
The Adult Phenylketonuria (PKU) Gut Microbiome


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Isolation and Quantification
2.3. 16S rRNA Gene Amplification
2.4. Library Preparation and Sequencing
2.5. Sequencing Data Analysis
3. Results
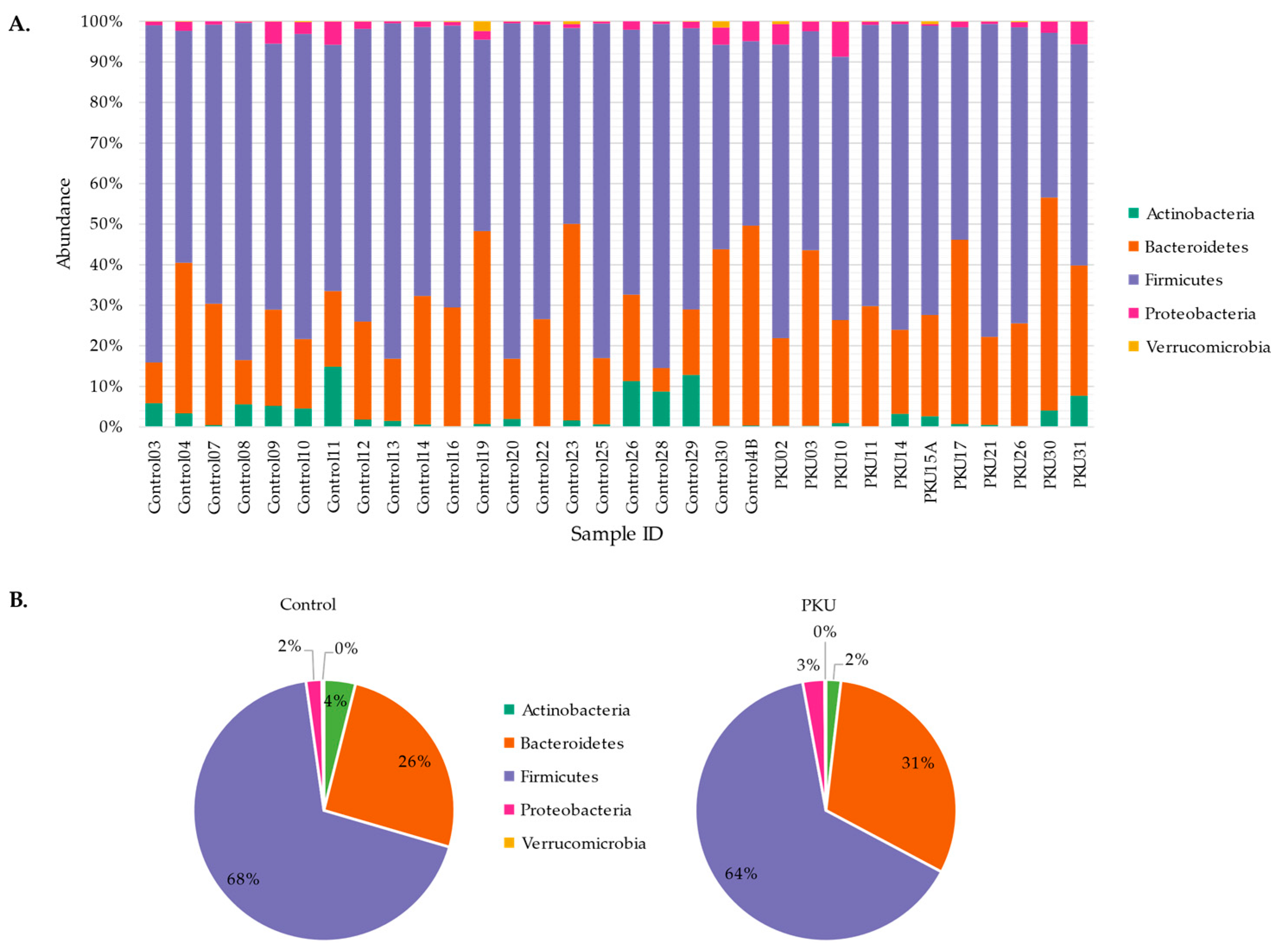
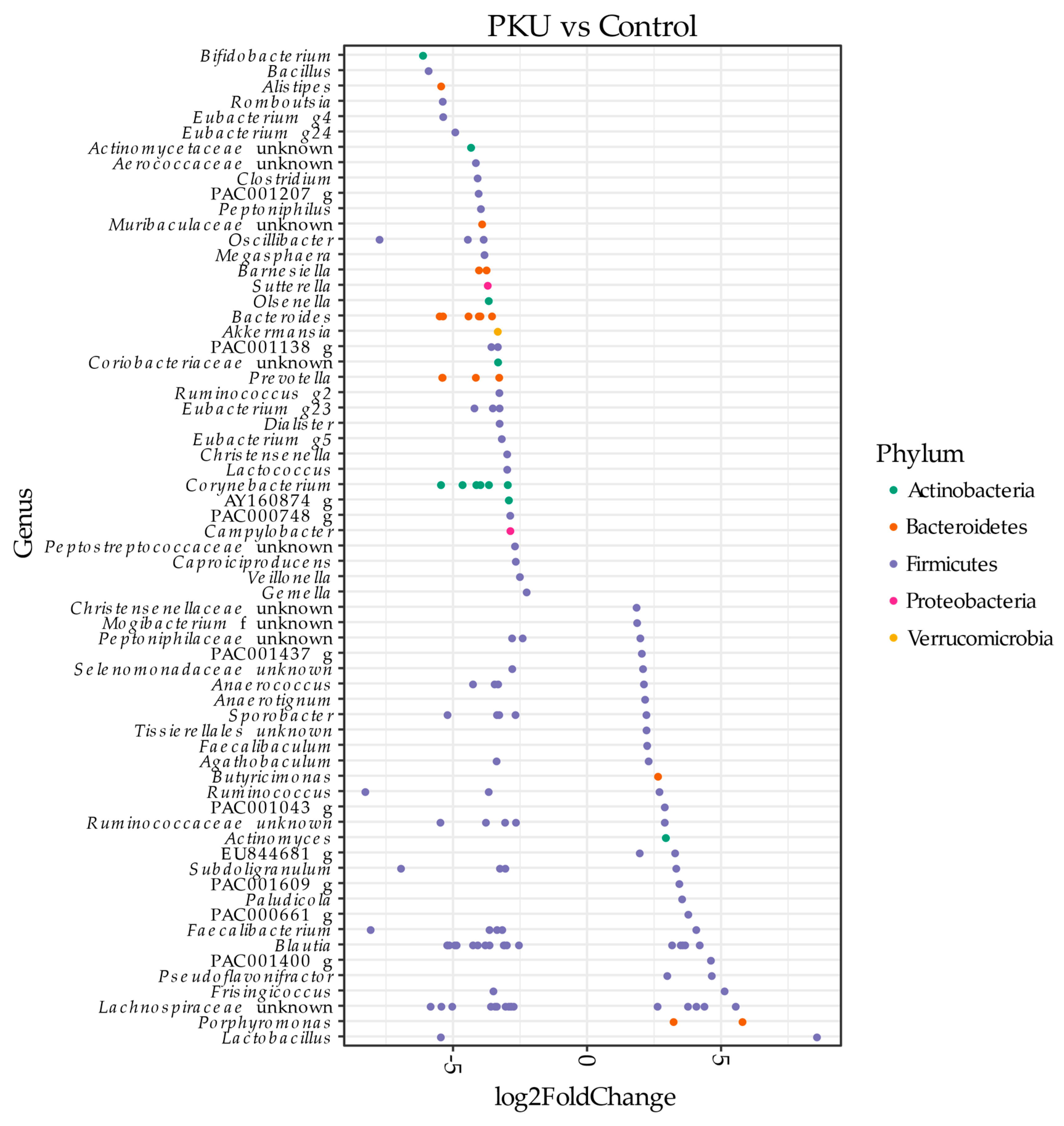
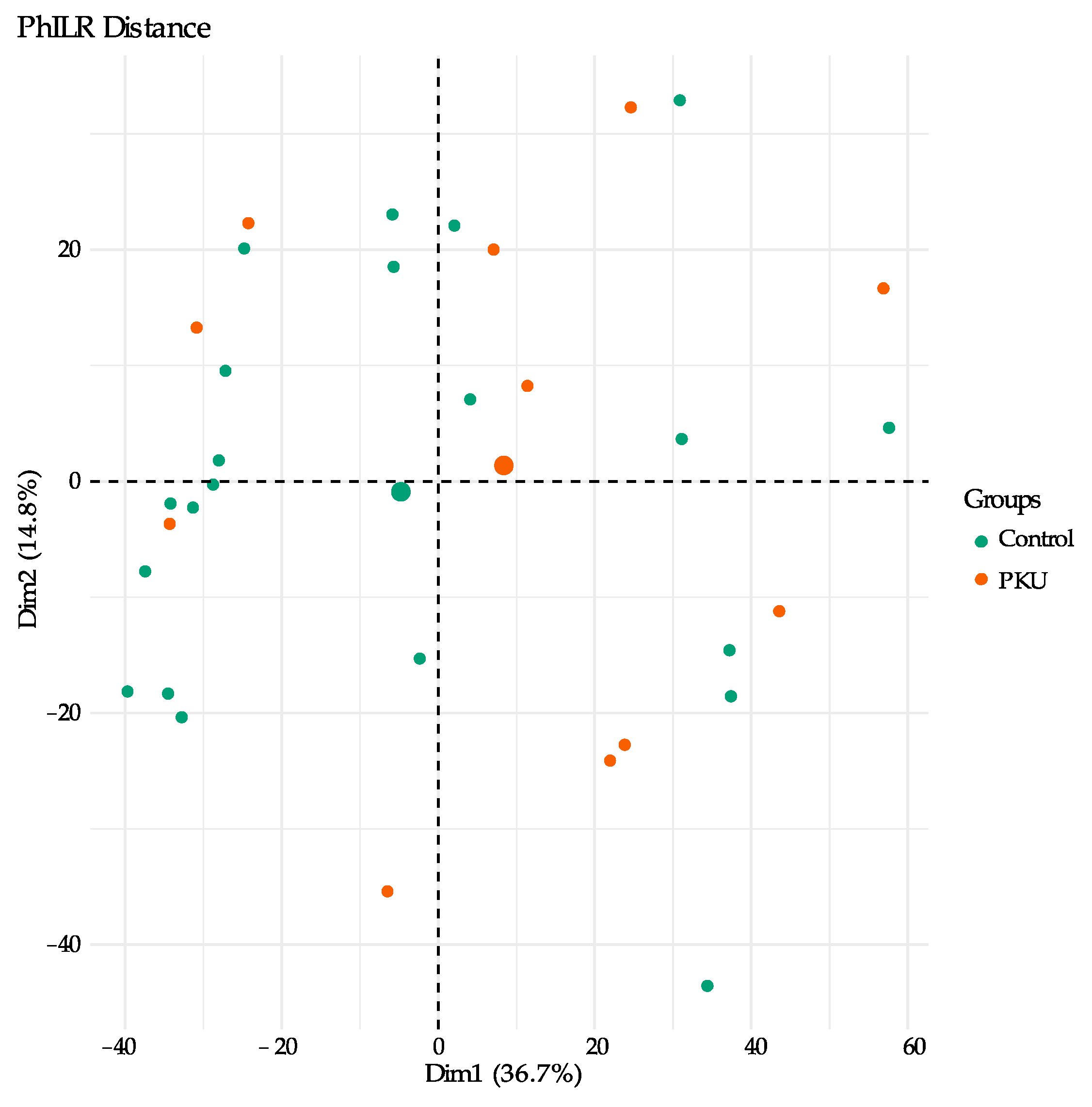
3.1. Gut Microbial Composition of PKU Patients Diverges from Control Individuals
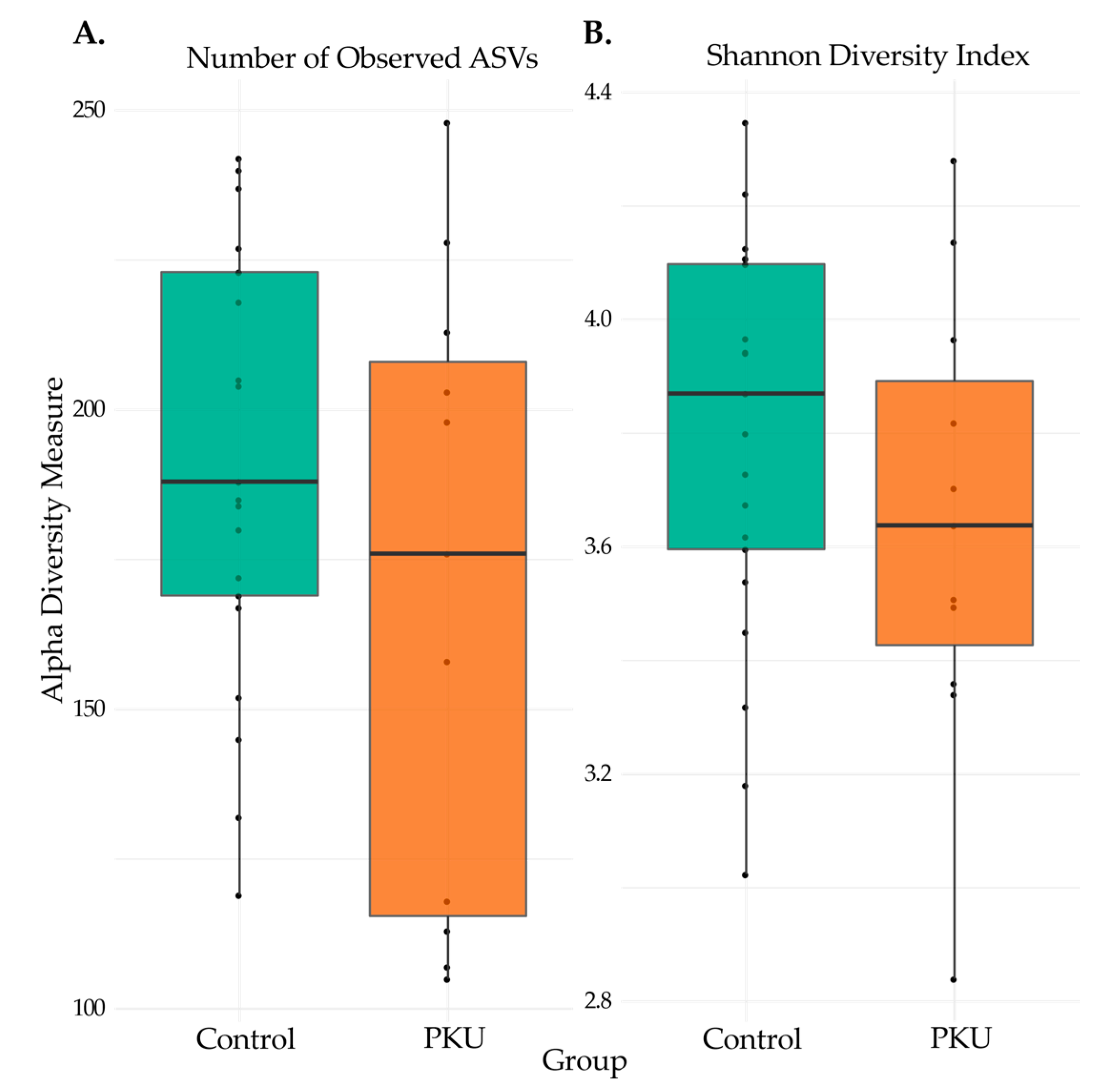
3.2. Comparison of Microbial Diversity of PKU and Control Samples
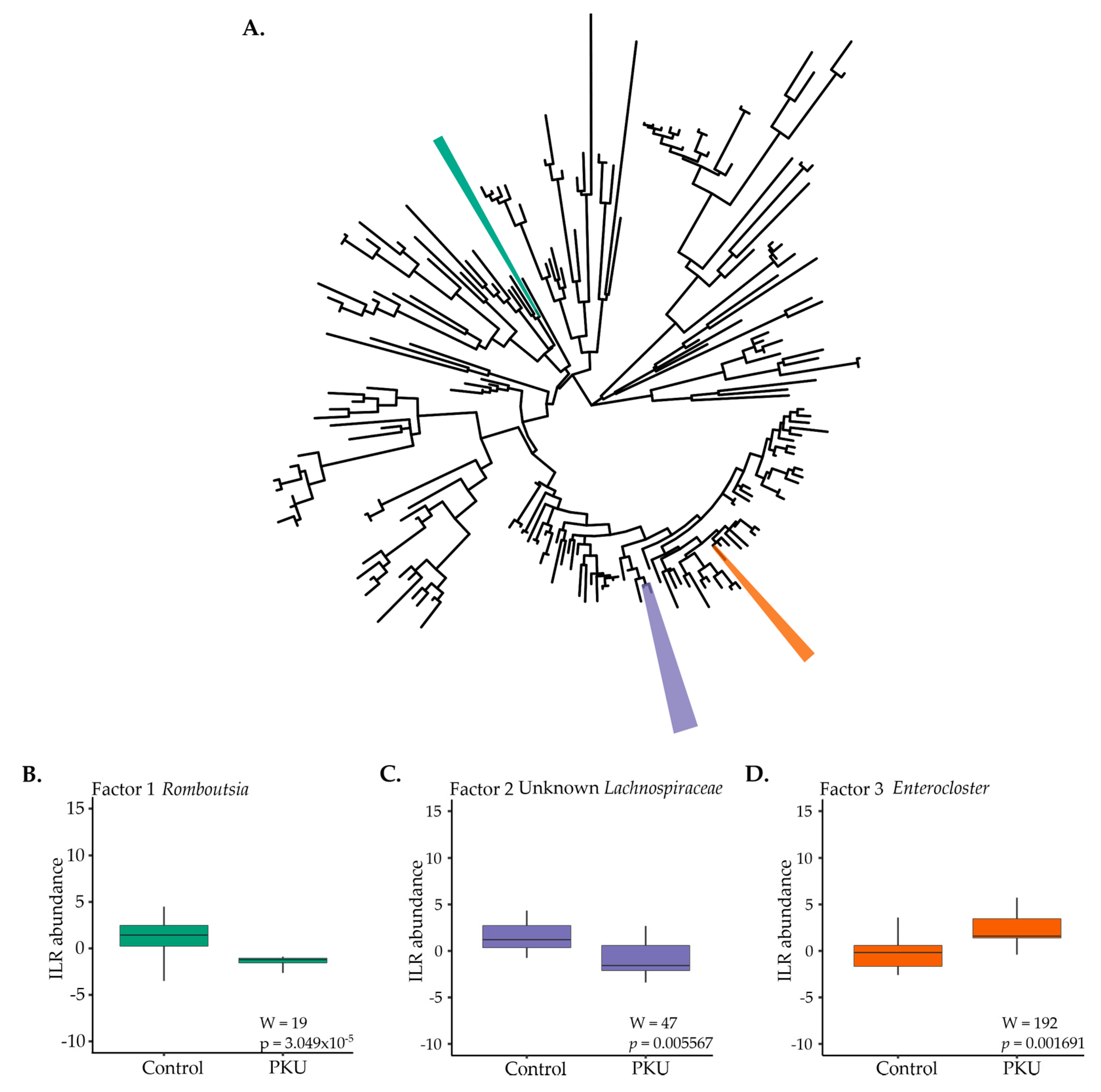
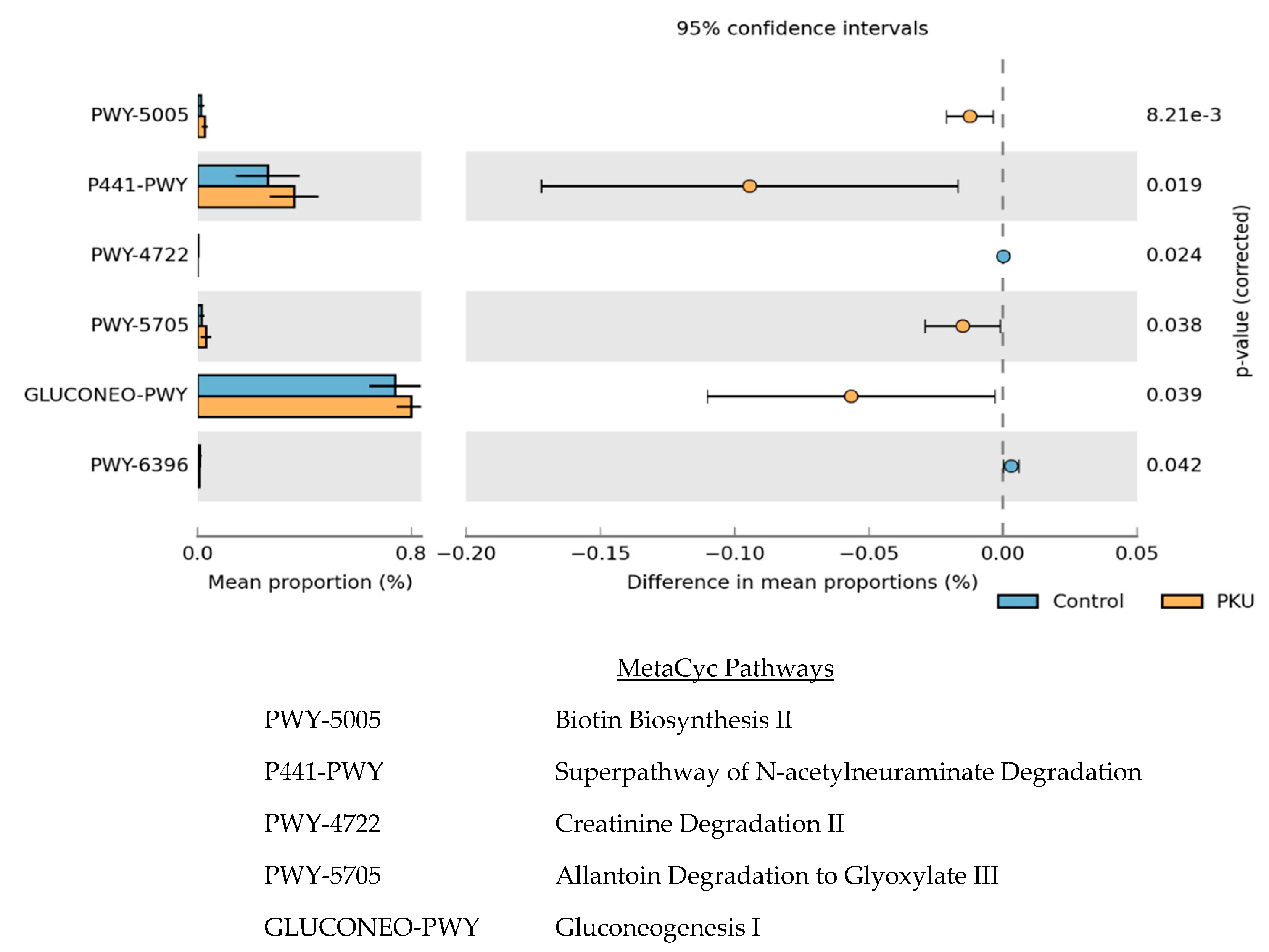
3.3. Metagenome Imputation Analysis
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sullivan, J.E.; Chang, P. Review: Emotional and behavioral functioning in phenylketonuria. J. Pediatr. Psychol. 1999, 24, 281–299. [Google Scholar] [CrossRef] [Green Version]
- Kure, S.; Hou, D.-C.; Ohura, T.; Iwamoto, H.; Suzuki, S.; Sugiyama, N.; Sakamoto, O.; Fujii, K.; Matsubara, Y.; Narisawa, K. Tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency. J. Pediatr. 1999, 135, 375–378. [Google Scholar] [CrossRef]
- Therrell, B.L.; Padilla, C.D.; Loeber, J.G.; Kneisser, I.; Saadallah, A.; Borrajo, G.J.; Adams, J. Current status of newborn screening worldwide: 2015. Semin. Perinatol. 2015, 39, 171–187. [Google Scholar] [CrossRef] [Green Version]
- Hanley, W.B. Adult phenylketonuria. Am. J. Med. 2004, 117, 590–595. [Google Scholar] [CrossRef]
- Hoeks, M.P.A.; Heijer, M.D.; Janssen, M.C.H. Adult issues in phenylketonuria. Neth. J. Med. 2009, 67, 2–7. [Google Scholar]
- Hanley, W.B.; Feigenbaum, A.S.; Clarke, J.T.; Schoonheyt, W.E.; Austin, V.J. Vitamin B12 deficiency in adolescents and young adults with phenylketonuria. Eur. J. Pediatr. 1996, 155 (Suppl. 1), S145–S147. [Google Scholar] [CrossRef]
