
The Disordered C-Terminus of the Chaperone DnaK Increases the Competitive Fitness of Pseudomonas putida and Facilitates the Toxicity of GraT


Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains, Plasmids, and Growth Conditions
2.2. Construction of Plasmids and Strains
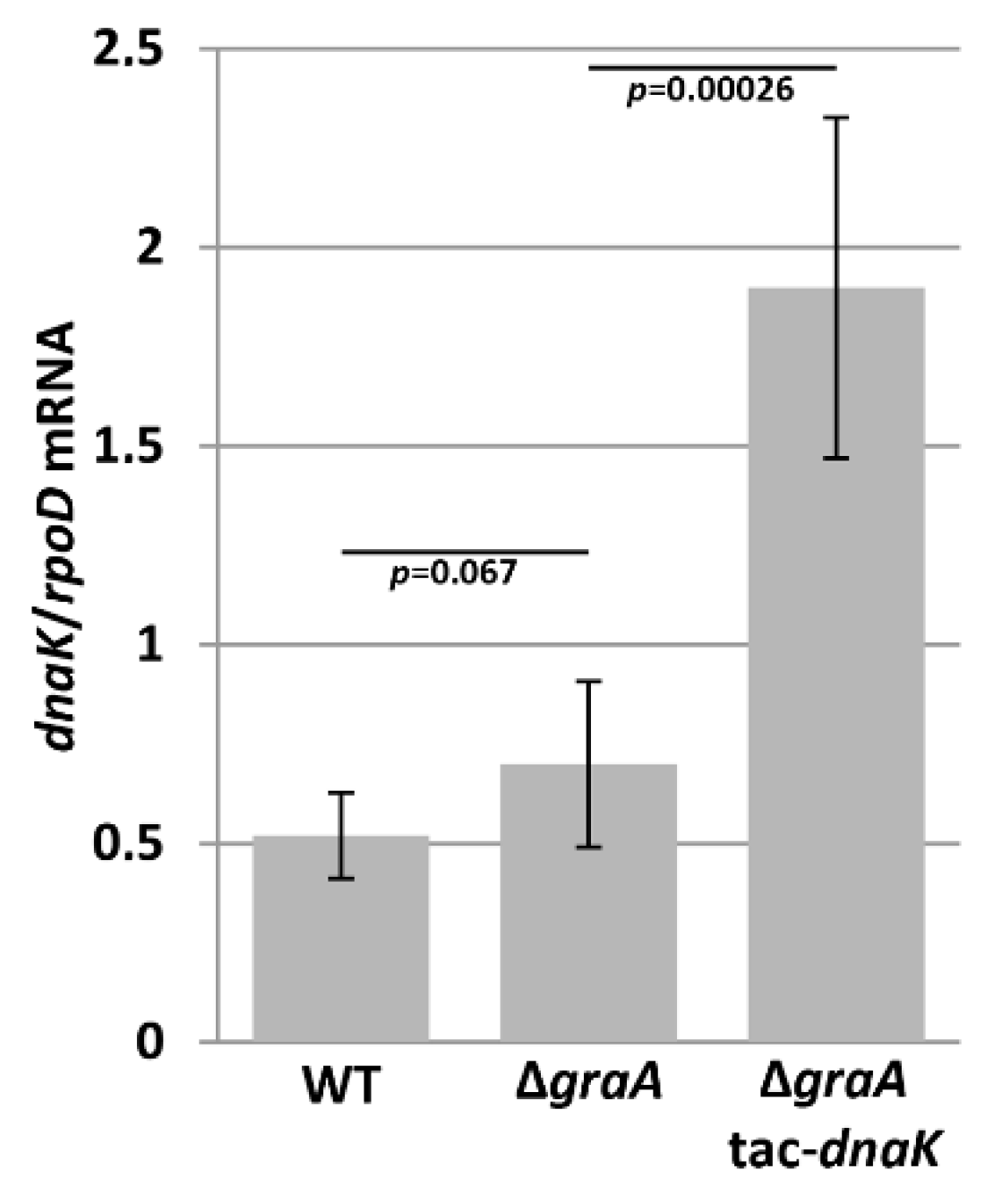
2.3. qRT-PCR
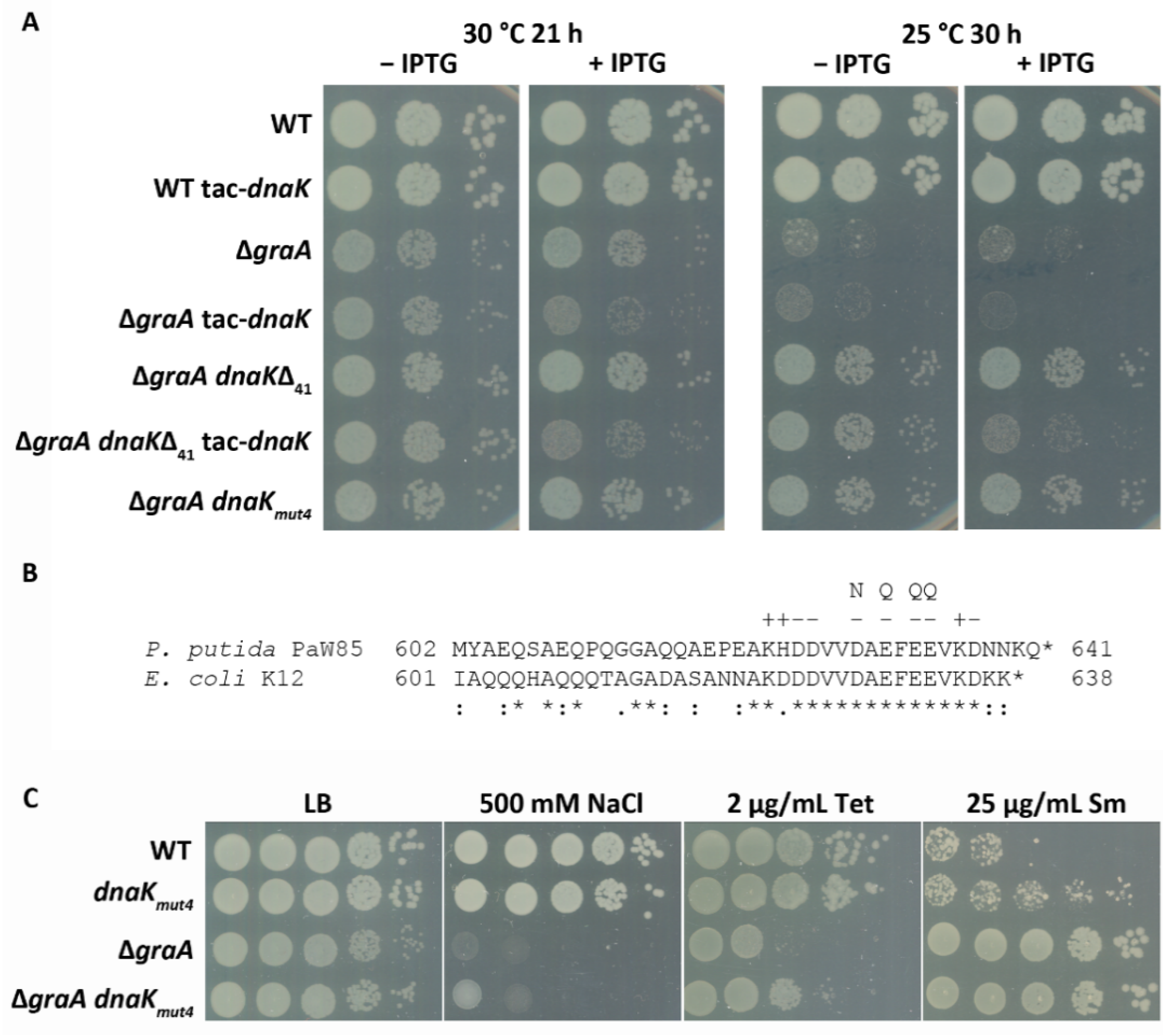
2.4. Temperature and Stress Tolerance Assays
2.5. Growth Curves
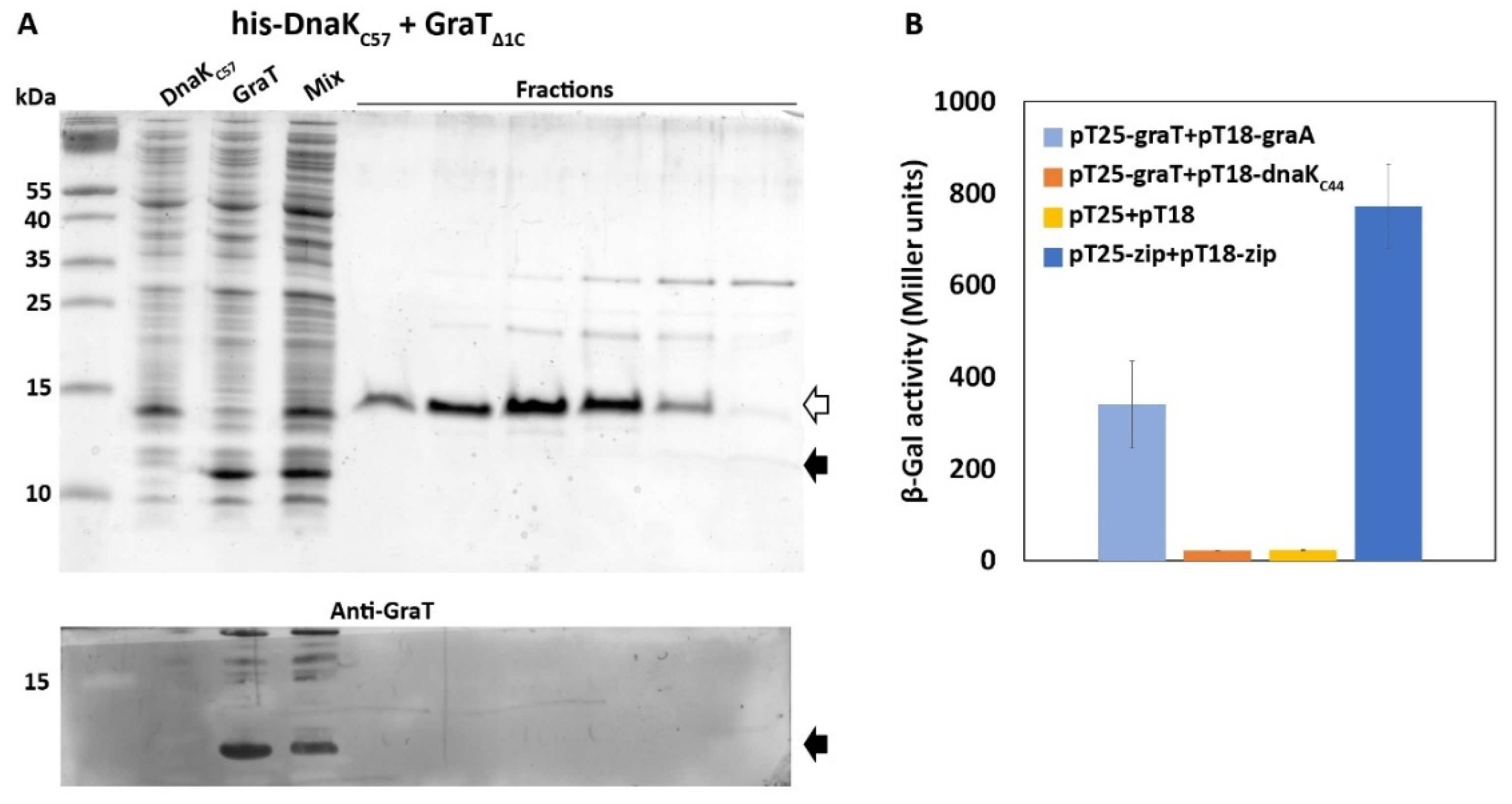
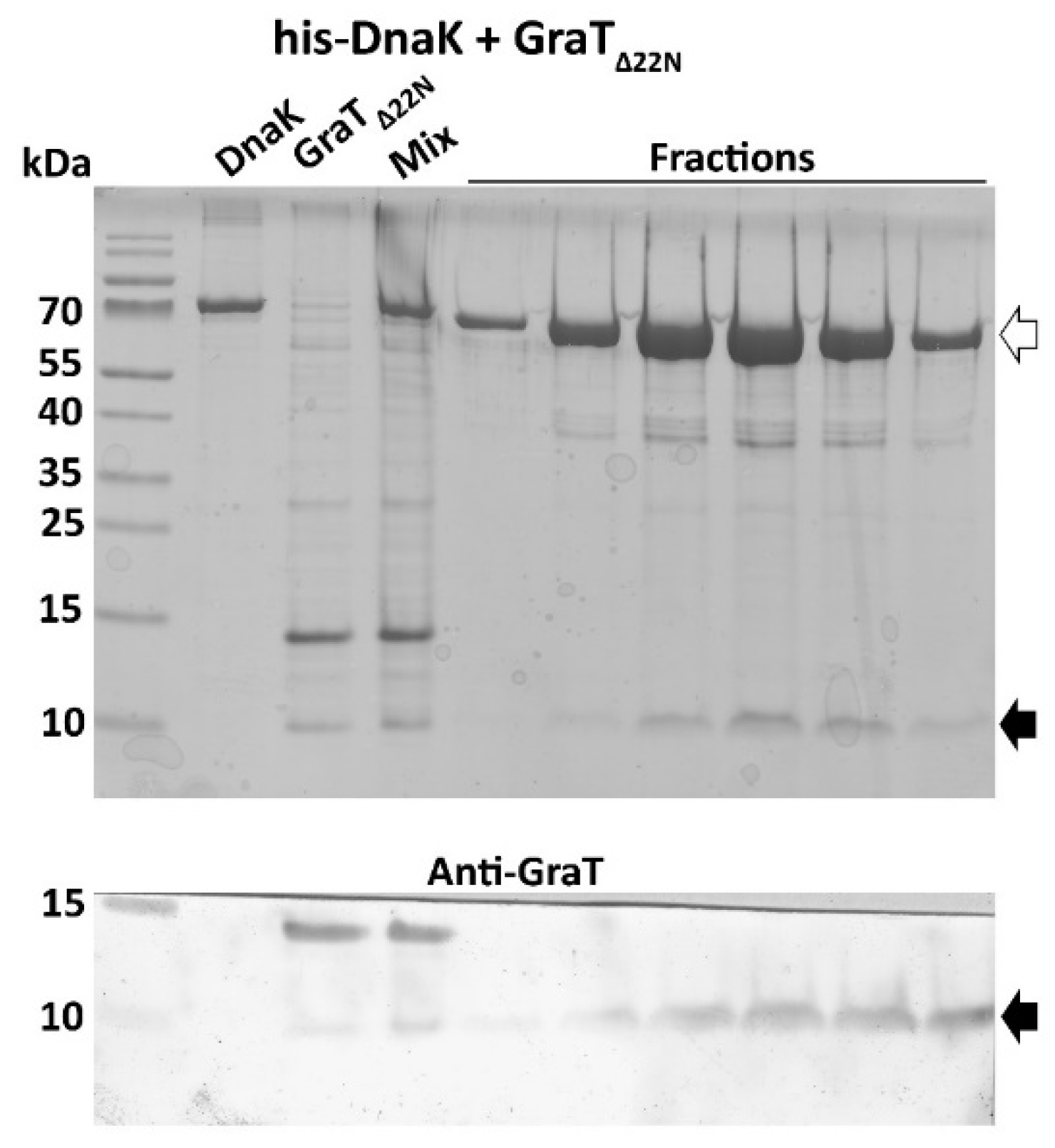
2.6. Pull-Down Assay
2.7. Bacterial Two-Hybrid Assay (BACTH)
2.8. Measurement of Heat Shock Response
2.9. Competition Assay
3. Results
3.1. DnaK mRNA Levels Are Unaltered in the ΔgraA Strain
3.2. DnaK Enhances the Toxicity of GraT
3.3. A Negatively Charged Motif in the DnaK C-Terminus Is Important for GraT Toxicity
3.4. The Disordered N-Terminus of GraT Is Not Essential for Binding to DnaK
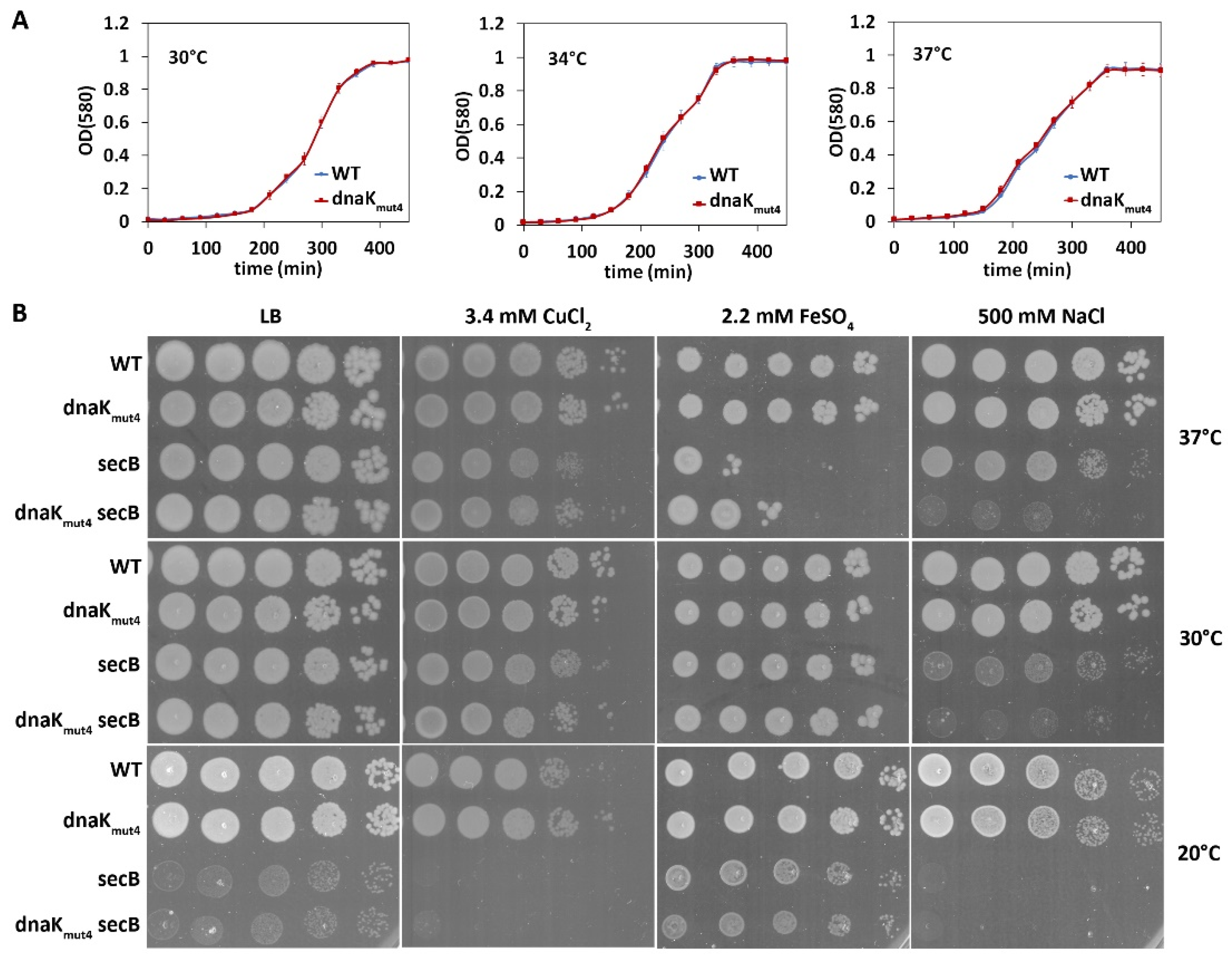
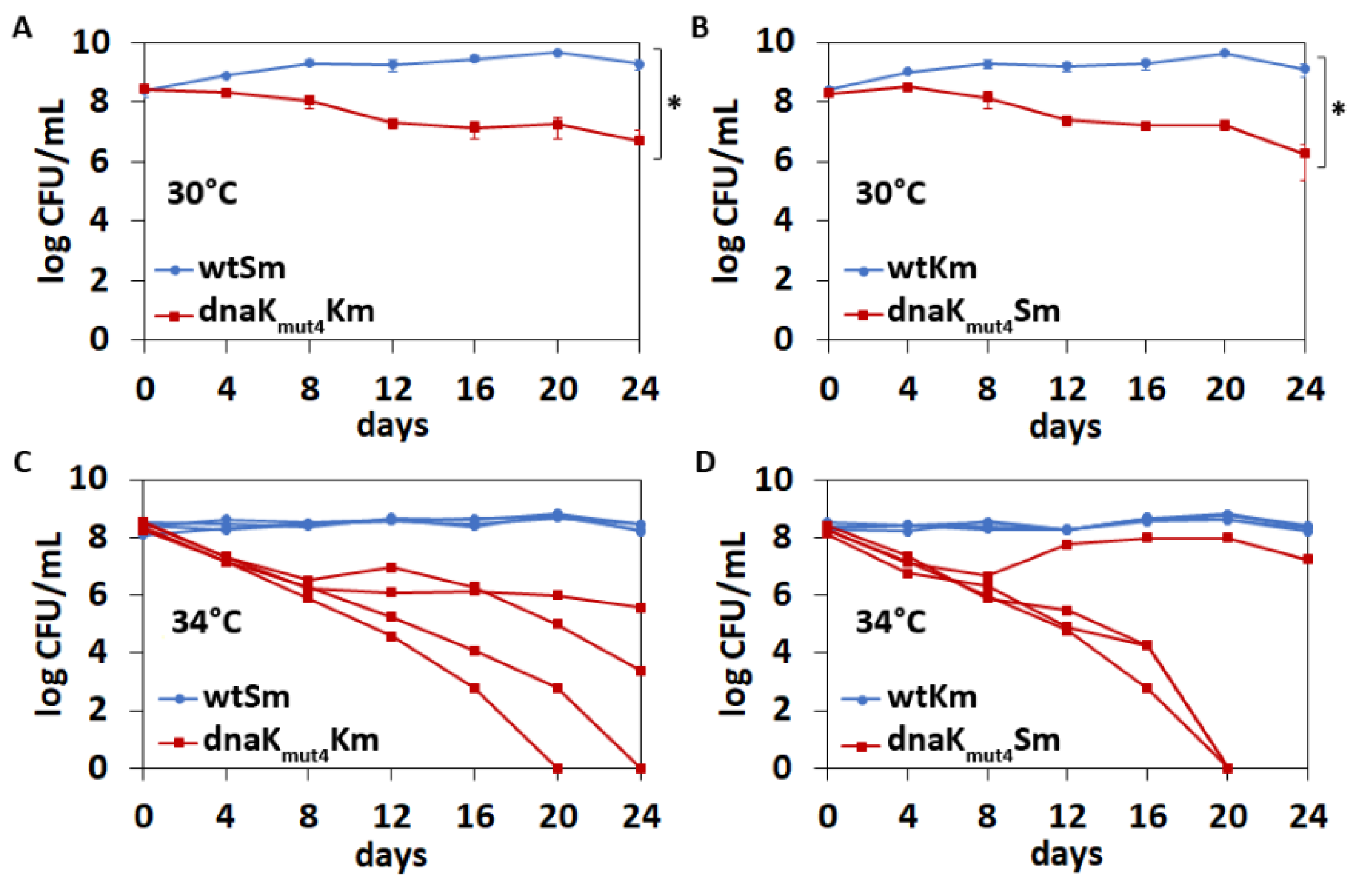
3.5. The Negatively Charged Motif in the DnaK C-Terminus Contributes to Competitive Fitness of P. putida Not Only at High but Also at Optimal Temperature
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Balchin, D.; Hayer-Hartl, M.; Hartl, F.U. In vivo aspects of protein folding and quality control. Science 2016, 353. [Google Scholar] [CrossRef] [PubMed]
- Finka, A.; Mattoo, R.U.; Goloubinoff, P. Experimental milestones in the discovery of molecular chaperones as polypeptide unfolding enzymes. Annu. Rev. Biochem. 2016, 85, 715–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bukau, B.; Walker, G.C. Cellular defects caused by deletion of the Escherichia coli DnaK gene indicate roles for heat shock protein in normal metabolism. J. Bacteriol. 1989, 171, 2337–2346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkler, J.; Seybert, A.; Konig, L.; Pruggnaller, S.; Haselmann, U.; Sourjik, V.; Weiss, M.; Frangakis, A.S.; Mogk, A.; Bukau, B. Quantitative and spatio-temporal features of protein aggregation in Escherichia coli and consequences on protein quality control and cellular ageing. EMBO J. 2010, 29, 910–923. [Google Scholar] [CrossRef] [Green Version]
- Calloni, G.; Chen, T.; Schermann, S.M.; Chang, H.C.; Genevaux, P.; Agostini, F.; Tartaglia, G.G.; Hayer-Hartl, M.; Hartl, F.U. DnaK functions as a central hub in the E. coli chaperone network. Cell Rep. 2012, 1, 251–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, M.P.; Gierasch, L.M. Recent advances in the structural and mechanistic aspects of hsp70 molecular chaperones. J. Biol. Chem. 2019, 294, 2085–2097. [Google Scholar] [CrossRef] [Green Version]
