
Mind the Gap: New Full-Length Sequences of Blastocystis Subtypes Generated via Oxford Nanopore Minion Sequencing Allow for Comparisons between Full-Length and Partial Sequences of the Small Subunit of the Ribosomal RNA Gene


Abstract
:1. Introduction
2. Materials and Methods
2.1. Source of Blastocystis Isolates
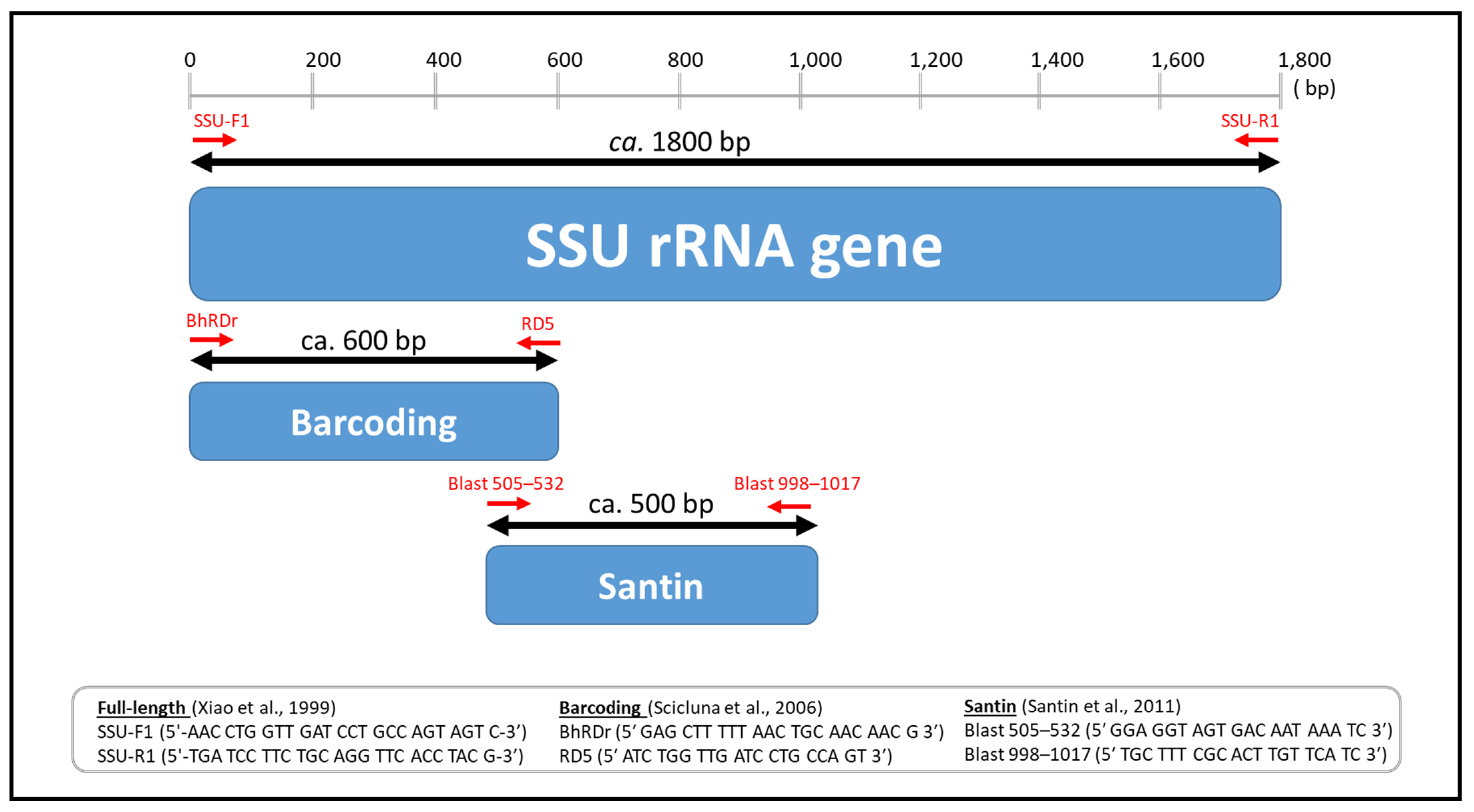
2.2. PCR Amplification of the Full-Length SSU rRNA Gene
2.3. Phylogenetic and Pairwise Distance Analyses
3. Results
3.1. Full-Length SSU rRNA Gene for ST21, ST23, ST24, ST25, ST26, ST27, and ST28
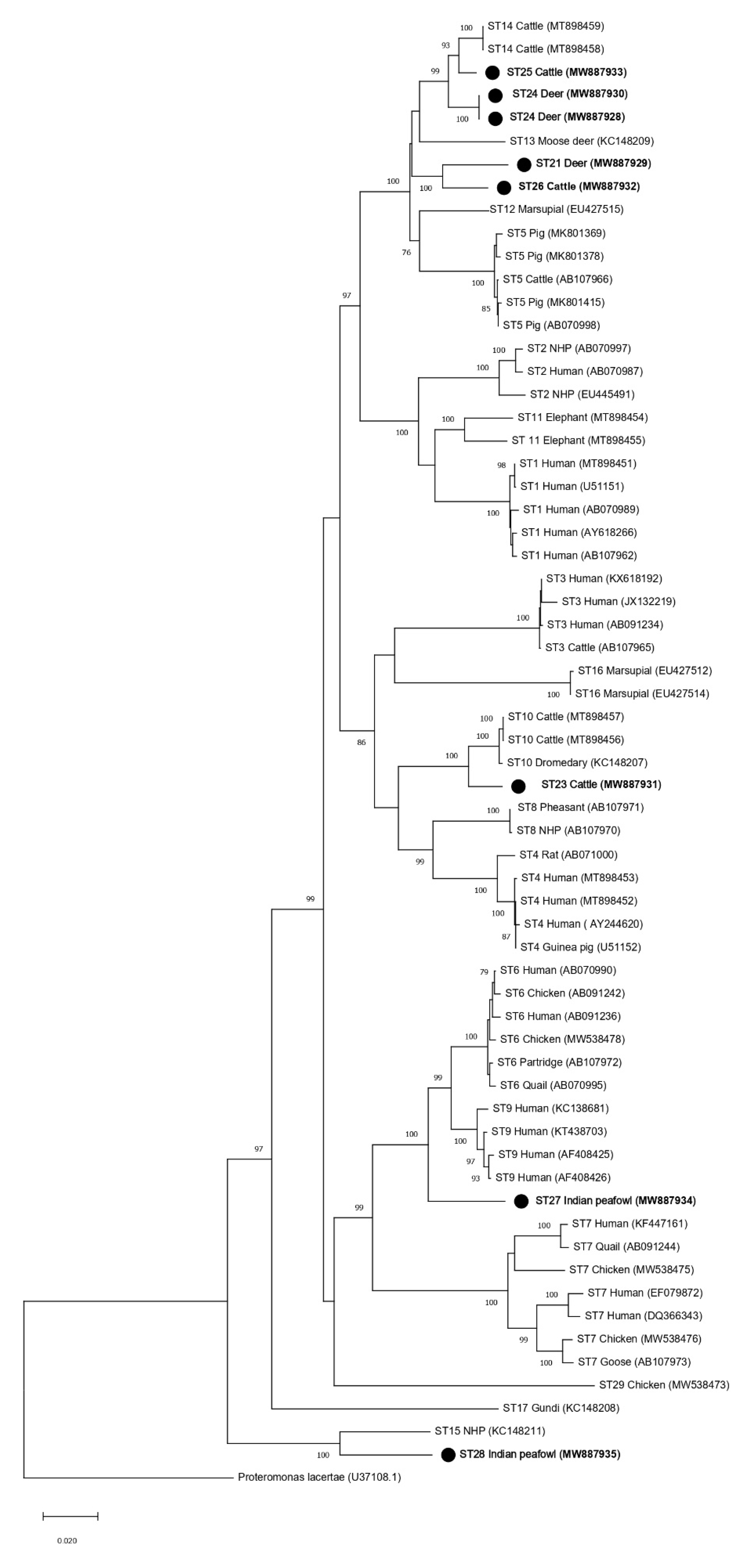
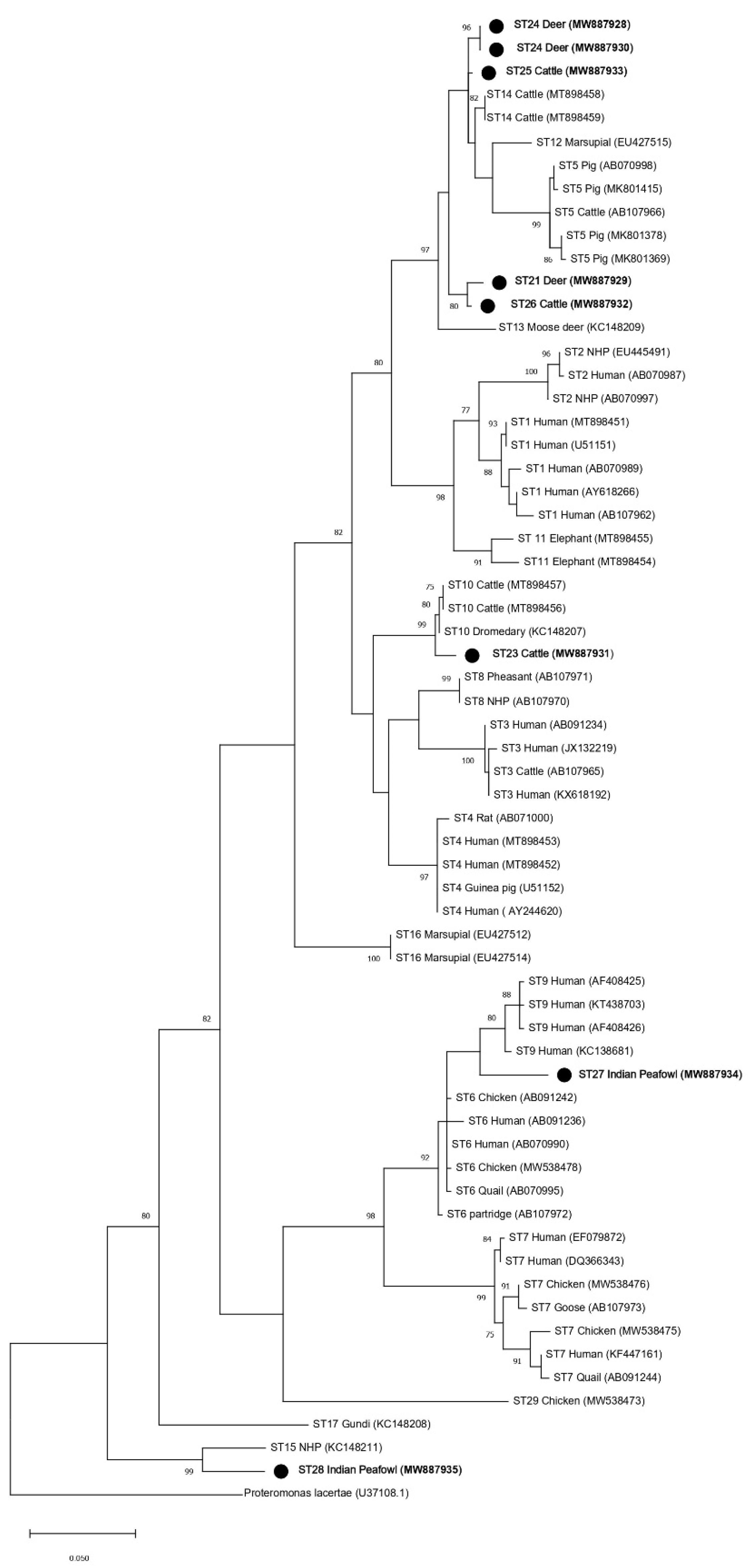