- Acosta, P.B.; Yannicelli, S.; Singh, R.H.; Elsas, L.J., II; Mofidi, S.; Steiner, R.D. Iron status of children with phenylketonuria undergoing nutrition therapy assessed by transferrin receptors. Genet Med. 2004, 6, 96–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindeler, S.; Ghosh-Jerath, S.; Thompson, S.; Rocca, A.; Joy, P.; Kemp, A.; Rae, C.; Green, K.; Wilcken, B.; Christodoulou, J. The effects of large neutral amino acid supplements in PKU: An MRS and neuropsychological study. Mol. Genet. Metab. 2007, 91, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Belanger, A.M.; Przybylska, M.; Gefteas, E.; Furgerson, M.; Geller, S.; Kloss, A.; Cheng, S.H.; Zhu, Y.; Yew, N.S. Inhibiting neutral amino acid transport for the treatment of phenylketonuria. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Longo, N.; Harding, C.O.; Burton, B.K.; Grange, R.K.; Vockley, J.; Wasserstein, M.; Rice, G.M.; Dorenbaum, A.; Neuenburg, J.K.; Musson, D.G.; et al. Single-dose, subcutaneous recombinant phenylalanine ammonia lyase conjugated with polyethylene glycol in adult patients with phenylketonuria: An open-label, multicentre, phase 1 dose-escalation trial. Lancet 2014, 384, 37–44. [Google Scholar] [CrossRef] [Green Version]
- Durrer, K.E.; Allen, M.S.; Hunt von Herbing, I. Genetically engineered probiotic for the treatment of phenylketonuria (PKU); assessment of a novel treatment in vitro and in the PAHenu2 mouse model of PKU. PLoS ONE 2017, 12, e0176286. [Google Scholar]