- Smock, R.G.; Blackburn, M.E.; Gierasch, L.M. Conserved, disordered c terminus of DnaK enhances cellular survival upon stress and DnaK in vitro chaperone activity. J. Biol. Chem. 2011, 286, 31821–31829. [Google Scholar] [CrossRef] [Green Version]
- Aponte, R.A.; Zimmermann, S.; Reinstein, J. Directed evolution of the DnaK chaperone: Mutations in the lid domain result in enhanced chaperone activity. J. Mol. Biol. 2010, 399, 154–167. [Google Scholar] [CrossRef]
- Rosenzweig, R.; Nillegoda, N.B.; Mayer, M.P.; Bukau, B. The hsp70 chaperone network. Nat. Rev. Mol. Cell Biol. 2019, 20, 665–680. [Google Scholar] [CrossRef]
- Kityk, R.; Vogel, M.; Schlecht, R.; Bukau, B.; Mayer, M.P. Pathways of allosteric regulation in hsp70 chaperones. Nat. Commun. 2015, 6, 8308. [Google Scholar] [CrossRef] [Green Version]
- English, C.A.; Sherman, W.; Meng, W.; Gierasch, L.M. The hsp70 interdomain linker is a dynamic switch that enables allosteric communication between two structured domains. J. Biol. Chem. 2017, 292, 14765–14774. [Google Scholar] [CrossRef] [Green Version]
- Buchberger, A.; Theyssen, H.; Schroder, H.; McCarty, J.S.; Virgallita, G.; Milkereit, P.; Reinstein, J.; Bukau, B. Nucleotide-induced conformational changes in the atpase and substrate binding domains of the DnaK chaperone provide evidence for interdomain communication. J. Biol. Chem. 1995, 270, 16903–16910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moro, F.; Fernandez, V.; Muga, A. Interdomain interaction through helices a and b of DnaK peptide binding domain. FEBS Lett. 2003, 533, 119–123. [Google Scholar] [CrossRef] [Green Version]
- Swain, J.F.; Schulz, E.G.; Gierasch, L.M. Direct comparison of a stable isolated hsp70 substrate-binding domain in the empty and substrate-bound states. J. Biol. Chem. 2006, 281, 1605–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordes, P.; Cirinesi, A.M.; Ummels, R.; Sala, A.; Sakr, S.; Bitter, W.; Genevaux, P. Secb-like chaperone controls a toxin-antitoxin stress-responsive system in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2011, 108, 8438–8443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordes, P.; Sala, A.J.; Ayala, S.; Texier, P.; Slama, N.; Cirinesi, A.M.; Guillet, V.; Mourey, L.; Genevaux, P. Chaperone addiction of toxin-antitoxin systems. Nat. Commun. 2016, 7, 13339. [Google Scholar] [CrossRef] [Green Version]
