
Effects of Clinical Wastewater on the Bacterial Community Structure from Sewage to the Environment

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling Campaign
2.2. DNA Extraction and 16S rRNA Gene Amplicon Sequencing
2.3. Data Processing and Visualization
2.3.1. Selection of Target Taxa for Fate Monitoring through Wastewater Pathway
2.3.2. Indication of Temporal Effects on Phylum Abundance
3. Results and Discussion
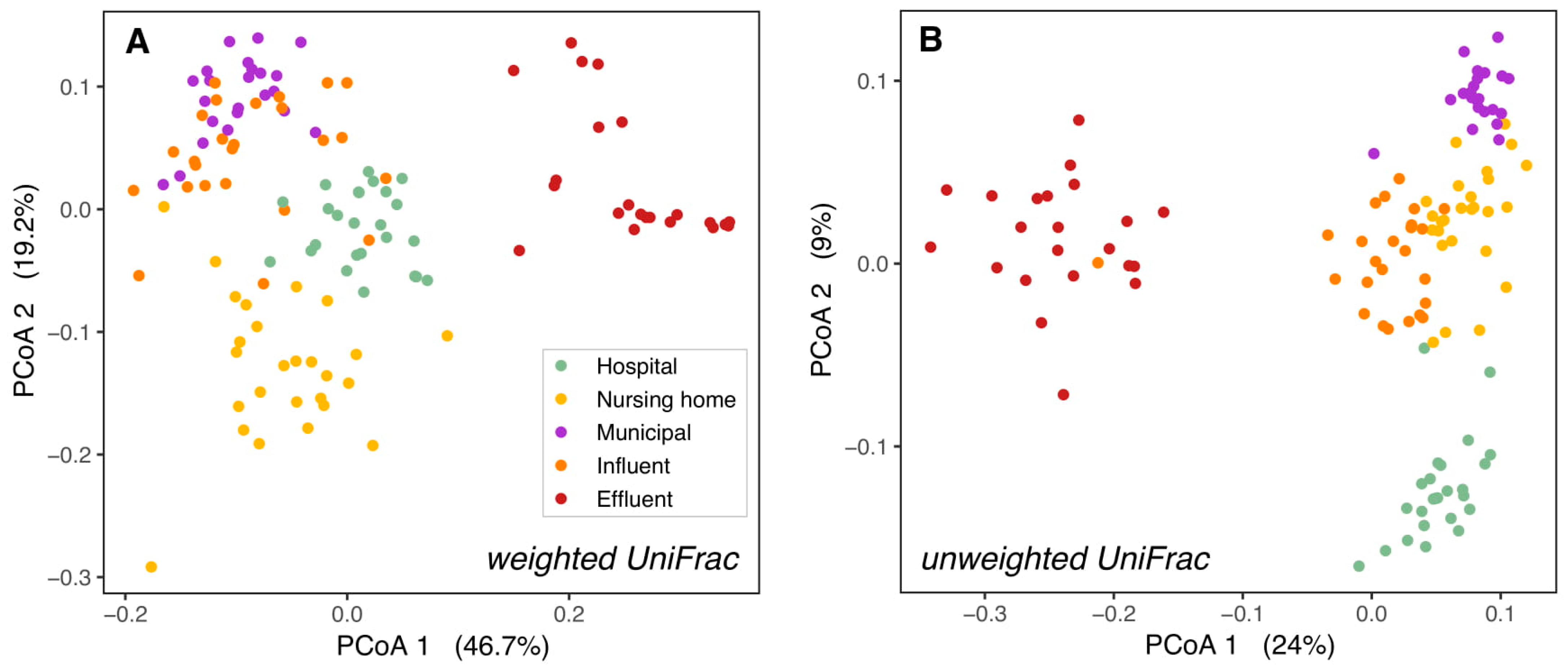
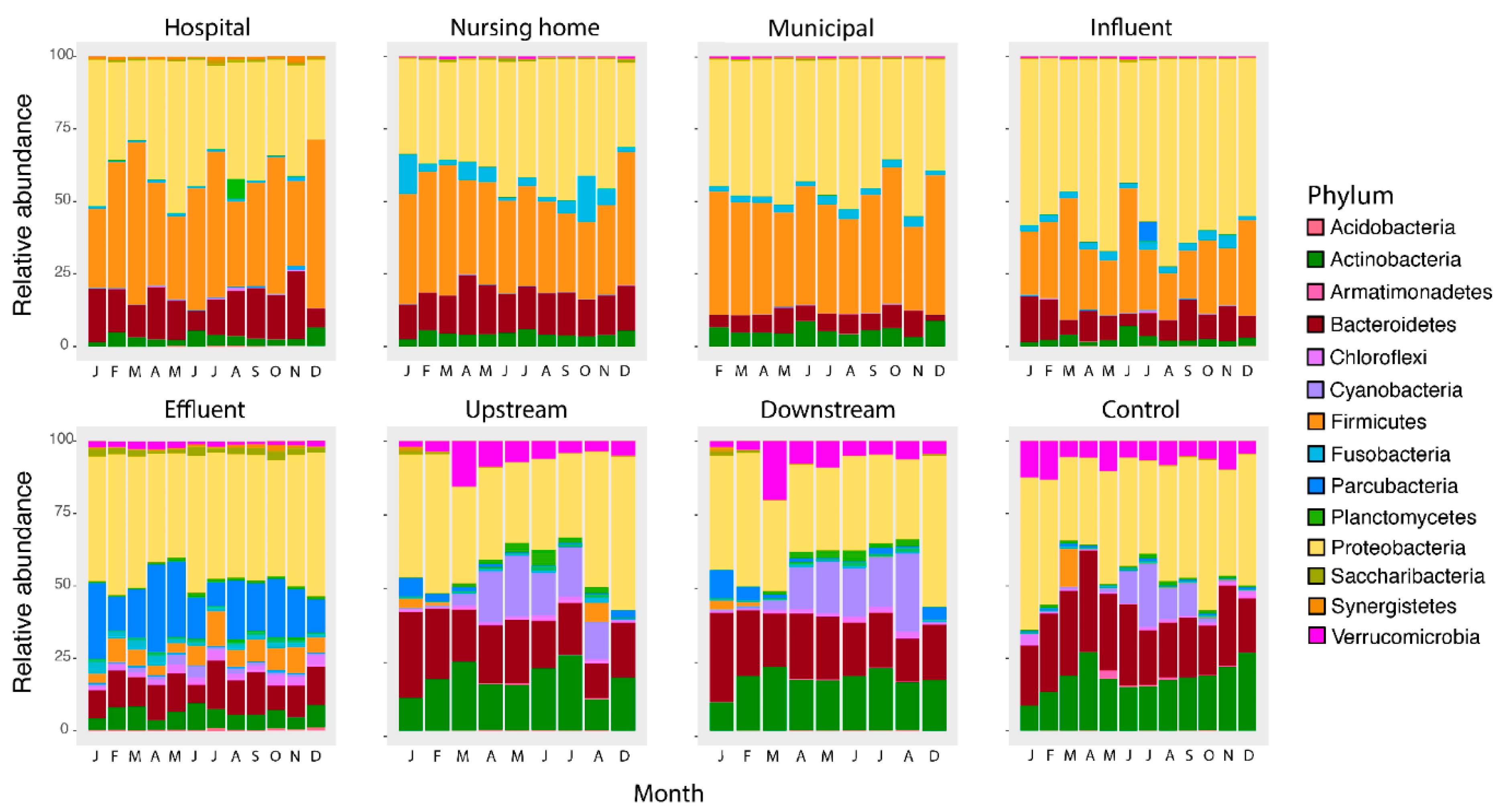
3.1. Bacterial Composition Differs between Sources and along the Studied Wastewater Pathway
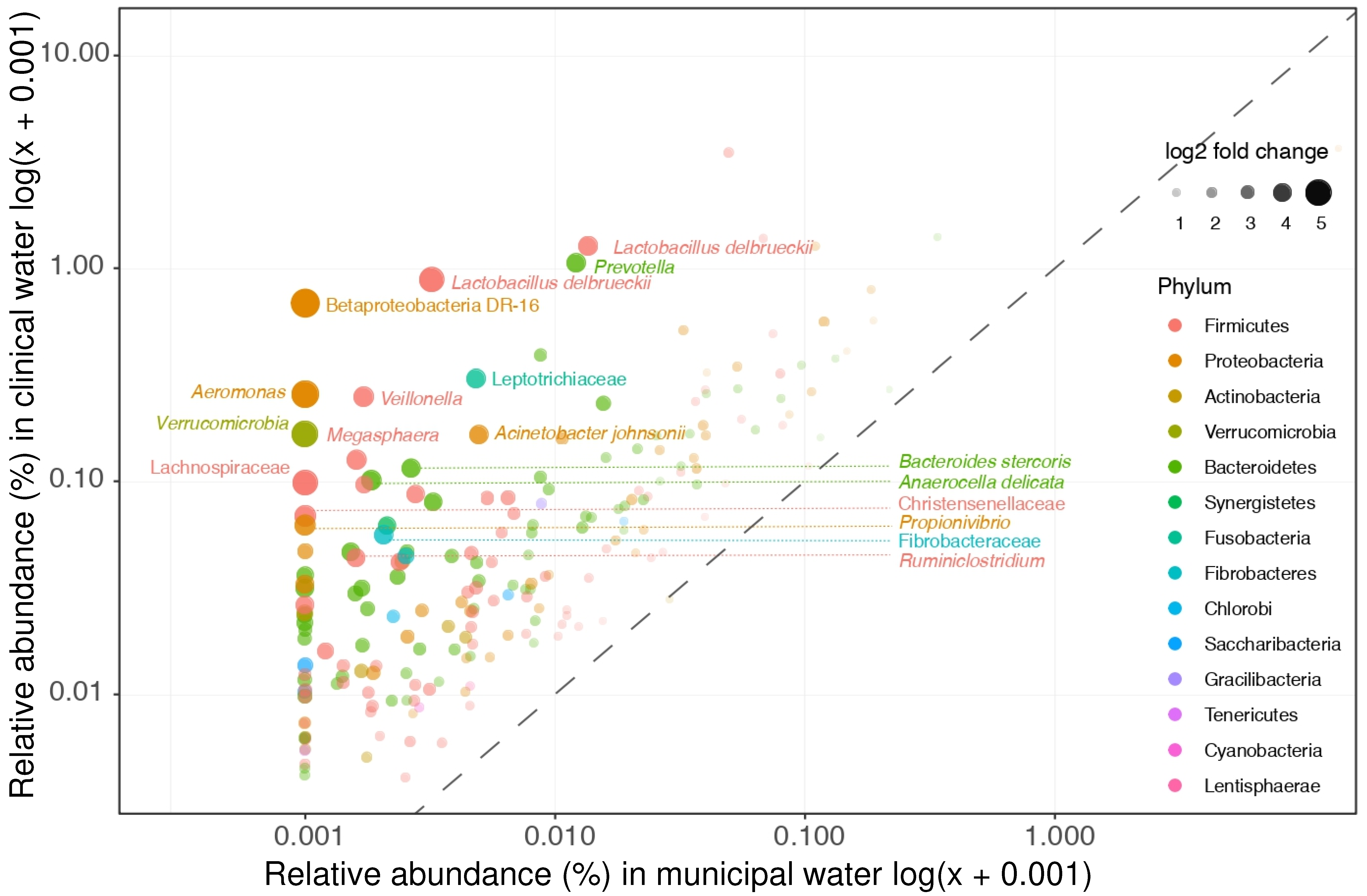
3.2. Microbial Community Differences between Clinical and Non-Clinical Wastewaters
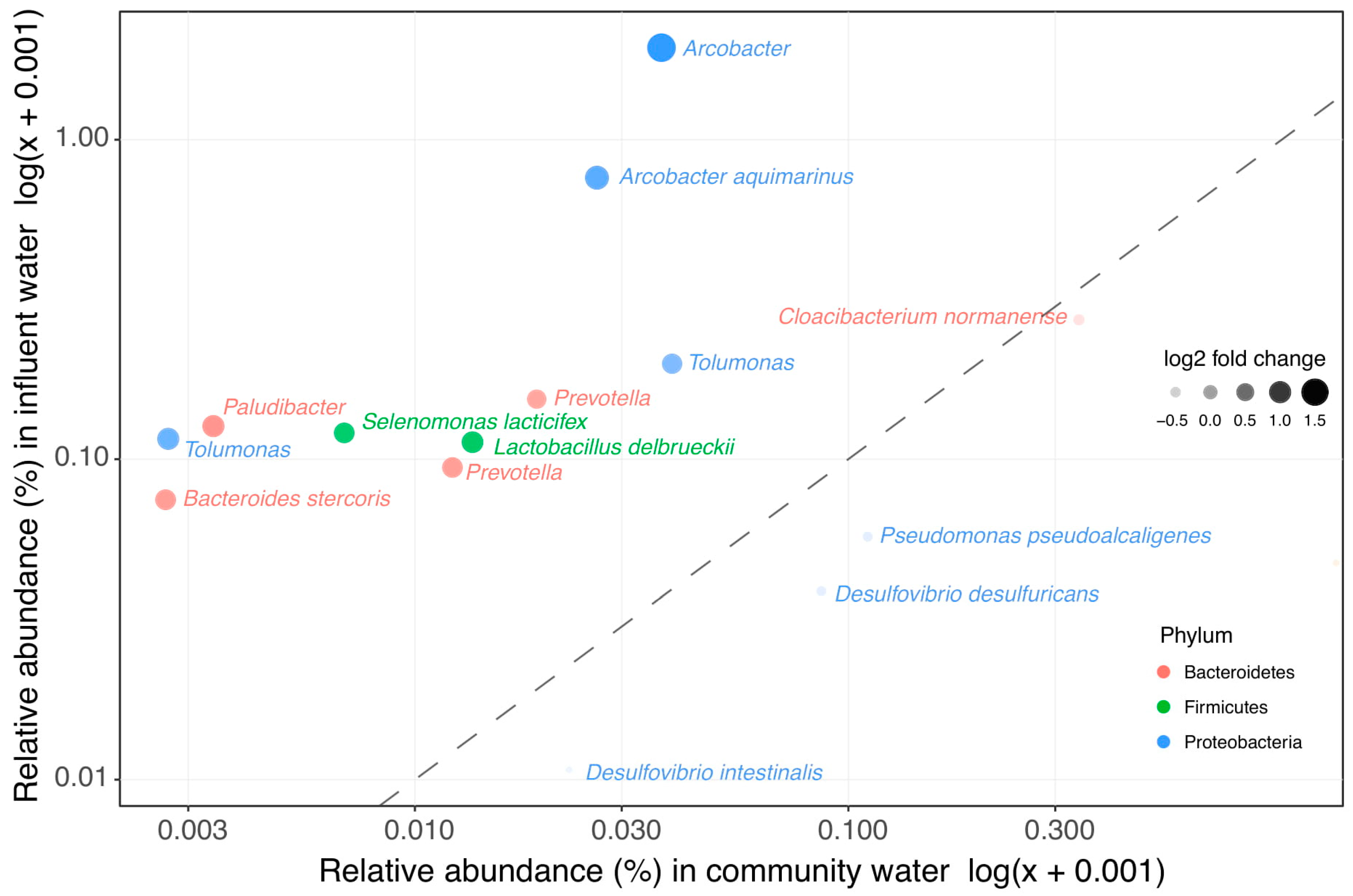