- Isabella, V.M.; Ha, B.N.; Castillo, M.J.; Lubkowicz, D.J.; Rowe, S.E.; Millet, Y.A.; Anderson, C.L.; Li, N.; Fisher, A.B.; West, K.A.; et al. Development of a synthetic live bacterial therapeutic for the human metabolic disease phenylketonuria. Nat. Biotechnol. 2018, 36, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Sarkissian, C.N.; Shao, Z.; Blain, F.; Peevers, R.; Su, H.; Heft, R.; Chang, T.M.; Scriver, C.R. A different approach to treatment of phenylketonuria: Phenylalanine degradation with recombinant phenylalanine ammonia lyase. Proc. Natl. Acad. Sci. USA 1999, 96, 2339–2344. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Manichanh, C.; Borruel, N.; Casellas, F.; Guarner, F. The gut microbiota in IBD. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.L.; Gold, M.J.; Hartmann, M.; Willing, B.P.; Thorson, L.; Wlodarska, M.; Gill, N.; Blanchet, M.; Mohn, W.W.; McNagny, K.M.; et al. Early life antibiotic-driven changes in microbiota enhance susceptibility to allergic asthma. EMBO Rep. 2012, 13, 440–447. [Google Scholar] [CrossRef]
- Braniste, V.; Al-Asmakh, M.; Kowal, C.; Anuar, F.; Abbaspour, A.; Tóth, M.; Korecka, A.; Bakocevic, N.; Ng, L.G.; Kundu, P.; et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci. Transl. Med. 2014, 6, 263ra158. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Xu, X.; Li, J.; Li, F. Association Between Gut Microbiota and Autism Spectrum Disorder: A Systematic Review and Meta-Analysis. Front. Psychiatry 2019, 10, 473. [Google Scholar] [CrossRef]
- Dickerson, F.; Severance, E.; Yolken, R. The microbiome, immunity, and schizophrenia and bipolar disorder. Brain, Behav. Immun. 2017, 62, 46–52. [Google Scholar] [CrossRef] [Green Version]
- Bercik, P.; Verdu, E.F.; Foster, J.A.; Macri, J.; Potter, M.; Huang, X.; Malinowski, P.; Jackson, W.; Blennerhassett, P.; Neufeld, K.A.; et al. Chronic Gastrointestinal Inflammation Induces Anxiety-Like Behavior and Alters Central Nervous System Biochemistry in Mice. Gastroenterology 2010, 139, 2102–2112.e1. [Google Scholar] [CrossRef] [Green Version]