- Ainelo, A.; Tamman, H.; Leppik, M.; Remme, J.; Hõrak, R. The toxin grat inhibits ribosome biogenesis. Mol. Microbiol. 2016, 100, 719–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Comprehensive comparative-genomic analysis of type 2 toxin-antitoxin systems and related mobile stress response systems in prokaryotes. Biol. Direct 2009, 4, 19. [Google Scholar] [CrossRef] [Green Version]
- Leplae, R.; Geeraerts, D.; Hallez, R.; Guglielmini, J.; Dreze, P.; Van Melderen, L. Diversity of bacterial type II toxin-antitoxin systems: A comprehensive search and functional analysis of novel families. Nucleic Acids Res. 2011, 39, 5513–5525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuster, C.F.; Bertram, R. Toxin-antitoxin systems are ubiquitous and versatile modulators of prokaryotic cell fate. FEMS Microbiol. Lett. 2013, 340, 73–85. [Google Scholar] [CrossRef]
- Chan, W.T.; Espinosa, M.; Yeo, C.C. Keeping the wolves at bay: Antitoxins of prokaryotic type ii toxin-antitoxin systems. Front. Mol. Biosci. 2016, 3, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraikin, N.; Goormaghtigh, F.; Van Melderen, L. Type II toxin-antitoxin systems: Evolution and revolutions. J. Bacteriol. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harms, A.; Brodersen, D.E.; Mitarai, N.; Gerdes, K. Toxins, targets, and triggers: An overview of toxin-antitoxin biology. Mol. Cell 2018, 70, 768–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, R.B.; Gerdes, K. Programmed cell death in bacteria: Proteic plasmid stabilization systems. Mol. Microbiol. 1995, 17, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Van Melderen, L.; Bernard, P.; Couturier, M. Lon-dependent proteolysis of CcdA is the key control for activation of CcdB in plasmid-free segregant bacteria. Mol. Microbiol. 1994, 11, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Loris, R.; Marianovsky, I.; Lah, J.; Laeremans, T.; Engelberg-Kulka, H.; Glaser, G.; Muyldermans, S.; Wyns, L. Crystal structure of the intrinsically flexible addiction antidote maze. J. Biol. Chem. 2003, 278, 28252–28257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loris, R.; Garcia-Pino, A. Disorder- and dynamics-based regulatory mechanisms in toxin-antitoxin modules. Chem. Rev. 2014, 114, 6933–6947. [Google Scholar] [CrossRef]
- Sala, A.; Calderon, V.; Bordes, P.; Genevaux, P. Tac from Mycobacterium tuberculosis: A paradigm for stress-responsive toxin-antitoxin systems controlled by SecB-like chaperones. Cell Stress Chaperones 2013, 18, 126–135. [Google Scholar] [CrossRef] [Green Version]
- Talavera, A.; Tamman, H.; Ainelo, A.; Konijnenberg, A.; Hadzi, S.; Sobott, F.; Garcia-Pino, A.; Hõrak, R.; Loris, R. A dual role in regulation and toxicity for the disordered n-terminus of the toxin grat. Nat. Commun. 2019, 10, 972. [Google Scholar] [CrossRef] [PubMed]
