Sediment |
| L2 | 33.92297 | −37.5103 | −1072 | Sediment |
| L3 | 33.92297 start 33.91748 end | −37.5103 start −37.5053 end | −1065 to −1073 | Composite Sediment |
| L4 | 33.91748 | −37.5053 | −1067 | Sediment |
| Isolate | Closest Identification * | Similarity (%) | Sequence Length (bp) | GenBank Acession Number | |
|---|---|---|---|---|---|
| L1 | DSH 1 | Achromobacter sp. | 100 | 1384 | MN960488 |
| DSH 2 | Sphingobium sp. | 99.71 | 1355 | MN960489 | |
| DSH 4 | Achromobacter sp. | 99.93 | 1377 | MN960490 | |
| DSH 5 | Pseudomonas sp. | 100 | 1386 | MN960491 | |
| DSH 6 | Rheinheimera aquimaris | 99.72 | 1406 | MN960492 | |
| DSH 7 | Acinetobacter venetianus | 99.86 | 1397 | MN960493 | |
| DSH 8 | Pseudomonas balearica | 99.64 | 1401 | MN960494 | |
| DSH 9 | Microbacterium testaceum | 100 | 1362 | MN960495 | |
| L2 | DSH 11 | Pseudomonas balearica | 99.86 | 1399 | MN960496 |
| DSH 12 | Pseudomonas sp. | 100 | 1367 | MN960497 | |
| DSH 13 | Pseudomonas sp. | 99.71 | 1402 | MN960498 | |
| DSH 16 | Pseudomonas sp. | 100 | 1402 | MN960499 | |
| DSH 19 | Pseudomonas sp. | 99.93 | 1402 | MN960500 | |
| L3 | DSH 20 | Pseudomonas pseudoalcaligenes | 99.86 | 1402 | MN960501 |
| DSH 21 | Pseudomonas zhaodongensis | 99.93 | 1400 | MN960502 | |
| DSH 22 | Microbacterium testaceum | 99.35 | 1378 | MN960503 | |
| L4 | DSH 26 | Agrobacterium sp. | 100 | 1350 | MN960504 |
| DSH 27 | Microbacterium testaceum | 99.93 | 1386 | MN960505 | |
| DSH 30 | Pseudomonas sp. | 99.79 | 1402 | MN960506 | |
| DSH 31 | Pseudomonas sp. | 100 | 1400 | MN960507 | |
| DSH 32 | Sphingobium sp. | 99.78 | 1353 | MN960508 | |
| DSH 33 | Pseudomonas sp. | 99.86 | 1398 | MN960509 | |
| DSH 34 | Staphylococcus hominis subsp. novobiosepticus | 99.93 | 1422 | MN960510 | |
| DSH 35 | Achromobacter sp. | 100 | 1396 | MN960511 | |
| DSH 36 | Achromobacter sp. | 99.93 | 1384 | MN960512 | |
| DSH 38 | Pseudomonas sp. | 99.93 | 1402 | MN960513 | |
| Bioaugmentation With Inoculum Pre-Grown In Acetate (BAa) | BAM 1 | Pseudomonas sp. | 99.93 | 1397 | MZ352111 |
| BAM 2 | Acinetobacter sp. | 99.93 | 1390 | MZ352118 | |
| BAM 3 | Microbacterium testaceum | 99.86 | 1382 | MZ352120 | |
| BAM 4 | Leeuwenhoekiella marinoflava | 99.57 | 1385 | MZ352121 | |
| BAM 7 | Pseudomonas sp. | 99.93 | 1403 | MZ352122 | |
| BAM 8 | Rhodobacteraceae | 100 | 1323 | MZ352123 | |
| BAM 12 | Microbacterium testaceum | 99.78 | 1391 | MZ352112 | |
| BAM 13 | Acinetobacter sp. | 99.93 | 1389 | MZ352113 | |
| BAM 15 | Achromobacter sp. | 99.93 | 1391 | MZ352114 | |
| BAM 16 | Sulfitobacter sp. | 99.95 | 1331 | MZ352115 | |
| BAM 17 | Pseudomonas sp. | 99.93 | 1403 | MZ352116 | |
| BAM 18 | Pseudomonas balearica | 99.93 | 1403 | MZ352117 | |
| BAM 23 | Pseudomonas sp. | 99.93 | 1391 | MZ352119 | |
| Bioaugmentation With Inoculum Pre-Grown In Petroleum (BAp) | BPM 1 | Achromobacter sp. | 100 | 1380 | MZ352124 |
| BPM 2 | Microbacterium testaceum | 99.85 | 1377 | MZ352131 | |
| BPM 3 | Aquicoccus sp. | 96.97 | 1321 | MZ352132 | |
| BPM 4 | Frondibacter sp. | 95.71 | 1375 | MZ352133 | |
| BPM 6 | Pseudomonas sp. | 100 | 1394 | MZ352134 | |
| BPM 7 | Pseudomonas sp. | 100 | 1396 | MZ352135 | |
| BPM 8 | Microbacterium testaceum | 99.93 | 1377 | MZ352136 | |
| BPM 9 | Acinetobacter sp. | 99.93 | 1399 | MZ352137 | |
| BPM 10 | Rhodobacteraceae | 100 | 1329 | MZ352125 | |
| BPM 11 | Achromobacter sp. | 99.31 | 1394 | MZ352126 | |
| BPM 12 | Microbacterium testaceum | 99.93 | 1384 | MZ352127 | |
| BPM 13 | Pseudooceanicola marinus | 99.92 | 1328 | MZ352128 | |
| BPM 14 | Acinetobacter sp. | 99.93 | 1402 | MZ352129 | |
| BPM 15 | Acinetobacter sp. | 99.93 | 1403 | MZ352130 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomasino, M.P.; Aparício, M.; Ribeiro, I.; Santos, F.; Caetano, M.; Almeida, C.M.R.; de Fátima Carvalho, M.; Mucha, A.P. Diversity and Hydrocarbon-Degrading Potential of Deep-Sea Microbial Community from the Mid-Atlantic Ridge, South of the Azores (North Atlantic Ocean). Microorganisms 2021, 9, 2389. https://doi.org/10.3390/microorganisms9112389
Tomasino MP, Aparício M, Ribeiro I, Santos F, Caetano M, Almeida CMR, de Fátima Carvalho M, Mucha AP. Diversity and Hydrocarbon-Degrading Potential of Deep-Sea Microbial Community from the Mid-Atlantic Ridge, South of the Azores (North Atlantic Ocean). Microorganisms. 2021; 9(11):2389. https://doi.org/10.3390/microorganisms9112389
Chicago/Turabian StyleTomasino, Maria Paola, Mariana Aparício, Inês Ribeiro, Filipa Santos, Miguel Caetano, C. Marisa R. Almeida, Maria de Fátima Carvalho, and Ana P. Mucha. 2021. "Diversity and Hydrocarbon-Degrading Potential of Deep-Sea Microbial Community from the Mid-Atlantic Ridge, South of the Azores (North Atlantic Ocean)" Microorganisms 9, no. 11: 2389. https://doi.org/10.3390/microorganisms9112389
APA StyleTomasino, M. P., Aparício, M., Ribeiro, I., Santos, F., Caetano, M., Almeida, C. M. R., de Fátima Carvalho, M., & Mucha, A. P. (2021). Diversity and Hydrocarbon-Degrading Potential of Deep-Sea Microbial Community from the Mid-Atlantic Ridge, South of the Azores (North Atlantic Ocean). Microorganisms, 9(11), 2389. https://doi.org/10.3390/microorganisms9112389