- Turnbaugh, P.J.; Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Knight, R.; Gordon, J.I. The Effect of Diet on the Human Gut Microbiome: A Metagenomic Analysis in Humanized Gnotobiotic Mice. Sci. Transl. Med. 2009, 1, 6ra14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, G.R.; Probert, H.M.; Van Loo, J.; Rastall, R.A.; Roberfroid, M.B. Dietary modulation of the human colonic microbiota: Updating the concept of prebiotics. Nutr. Res. Rev. 2004, 17, 259–275. [Google Scholar] [CrossRef] [Green Version]
- Bassanini, G.; Ceccarani, C.; Borgo, F.; Severgnini, M.; Rovelli, V.; Morace, G.; Verduci, E.; Borghi, E. Phenylketonuria Diet Promotes Shifts in Firmicutes Populations. Front. Cell. Infect. Microbiol. 2019, 9, 101. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, F.P.; Mendes, R.H.; Dobbler, P.T.; Mai, V.; Pylro, V.S.; Waugh, S.G.; Vairo, F.; Refosco, L.F.; Roesch, L.F.; Schwartz, I.V. Phenylketonuria and Gut Microbiota: A Controlled Study Based on Next-Generation Sequencing. PLoS ONE 2016, 11, e0157513. [Google Scholar] [CrossRef] [PubMed]
- Hemarajata, P.; Versalovic, J. Effects of probiotics on gut microbiota: Mechanisms of intestinal immunomodulation and neuromodulation. Ther. Adv. Gastroenterol. 2012, 6, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4516–4522. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silverman, J.D.; Washburne, A.D.; Mukherjee, S.; David, L.A. A phylogenetic transform enhances analysis of compositional microbiota data. eLife 2017, 6, e21887. [Google Scholar] [CrossRef]
- Oksanen, J.B.F.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P. vegan: Community Ecology Package; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]