- Tamman, H.; Ainelo, A.; Ainsaar, K.; Hõrak, R. A moderate toxin, GraT, modulates growth rate and stress tolerance of Pseudomonas putida. J. Bacteriol. 2014, 196, 157–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ainelo, A.; Porosk, R.; Kilk, K.; Rosendahl, S.; Remme, J.; Hõrak, R. Pseudomonas putida responds to the toxin GraT by inducing ribosome biogenesis factors and repressing tca cycle enzymes. Toxins 2019, 11, 103. [Google Scholar] [CrossRef] [Green Version]
- Brzozowska, I.; Zielenkiewicz, U. Regulation of toxin-antitoxin systems by proteolysis. Plasmid 2013, 70, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Tamman, H.; Ainelo, A.; Tagel, M.; Hõrak, R. Stability of the GraA antitoxin depends on the growth phase, ATP level, and global regulator mext. J. Bacteriol. 2016, 198, 787–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schureck, M.A.; Maehigashi, T.; Miles, S.J.; Marquez, J.; Cho, S.E.; Erdman, R.; Dunham, C.M. Structure of the Proteus vulgaris higb-(higa)2-higb toxin-antitoxin complex. J. Biol. Chem. 2014, 289, 1060–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maki, J.A.; Schnobrich, D.J.; Culver, G.M. The DnaK chaperone system facilitates 30s ribosomal subunit assembly. Mol. Cell 2002, 10, 129–138. [Google Scholar] [CrossRef]
- Al Refaii, A.; Alix, J.H. Ribosome biogenesis is temperature-dependent and delayed in Escherichia coli lacking the chaperones DnaK or DnaJ. Mol. Microbiol. 2009, 71, 748–762. [Google Scholar] [CrossRef]
- Bayley, S.A.; Duggleby, C.J.; Worsey, M.J.; Williams, P.A.; Hardy, K.G.; Broda, P. Two modes of loss of the Tol function from Pseudomonas putida mt-2. Mol. Gen. Genet. 1997, 154, 203–204. [Google Scholar] [CrossRef] [PubMed]
- Rosendahl, S.; Tamman, H.; Brauer, A.; Remm, M.; Hõrak, R. Chromosomal toxin-antitoxin systems in Pseudomonas putida are rather selfish than beneficial. Sci. Rep. 2020, 10, 9230. [Google Scholar] [CrossRef] [PubMed]
- Regenhardt, D.; Heuer, H.; Heim, S.; Fernandez, D.U.; Strömpl, C.; Moore, E.R.; Timmis, K.N. Pedigree and taxonomic credentials of Pseudomonas putida strain kt2440. Environ. Microbiol. 2002, 4, 912–915. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.C.; Schimke, R.T. Preparation of electrocompetent e. Coli using salt-free growth medium. Biotechniques 1996, 20, 42–44. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Garcia, E.; de Lorenzo, V. Engineering multiple genomic deletions in gram-negative bacteria: Analysis of the multi-resistant antibiotic profile of Pseudomonas putida kt2440. Environ. Microbiol. 2011, 13, 2702–2716. [Google Scholar] [CrossRef] [PubMed]