3.3. Clinical Wastewater Does Not Impact the Overall Bacterial Composition of Influent
3.4. Decrease of Relative Abundance in Most Wastewater Taxa during WWTP Treatment
3.5. The Bacterial Composition Throughout the Year Only Differs in Surface Waters
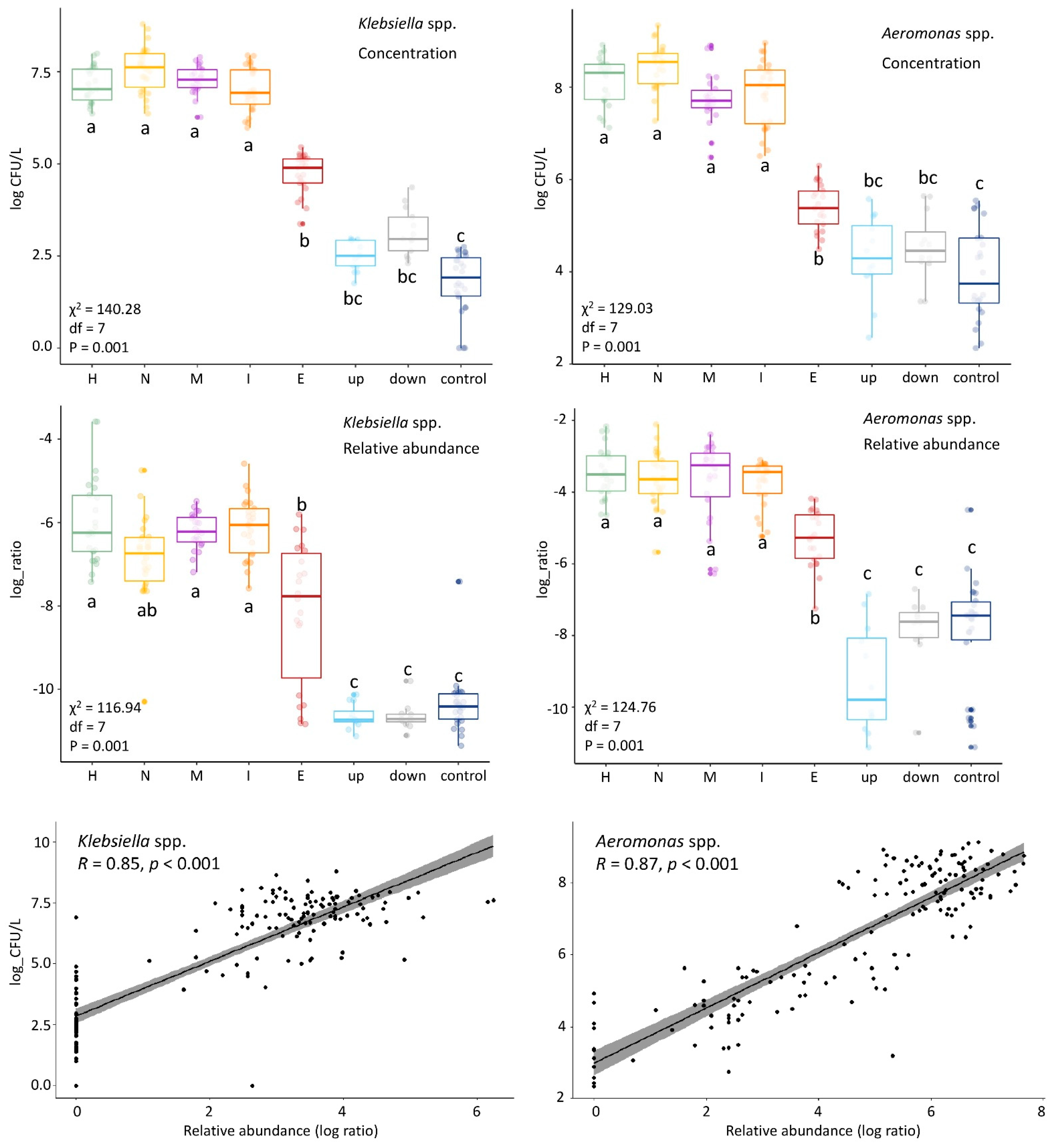
3.6. Culturing More Sensitive Than Sequencing to Detect Impact of This WWTP on Surface Water
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization (WHO). Antimicrobial Resistance: Global Report on Surveillance; World Health Organization: Geneva, Switzerland, 2014; Available online: https://scholar.google.com/scholar_lookup?title=Antimicrobial+Resistance+Global+Report+on+Surveillance&publication_year=2014& (accessed on 21 February 2018).