- Anderson, M.J. Permutational Multivariate Analysis of Variance (PERMANOVA). In Wiley StatsRef: Statistics Reference Online; Wiley: Hoboken, NJ, USA, 2017; pp. 1–15. [Google Scholar]
- Washburne, A.D.; Silverman, J.D.; Leff, J.W.; Bennett, D.J.; Darcy, J.L.; Mukherjee, S.; Fierer, N.; David, L.A. Phylogenetic factorization of compositional data yields lineage-level associations in microbiome datasets. PeerJ 2017, 5, e2969. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Parks, D.H.; Beiko, R.G. Identifying biologically relevant differences between metagenomic communities. Bioinformatics 2010, 26, 715–721. [Google Scholar] [CrossRef] [Green Version]
- Haas, K.N.; Blanchard, J.L. Reclassification of the Clostridium clostridioforme and Clostridium sphenoides clades as Enterocloster gen. nov. and Lacrimispora gen. nov., including reclassification of 15 taxa. Int. J. Syst. Evol. Microbiol. 2020, 70, 23–34. [Google Scholar] [CrossRef]
- Pryde, S.E.; Duncan, S.H.; Hold, G.L.; Stewart, C.S.; Flint, H.J. The microbiology of butyrate formation in the human colon. FEMS Microbiol. Lett. 2002, 217, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Roediger, W.E. Role of anaerobic bacteria in the metabolic welfare of the colonic mucosa in man. Gut 1980, 21, 793–798. [Google Scholar] [CrossRef] [Green Version]
- McIntyre, A.; Gibson, P.R.; Young, G.P. Butyrate production from dietary fibre and protection against large bowel cancer in a rat model. Gut 1993, 34, 386–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matt, S.M.; Allen, J.M.; Lawson, M.A.; Mailing, L.J.; Woods, J.A.; Johnson, R.W. Butyrate and Dietary Soluble Fiber Improve Neuroinflammation Associated With Aging in Mice. Front. Immunol. 2018, 9, 1832. [Google Scholar] [CrossRef]