- Studier, F.W.; Moffatt, B.A. Use of bacteriophage t7 rna polymerase to direct selective high-level expression of cloned genes. J. Mol. Biol. 1986, 189, 113–130. [Google Scholar] [CrossRef]
- Koch, B.; Jensen, L.E.; Nybroe, O. A panel of tn7-based vectors for insertion of the gfp marker gene or for delivery of cloned DNA into gram-negative bacteria at a neutral chromosomal site. J. Microbiol. Methods 2001, 45, 187–195. [Google Scholar] [CrossRef]
- Bao, Y.; Lies, D.P.; Fu, H.; Roberts, G.P. An improved tn7-based system for the single-copy insertion of cloned genes into chromosomes of gram-negative bacteria. Gene 1991, 109, 167–168. [Google Scholar] [CrossRef]
- Wong, S.M.; Mekalanos, J.J. Genetic footprinting with mariner-based transposition in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2000, 97, 10191–10196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karimova, G.; Pidoux, J.; Ullmann, A.; Ladant, D. A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 5752–5756. [Google Scholar] [CrossRef] [Green Version]
- De Lorenzo, V.; Herrero, M.; Jakubzik, U.; Timmis, K.N. Mini-tn5 transposon derivatives for insertion mutagenesis, promoter probing, and chromosomal insertion of cloned DNA in gram-negative eubacteria. J. Bacteriol. 1990, 172, 6568–6572. [Google Scholar] [CrossRef] [Green Version]
- Pavel, H.; Forsman, M.; Shingler, V. An aromatic effector specificity mutant of the transcriptional regulator DmpR overcomes the growth constraints of Pseudomonas sp. strain CF600 on para-substituted methylphenols. J. Bacteriol. 1994, 176, 7550–7557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figurski, D.H.; Helinski, D.R. Replication of an origin-containing derivative of plasmid rk2 dependent on a plasmid function provided in trans. Proc. Natl. Acad. Sci. USA 1979, 76, 1648–1652. [Google Scholar] [CrossRef] [Green Version]
- Gawin, A.; Peebo, K.; Hans, S.; Ertesvag, H.; Irla, M.; Neubauer, P.; Brautaset, T. Construction and characterization of broad-host-range reporter plasmid suitable for on-line analysis of bacterial host responses related to recombinant protein production. Microb. Cell. Fact. 2019, 18, 80. [Google Scholar] [CrossRef] [Green Version]