- Rice, L.B. Federal Funding for the Study of Antimicrobial Resistance in Nosocomial Pathogens: No ESKAPE. J. Infect. Dis. 2008, 197, 1079–1081. [Google Scholar] [CrossRef]
- Pendleton, J.N.; Gorman, S.P.; Gilmore, B.F. Clinical relevance of the ESKAPE pathogens. Expert Rev. Anti-Infect. Ther. 2013, 11, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef]
- Raymond, F.; Ouameur, A.A.; Déraspe, M.; Iqbal, N.; Gingras, H.; Dridi, B.; Leprohon, P.; Plante, P.-L.; Giroux, R.; Bérubé, È.; et al. The initial state of the human gut microbiome determines its reshaping by antibiotics. ISME J. 2016, 10, 707–720. [Google Scholar] [CrossRef]
- Hocquet, D.; Muller, A.; Bertrand, X. What happens in hospitals does not stay in hospitals: Antibiotic-resistant bacteria in hospital wastewater systems. J. Hosp. Infect. 2016, 93, 395–402. [Google Scholar] [CrossRef]
- Sandegren, L. Selection of antibiotic resistance at very low antibiotic concentrations. Upsala J. Med. Sci. 2014, 119, 103–107. [Google Scholar] [CrossRef]
- Pituch, H.; Obuch-Woszczatyński, P.; Łuczak, M.; Meisel-Mikołajczyk, F. Clostridium difficile and enterotoxigenic Bacteroides fragilis strains isolated from patients with antibiotic associated diarrhoea. Anaerobe 2003, 9, 161–163. [Google Scholar] [CrossRef]
- Panda, S.; El Khader, I.; Casellas, F.; Vivancos, J.L.; Cors, M.G.; Santiago, A.; Cuenca, S.; Guarner, F.; Manichanh, C. Short-Term Effect of Antibiotics on Human Gut Microbiota. PLoS ONE 2014, 9, e95476. [Google Scholar] [CrossRef] [PubMed]
- Larcombe, S.; Hutton, M.L.; Lyras, D. Involvement of Bacteria Other Than Clostridium difficile in Antibiotic-Associated Diar-rhoea. Trends Microbiol. 2016, 24, 463–476. [Google Scholar] [CrossRef]
- SWAB (Dutch Working Party on Antibiotic Policy). Nethmap 2018—Consumption on Antimicrobial Agents and Antimicrobial Resistance among Medically Important Bacteria in The Netherlands; RIVM: Bilthoven, The Netherlands, 2018.
- Verburg, I.; García-Cobos, S.; Leal, L.H.; Waar, K.; Friedrich, A.W.; Schmitt, H. Abundance and Antimicrobial Resistance of Three Bacterial Species along a Complete Wastewater Pathway. Microorganisms 2019, 7, 312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulus, G.K.; Hornstra, L.M.; Alygizakis, N.; Slobodnik, J.; Thomaidis, N.; Medema, G. The impact of on-site hospital wastewater treatment on the downstream communal wastewater system in terms of antibiotics and antibiotic resistance genes. Int. J. Hyg. Environ. Health 2019, 222, 635–644. [Google Scholar] [CrossRef]
- Buelow, E.; Bayjanov, J.R.; Majoor, E.; Willems, R.J.L.; Bonten, M.J.M.; Schmitt, H.; Van Schaik, W. Limited influence of hospital wastewater on the microbiome and resistome of wastewater in a community sewerage system. FEMS Microbiol. Ecol. 2018, 94. [Google Scholar] [CrossRef]
- Amador, P.P.; Fernadez, R.M.; Prudêncio, M.C.; Barreto, M.P.; Duarte, I.M. Antibiotic resistance in wastewater: Occurrence and fate of Enterobacteriaceae producers of class A and class C beta-lactamases. J. Environ. Sci. Health A Tox Hazard. Subst. Environ. Eng. 2015, 50, 26–39. [Google Scholar] [CrossRef]
- Cai, L.; Ju, F.; Zhang, T. Tracking human sewage microbiome in a municipal wastewater treatment plant. Appl. Microbiol. Biotechnol. 2013, 98, 3317–3326. [Google Scholar] [CrossRef]
- Numberger, D.; Ganzert, L.; Zoccarato, L.; Mühldorfer, K.; Sauer, S.; Grossart, H.-P.; Greenwood, A.D. Characterization of bacterial communities in wastewater with enhanced taxonomic resolution by full-length 16S rRNA sequencing. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Pruden, A.; Ashbolt, N.; Miller, J. Overview of issues for water bacterial pathogens. In Water and Sanitation for the 21st Century: Health and Microbiological Aspects of Excreta and Wastewater Management (Global Water Pathogen Project); Michigan State University Press: East Lansing, MI, USA, 2019. [Google Scholar]