- Breyner, N.M.; Michon, C.; De Sousa, C.S.; Boas, P.C.B.V.; Chain, F.; Azevedo, V.A.; Langella, P.; Chatel, J.-M. Microbial Anti-Inflammatory Molecule (MAM) from Faecalibacterium prausnitzii Shows a Protective Effect on DNBS and DSS-Induced Colitis Model in Mice through Inhibition of NF-κB Pathway. Front. Microbiol. 2017, 8, 114. [Google Scholar] [CrossRef] [Green Version]
- Fujimoto, T.; Imaeda, H.; Takahashi, K.; Kasumi, E.; Bamba, S.; Fujiyama, Y.; Andoh, A. Decreased abundance of Faecalibacterium prausnitzii in the gut microbiota of Crohn’s disease. J. Gastroenterol. Hepatol. 2013, 28, 613–619. [Google Scholar] [CrossRef]
- Sokol, H.; Seksik, P.; Furet, J.P.; Firmesse, O.; Nion-Larmurier, I.; Beaugerie, L.; Cosnes, J.; Corthier, G.; Marteau, P.; Doré, J. Low counts of Faecalibacterium prausnitzii in colitis microbiota. Inflamm. Bowel. Dis. 2009, 15, 1183–1189. [Google Scholar] [CrossRef]
- Schirmer, M.; Franzosa, E.A.; Lloyd-Price, J.; McIver, L.J.; Schwager, R.; Poon, T.W.; Ananthakrishnan, A.N.; Andrews, E.; Barron, G.; Lake, K.; et al. Dynamics of metatranscription in the inflammatory bowel disease gut microbiome. Nat. Microbiol. 2018, 3, 337–346. [Google Scholar] [CrossRef]
- Statovci, D.; Aguilera, M.; MacSharry, J.; Melgar, S. The Impact of Western Diet and Nutrients on the Microbiota and Immune Response at Mucosal Interfaces. Front. Immunol. 2017, 8, 838. [Google Scholar] [CrossRef] [Green Version]
- Gerritsen, J.; Umanets, A.; Staneva, I.; Hornung, B.; Ritari, J.; Paulin, L.; Rijkers, G.T.; De Vos, W.M.; Smidt, H. Romboutsia hominis sp. nov., the first human gut-derived representative of the genus Romboutsia, isolated from ileostoma effluent. Int. J. Syst. Evol. Microbiol. 2018, 68, 3479–3486. [Google Scholar] [CrossRef] [PubMed]