- Ruijter, J.M.; Ramakers, C.; Hoogaars, W.M.; Karlen, Y.; Bakker, O.; van den Hoff, M.J.; Moorman, A.F. Amplification efficiency: Linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res. 2009, 37, e45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullers, R.S.; Ang, D.; Schwager, F.; Georgopoulos, C.; Genevaux, P. Trigger factor can antagonize both SecB and DnaK/DnaJ chaperone functions in Escherichia coli. Proc. Natl. Acad. Sci. USA 2007, 104, 3101–3106. [Google Scholar] [CrossRef] [Green Version]
- Schureck, M.A.; Dunkle, J.A.; Maehigashi, T.; Miles, S.J.; Dunham, C.M. Defining the mRNA recognition signature of a bacterial toxin protein. Proc. Natl. Acad. Sci. USA 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schureck, M.A.; Maehigashi, T.; Miles, S.J.; Marquez, J.; Dunham, C.M. mRNA bound to the 30s subunit is a higb toxin substrate. RNA 2016, 22, 1261–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bukau, B.; Walker, G.C. Mutations altering heat shock specific subunit of RNA polymerase suppress major cellular defects of E. coli mutants lacking the DnaK chaperone. EMBO J. 1990, 9, 4027–4036. [Google Scholar] [CrossRef]
- Ullers, R.S.; Luirink, J.; Harms, N.; Schwager, F.; Georgopoulos, C.; Genevaux, P. SecB is a bona fide generalized chaperone in Escherichia coli. Proc. Natl. Acad. Sci. USA 2004, 101, 7583–7588. [Google Scholar] [CrossRef] [Green Version]
- Gong, W.; Hu, W.; Xu, L.; Wu, H.; Wu, S.; Zhang, H.; Wang, J.; Jones, G.W.; Perrett, S. The c-terminal GGAP motif of Hsp70 mediates substrate recognition and stress response in yeast. J. Biol. Chem. 2018, 293, 17663–17675. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain or Plasmid | Genotype or Characteristic(s) | Source or Reference |
|---|---|---|
| E. coli strains | ||
| DH5α λpir | λpir lysogen of DH5α | [41] |
| BL21(DE3) | hsdS gal (λcI ts857 ind-1 Sam7 nin-5 lacUV5-T7 gene 1) | [42] |
| P. putida strains | ||
| PaW85 | Wild-type, isogenic to KT2440 | [37] |
| ∆graA | PaW85 ∆graA | [30] |
| tac-dnaK | PaW85 with lacIq-Ptac-dnaK expression cassette in glmS locus (Smr) | This study |
| ∆graA tac-dnaK | ∆graA with lacIq-Ptac-dnaK expression cassette in glmS locus (Smr) | This study |
| ∆graA dnaKΔ41 | ∆graA with 41 C-terminal amino acids truncated from DnaK | [17] |
| dnaKmut4 | DnaK carries substitution mutations D629N, E631Q, E633Q and E634Q | This study |
| ∆graA dnaKmut4 | ∆graA with DnaK carrying mutations D629N, E631Q, E633Q and E634Q | This study |
| secB | PaW85 secB::Sm (Smr) | This study |
| dnaKmut4 secB | dnaKmut4 with secB::Sm (Smr) | This study |
| wtSm | PaW85 with miniTn7-ΩSm in glmS locus | [38] |
| wtKm | PaW85 with miniTn7-Km in glmS locus | [38] |
| dnaKmut4Sm | dnaKmut4 with miniTn7-ΩSm in glmS locus | This study |
| dnaKmut4Km | dnaKmut4 with miniTn7-Km in glmS locus | This study |