- Li, T.; Abebe, L.S.; Cronk, R.; Bartram, J. A systematic review of waterborne infections from nontuberculous mycobacteria in health care facility water systems. Int. J. Hyg. Environ. Health 2017, 220, 611–620. [Google Scholar] [CrossRef] [PubMed]
- O’Dwyer, J.; Dowling, A.; Adley, C. The Impact of Climate Change on the Incidence of Infectious Waterborne Disease. Urban Water Reuse Handbook; CRC Press: Boca Raton, FL, USA, 2016; pp. 1053–1062. [Google Scholar]
- Falkinham, J.O.; Pruden, A.; Edwards, M. Opportunistic Premise Plumbing Pathogens: Increasingly Important Pathogens in Drinking Water. Pathogens 2015, 4, 373–386. [Google Scholar] [CrossRef] [Green Version]
- Cann, K.F.; Thomas, D.R.; Salmon, R.L.; Wyn-Jones, A.P.; Kay, D. Extreme water-related weather events and waterborne disease. Epidemiol. Infect. 2012, 141, 671–686. [Google Scholar] [CrossRef] [PubMed]
- Veeningen, R. and F. van der Meer, Geneesmiddelen in het oppervlaktewater: Standaarden bij de monitoring en prioritering van ’ hotspots’: [thema] nieuwe verontreinigingen. Bodem Kwart. Inf. Uitwisseling Discuss. Bodem-Bescherm. Bodemsaner. 2015, 25, 31–34. [Google Scholar]
- Illumina, 16S Metagenomic Sequencing Library Preparation; Preparing 16S Ribosomal RNA Gene Amplicons for the Illumina MiSeq System. 2013. Available online: https://www.illumina.com/content/dam/illumina-support/documents/documentation/chemistry_documentation/16s/16s-metagenomic-library-prep-guide-15044223-b.pdf (accessed on 21 February 2018).
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozupone, C.; Knight, R. UniFrac: A New Phylogenetic Method for Comparing Microbial Communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [Green Version]
- Lozupone, C.A.; Hamady, M.; Kelley, S.T.; Knight, R. Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol. 2007, 73, 1576–1585. [Google Scholar] [CrossRef] [Green Version]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package. R Package Version 2.4-2. 2017. Available online: https://cran.r-project.org/package=vegan (accessed on 30 March 2021).
- Hervé, M. RVAideMemoire: Testing and Plotting Procedures for Biostatistics. R Package Version 0.9-73. 2019. Available online: https://CRAN.R-project.org/package=RVAideMemoire (accessed on 23 January 2020).
- Pohlert, T. The Pairwise Multiple Comparison of Mean Ranks Package (PMCMR). R Package. 2014. Available online: http://CRAN.R-project.org/package=PMCMR (accessed on 23 January 2020).
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate—A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMurdie, P.J.; Holmes, S. Waste Not, Want Not: Why Rarefying Microbiome Data Is Inadmissible. PLoS Comput. Biol. 2014, 10, e1003531. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Bougouffa, S.; Wang, Y.; Lee, O.O.; Yang, J.; Chan, C.; Song, X.; Qian, P.-Y. Toward Understanding the Dynamics of Microbial Communities in an Estuarine System. PLoS ONE 2014, 9, e94449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morton, J.T.; Marotz, C.; Washburne, A.; Silverman, J.; Zaramela, L.S.; Edlund, A.; Zengler, K.; Knight, R. Establishing microbial composition measurement standards with reference frames. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Quintela-Baluja, M.; Abouelnaga, M.; Romalde, J.; Su, J.-Q.; Yu, Y.; Gomez-Lopez, M.; Smets, B.; Zhu, Y.-G.; Graham, D.W. Spatial ecology of a wastewater network defines the antibiotic resistance genes in downstream receiving waters. Water Res. 2019, 162, 347–357. [Google Scholar] [CrossRef] [PubMed]
- McLellan, S.L.; Roguet, A. The unexpected habitat in sewer pipes for the propagation of microbial communities and their imprint on urban waters. Curr. Opin. Biotechnol. 2019, 57, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Newton, R.J.; McLellan, S.L.; Dila, D.K.; Vineis, J.H.; Morrison, H.G.; Eren, A.M.; Sogin, M.L. Sewage Reflects the Microbiomes of Human Populations. mBio 2015, 6, e02574. [Google Scholar] [CrossRef] [Green Version]