- Gerritsen, J.; Hornung, B.; Ritari, J.; Paulin, L.; Rijkers, G.T.; Schaap, P.J.; de Vos, W.M.; Smidt, H. A comparative and functional genomics analysis of the genus Romboutsia provides insight into adaptation to an intestinal lifestyle. BioRxiv 2019, 845511. [Google Scholar]
- Vacca, M.; Celano, G.; Calabrese, F.M.; Portincasa, P.; Gobbetti, M.; De Angelis, M. The Controversial Role of Human Gut Lachnospiraceae. Microorganisms 2020, 8, 573. [Google Scholar] [CrossRef] [PubMed]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Carding, S.; Verbeke, K.; Vipond, D.T.; Corfe, B.M.; Owen, L.J. Dysbiosis of the gut microbiota in disease. Microb. Ecol. Health Dis. 2015, 26, 26191. [Google Scholar] [CrossRef] [PubMed]
- Burton, B.K.; Jones, K.B.; Cederbaum, S.; Rohr, F.; Waisbren, S.; Irwin, D.E.; Kim, G.; Lilienstein, J.; Alvarez, I.; Jurecki, E.; et al. Prevalence of comorbid conditions among adult patients diagnosed with phenylketonuria. Mol. Genet. Metab. 2018, 125, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Bilder, D.A.; Kobori, J.A.; Cohen-Pfeffer, J.L.; Johnson, E.M.; Jurecki, E.R.; Grant, M.L. Neuropsychiatric comorbidities in adults with phenylketonuria: A retrospective cohort study. Mol. Genet. Metab. 2017, 121, 1–8. [Google Scholar] [CrossRef]
- Clacy, A.; Sharman, R.; McGill, J. Depression, anxiety, and stress in young adults with phenylketonuria: Associations with biochemistry. J. Dev. Behav. Pediatr. 2014, 35, 388–391. [Google Scholar] [CrossRef]
- Kau, A.L.; Ahern, P.P.; Griffin, N.W.; Goodman, A.L.; Gordon, J.I. Human nutrition, the gut microbiome and the immune system. Nat. Cell Biol. 2011, 474, 327–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oleskin, A.V.; Shenderov, B.A. Probiotics and Psychobiotics: The Role of Microbial Neurochemicals. Probiotics Antimicrob. Proteins 2019, 11, 1071–1085. [Google Scholar] [CrossRef]