| Plasmids | ||
| pUCNot-lacItac | pUC18Not with lacIq-Ptac cassette (Ampr) | This study |
| pUCNotlacItac-dnaK | pUC18Not with lacIq-Ptac-dnaK expression cassette (Ampr) | This study |
| pBK-miniTn7-ΩSm | pUC19-based delivery plasmid for miniTn7-ΩSm (Ampr Smr) | [43] |
| pminiTn7lacItac-dnaK | pBK-miniTn7-ΩSm with lacIq-Ptac-dnaK expression cassette (Ampr Smr) | This study |
| pBK-miniTn7-Km | pUC19-based delivery plasmid for miniTn7-Km (Ampr Kmr) | [38] |
| pUXBF13 | Plasmid coding for the Tn7 transposition proteins (Ampr mob+) | [44] |
| pEMG | Suicide plasmid containing lacZα with two flanking I-SceI sites (Kmr) | [41] |
| pSW(I-SceI) | Plasmid for I-SceI expression (Apr) | [45] |
| pEMG-dnaKmut4 | pEMG with a PCR-designed 1040 bp XbaI-KpnI insert containing substitution mutations D629N, E631Q, E633Q and E634Q in the DnaK C-terminus | This study |
| pET11c | Protein expression vector (Apr) | Lab collection |
| pET-hisDnaK | pET11c for expression of DnaK with N-terminal His6 tag (Apr) | [17] |
| pET-hisDnaKC57 | pET11c for expression of His6-DnaKC57, 57 C-terminal amino acids of DnaK are fused with N-terminal His6 tag (Apr) | This study |
| pET-graTΔ1C | pET11c for expression of GraTΔ1C (Apr) | [17] |
| pET-Δ22graT | pET11c for expression of N-terminally truncated Δ22GraT (Apr) | [29] |
| pT25 | Plasmid encoding the T25 fragment (1–224 amino acids) of cyaA (Kmr) | [46] |
| pT18 | Plasmid encoding the T18 fragment (225–399 amino acids) of cyaA (Apr) | [46] |
| pT25-zip | Plasmid encoding the T25 fragment fused with leucine zipper (Kmr) | [46] |
| pT18-zip | Plasmid encoding the T18 fragment fused with leucine zipper (Apr) | [46] |
| pT25-graT | Plasmid encoding the T25 fragment fused with graT (Kmr) | This study |
| pT18-graA | Plasmid encoding the T18 fragment fused with graA (Apr) | This study |
| pT18-dnaKC44 | Plasmid encoding the T18 fragment fused with 44 C-terminal amino acids of DnaK (Apr) | This study |
| pKS-secB | pBluescript KS containing PCR-amplified secB (PP_5053) (Ampr) | This study |
| pUTmini-Tn5Sm/Sp | Delivery plasmid for mini-Tn5Sm/Sp (Ampr Smr) | [47] |
| pKS-secB::Sm | Central 280 bp region of secB in pKS-secB is replaced by Smr gene (Ampr Smr) | This study |
| pGP704L | Delivery plasmid for homologous recombination (Ampr) | [48] |
| p704L-secB::Sm | pGP704L with Acc65I-SacI fragment of secB::Sm from pKS-secB::Sm (Ampr Smr) | This study |
| pRK2013 | Helper plasmid for conjugal transfer of pGP704L | [49] |
| pAG032 | Broad-host-range reporter plasmid carrying Pibpfxs-cfp, PrpsJ-yfp and Pm-rfp promoter/reporter pairs | [50] |