- Zwart, G.; Crump, B.C.; Agterveld, M.P.K.-V.; Hagen, F.; Han, S.-K. Typical freshwater bacteria: An analysis of available 16S rRNA gene sequences from plankton of lakes and rivers. Aquat. Microb. Ecol. 2002, 28, 141–155. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.-M.; Wang, J.-Q.; Liu, T.-T.; Kong, W.-W.; Chen, N.; He, X.-Q.; Jin, Y. Bacterial communities of Beijing surface waters as revealed by 454 pyrosequencing of the 16S rRNA gene. Environ. Sci. Pollut. Res. 2015, 22, 12605–12614. [Google Scholar] [CrossRef]
- Jin, D.; Kong, X.; Cui, B.; Jin, S.; Xie, Y.; Wang, X.; Deng, Y. Bacterial communities and potential waterborne pathogens within the typical urban surface waters. Sci. Rep. 2018, 8, 13368. [Google Scholar] [CrossRef]
- Buelow, E.; Rico, A.; Gaschet, M.; Lourenco, J.; Kennedy, S.P.; Wiest, L.; Ploy, M.; Dagot, C. Classification of hospital and urban wastewater resistome and microbiota over time and their relationship to the eco-exposome. bioXriv 2019. biorxiv:10.1101/697433v2. [Google Scholar]
- Gupta, V.K.; Paul, S.; Dutta, C. Geography, Ethnicity or Subsistence-Specific Variations in Human Microbiome Composition and Diversity. Front. Microbiol. 2017, 8, 1162. [Google Scholar] [CrossRef] [Green Version]
- Vila, A.V.; Collij, V.; Sanna, S.; Sinha, T.; Imhann, F.; Bourgonje, A.R.; Mujagic, Z.; Jonkers, D.M.A.E.; Masclee, A.A.M.; Fu, J.; et al. Impact of commonly used drugs on the composition and metabolic function of the gut microbiota. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Otte, E.; Nielsen, H.L.; Hasman, H.; Fuglsang-Damgaard, D. First report of metronidazole resistant, nimD-positive, Bacteroides stercoris isolated from an abdominal abscess in a 70-year-old woman. Anaerobe 2017, 43, 91–93. [Google Scholar] [CrossRef]
- Collado, L.; Figueras, M.J. Taxonomy, Epidemiology, and Clinical Relevance of the Genus Arcobacter. Clin. Microbiol. Rev. 2011, 24, 174–192. [Google Scholar] [CrossRef] [Green Version]
- Tong, Y.; Marion, T.; Schett, G.; Luo, Y.; Liu, Y. Microbiota and metabolites in rheumatic diseases. Autoimmun. Rev. 2020, 19, 102530. [Google Scholar] [CrossRef]
- Lopetuso, L.R.; Scaldaferri, F.; Petito, V.; Gasbarrini, A. Commensal clostridia: Leading players in the maintenance of gut homeostasis. Gut Pathog. 2013, 5, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Pillai, S.D.; Widmer, K.W.; Dowd, S.E.; Ricke, S.C. Occurrence of airborne bacteria and pathogen indicators during land application of sewage sludge. Appl. Environ. Microbiol. 1996, 62, 296–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfyffer, G.E. Mycobacterium: General Characteristics, Laboratory Detection, and Staining Procedures; 15.6 Packing and Shipping Infectious Substances; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015; pp. 536–569. [Google Scholar]
- Guo, F.; Zhang, T. Profiling bulking and foaming bacteria in activated sludge by high throughput sequencing. Water Res. 2012, 46, 2772–2782. [Google Scholar] [CrossRef]
- Diederen, B. Legionella spp. and Legionnaires’ disease. J. Infect. 2008, 56, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lodder, W.J.; van den Berg, H.H.J.L.; van Leerdam, R.C.; de Roda Husman, A.M. Potentiële maatregelen tegen verspreiding van Legionella uit afvalwaterzuiveringsinstallaties. Rijksinst. Volksgezond. Milieu (RIVM) 2019. [Google Scholar] [CrossRef]
- Levett, P.N. Leptospirosis. Clin. Microbiol. Rev. 2001, 14, 296–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommer, F.; Anderson, J.M.; Bharti, R.; Raes, J.; Rosenstiel, P. The resilience of the intestinal microbiota influences health and disease. Nat. Rev. Microbiol. 2017, 15, 630–638. [Google Scholar] [CrossRef]
- Johnston, J.; LaPara, T.; Behrens, S. Composition and Dynamics of the Activated Sludge Microbiome during Seasonal Nitrifica-tion Failure. Sci. Rep. 2019, 9, 4565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battchikova, N.; Angeleri, M.; Aro, E.-M. Proteomic approaches in research of cyanobacterial photosynthesis. Photosynth. Res. 2014, 126, 47–70. [Google Scholar] [CrossRef] [PubMed]