- Byers, H.L.; Homer, K.A.; Tarelli, E.; Beighton, D. N-Acetylneuraminic acid transport by Streptococcus oralis strain AR3. J. Med. Microbiol. 1999, 48, 375–381. [Google Scholar] [CrossRef] [Green Version]
- Trefz, K.F.; Muntau, A.C.; Kohlscheen, K.M.; Altevers, J.; Jacob, C.; Braun, S.; Greiner, W.; Jha, A.; Jain, M.; Alvarez, I.; et al. Clinical burden of illness in patients with phenylketonuria (PKU) and associated comorbidities—A retrospective study of German health insurance claims data. Orphanet J. Rare Dis. 2019, 14. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Observations | PKU (n = 11) | Control (n = 21) |
|---|---|---|
| Sex | ||
| Female | 4 | 11 |
| Male | 6 | 10 |
| N/R 1 | 1 | 0 |
| Average Age (years) | 33 ± 1.98 | 29 ± 3.07 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mancilla, V.J.; Mann, A.E.; Zhang, Y.; Allen, M.S. The Adult Phenylketonuria (PKU) Gut Microbiome. Microorganisms 2021, 9, 530. https://doi.org/10.3390/microorganisms9030530
Mancilla VJ, Mann AE, Zhang Y, Allen MS. The Adult Phenylketonuria (PKU) Gut Microbiome. Microorganisms. 2021; 9(3):530. https://doi.org/10.3390/microorganisms9030530
Chicago/Turabian StyleMancilla, Viviana J., Allison E. Mann, Yan Zhang, and Michael S. Allen. 2021. "The Adult Phenylketonuria (PKU) Gut Microbiome" Microorganisms 9, no. 3: 530. https://doi.org/10.3390/microorganisms9030530
APA StyleMancilla, V. J., Mann, A. E., Zhang, Y., & Allen, M. S. (2021). The Adult Phenylketonuria (PKU) Gut Microbiome. Microorganisms, 9(3), 530. https://doi.org/10.3390/microorganisms9030530