| Name | Sequence (5’-3’) a | Use |
|---|---|---|
| DnaKBam | aaggatccaaagtagtcgctgctacc | construction of pUCNotlacItac-dnaK |
| dnaJdel | aatcacgcttggacataggt | construction of pUCNotlacItac-dnaK and pT18-dnaKC44 |
| dnaKKpn | ccggtaccattcacgtgctgcaagg | construction of pEMG-dnaKmut4 |
| dnaK-3E1D | tgtctttcacttGttGgaactGggcgtTaaccacgtcat | construction of pEMG-dnaKmut4 |
| K-3E | CagttcCaaCaagtgaaagacaacaacaag | construction of pEMG-dnaKmut4 |
| dnaJXba | atgcctgcaggtcgactctagatcgacacctgcatggccatact | construction of pEMG-dnaKmut4 |
| secBalg | ctgctcgagagcaagggcgt | construction of pKS-secB |
| secBlopp | ggctctagaacgggtttgccgggag | construction of pKS-secB |
| T25-graT | aaactgcagcgattcgaagctttagctgtgc | construction of pT25-graT |
| 1586Bam | cgggatccgttcttgagcatgatgc | construction of pT25-graT |
| T18-graA | aaagtcgacgctcaagaacggtatgcgtc | construction of pT18-graA |
| 1585Bam | atggatccgtttttcgatgtcagtc | construction of pT18-graA |
| T18-dnaKsaba | tctggtaccggttgcccagaagatgta | construction of pT18-dnaKC44 |
| his-dnaK(C57) | aatcatatgcatcaccaccaccatcacgacgccaaggttgaagagct | construction of pET-hisDnaKC57 |
| dnaKSmaBam | attggatcccccgggattactgcttgttgttg | construction of pET-hisDnaKC57 |
| dnaKqkeskFw | gtgccgaagtcctgaagaaa | qRT-PCR |
| dnaKqkeskRev | ctggctgtcgttgaagtagg | qRT-PCR |
| rpoDqFw | gcaacagcagtctcgtatca | qRT-PCR |
| rpoDqRev | atgatgtcttccacctgttcc | qRT-PCR |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosendahl, S.; Ainelo, A.; Hõrak, R. The Disordered C-Terminus of the Chaperone DnaK Increases the Competitive Fitness of Pseudomonas putida and Facilitates the Toxicity of GraT. Microorganisms 2021, 9, 375. https://doi.org/10.3390/microorganisms9020375
Rosendahl S, Ainelo A, Hõrak R. The Disordered C-Terminus of the Chaperone DnaK Increases the Competitive Fitness of Pseudomonas putida and Facilitates the Toxicity of GraT. Microorganisms. 2021; 9(2):375. https://doi.org/10.3390/microorganisms9020375
Chicago/Turabian StyleRosendahl, Sirli, Andres Ainelo, and Rita Hõrak. 2021. "The Disordered C-Terminus of the Chaperone DnaK Increases the Competitive Fitness of Pseudomonas putida and Facilitates the Toxicity of GraT" Microorganisms 9, no. 2: 375. https://doi.org/10.3390/microorganisms9020375
APA StyleRosendahl, S., Ainelo, A., & Hõrak, R. (2021). The Disordered C-Terminus of the Chaperone DnaK Increases the Competitive Fitness of Pseudomonas putida and Facilitates the Toxicity of GraT. Microorganisms, 9(2), 375. https://doi.org/10.3390/microorganisms9020375