- Kaevska, M.; Videnska, P.; Sedlar, K.; Slana, I. Seasonal changes in microbial community composition in river water studied using 454-pyrosequencing. SpringerPlus 2016, 5, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eiler, A.; Bertilsson, S. Composition of freshwater bacterial communities associated with cyanobacterial blooms in four Swedish lakes. Environ. Microbiol. 2014, 6, 1228–1243. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location | Number of Samples Sequenced | Average Number of Reads | Minimal Number of Reads | Maximal Number of Reads | Non-Rarefied ASV Count | Rarefied (to 18059) ASV Count | Average Detection Limit 1 (Log Ratio) |
|---|---|---|---|---|---|---|---|
| Hospital | 25 | 55,638 | 28,811 | 138,846 | 4920 | 4185 | −10.88 |
| Nursing home | 26 | 52,985 | 29,657 | 104,195 | 3913 | 3386 | −10.83 |
| Community | 23 | 56,871 | 31,794 | 101,117 | 3649 | 3156 | −10.91 |
| Influent | 25 | 63,326 | 34,958 | 97,559 | 6069 | 4932 | −11.01 |
| Effluent | 22 | 50,806 | 25,594 | 77,211 | 8499 | 7469 | −10.79 |
| Upstream | 12 | 44,054 | 25,061 | 68,055 | 4942 | 4359 | −10.65 |
| Downstream | 12 | 44,281 | 18,059 | 66,764 | 4980 | 4404 | −10.66 |
| Control | 26 | 37,912 | 20,363 | 85,529 | 6565 | 5851 | −10.47 |
| Average | 21 | 50,734 | 26,787 | 92,410 | 5442 | 4718 | −10.78 |
| Genus/Species | ESKAPE Pathogen | WHO Priority Pathogen for AMR 1 | AAD Associated/Increased after AB Treatment | Water-Based Pathogen | WWTP Increase |
|---|---|---|---|---|---|
| Escherichia spp./Shigella spp. 2 | [4] (a/c) | ||||
| Klebsiella spp. | [2] | [4] (a) | [12] | ||
| Enterobacter spp. | [2,1] | [4] (a) | |||
| Proteus spp. | [4] (a) | ||||
| Serratia spp. | [4] (a) | ||||
| Providencia spp. | [4] (a) | ||||
| Morganella spp. | [4] (a) | ||||
| Salmonella spp. | [4] (b) | ||||
| Mycobacterium spp. | [4] (a) | [23,25] | [15] | ||
| Enterococcus spp. | [2] | [4] (b) | |||
| Bacteroides spp. | [13,14] | ||||
| Acinetobacter baumanii | [2] | [4] (a) | |||
| Pseudomonas aeruginosa | [2] | [4] (a) | [25] | ||
| Staphylococcus aureus | [2] | [4] (b) | [12] | ||
| Helicobacter pylori | [4] (b) | [24] | |||
| Campylobacter spp. | [4] (b) | ||||
| Neisseria gonorrhoeae | [4] (b) | ||||
| Streptococcus pneumoniae | [4] (c) | ||||
| Haemophilus influenzae | [4] (c) | ||||
| Clostridium spp. | [12,14] | [15] | |||
| Aeromonas spp. | [24] | ||||
| Legionella spp. | [25] | [16] | |||
| Leptospira spp. | [26] | [16] | |||
| Vibrio spp. | [26] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verburg, I.; van Veelen, H.P.J.; Waar, K.; Rossen, J.W.A.; Friedrich, A.W.; Hernández Leal, L.; García-Cobos, S.; Schmitt, H. Effects of Clinical Wastewater on the Bacterial Community Structure from Sewage to the Environment. Microorganisms 2021, 9, 718. https://doi.org/10.3390/microorganisms9040718
Verburg I, van Veelen HPJ, Waar K, Rossen JWA, Friedrich AW, Hernández Leal L, García-Cobos S, Schmitt H. Effects of Clinical Wastewater on the Bacterial Community Structure from Sewage to the Environment. Microorganisms. 2021; 9(4):718. https://doi.org/10.3390/microorganisms9040718
Chicago/Turabian StyleVerburg, Ilse, H. Pieter J. van Veelen, Karola Waar, John W. A. Rossen, Alex W. Friedrich, Lucia Hernández Leal, Silvia García-Cobos, and Heike Schmitt. 2021. "Effects of Clinical Wastewater on the Bacterial Community Structure from Sewage to the Environment" Microorganisms 9, no. 4: 718. https://doi.org/10.3390/microorganisms9040718
APA StyleVerburg, I., van Veelen, H. P. J., Waar, K., Rossen, J. W. A., Friedrich, A. W., Hernández Leal, L., García-Cobos, S., & Schmitt, H. (2021). Effects of Clinical Wastewater on the Bacterial Community Structure from Sewage to the Environment. Microorganisms, 9(4), 718. https://doi.org/10.3390/microorganisms9040718