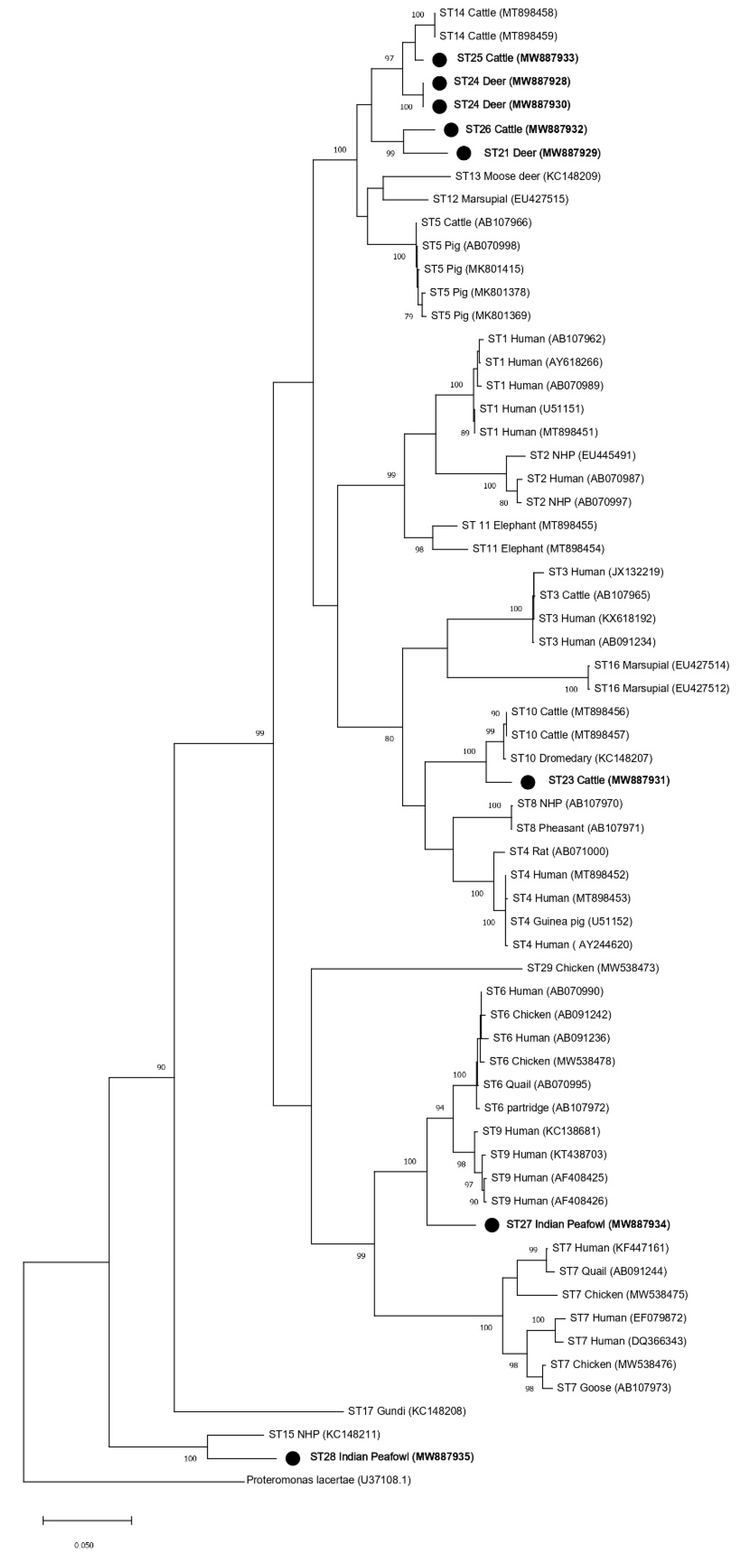
3.2. Phylogenetic Analyses
3.3. Pairwise Distance
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- El Safadi, D.; Gaayeb, L.; Meloni, D.; Cian, A.; Poirier, P.; Wawrzyniak, I.; Delbac, F.; Dabboussi, F.; Delhaes, L.; Seck, M.; et al. Children of Senegal River Basin show the highest prevalence of Blastocystis sp. ever observed worldwide. BMC Infect. Dis. 2014, 14, 164. [Google Scholar] [CrossRef]
- Hublin, J.S.Y.; Maloney, J.G.; Santin, M. Blastocystis in domesticated and wild mammals and birds. Res. Vet. Sci. 2021, 135, 260–282. [Google Scholar] [CrossRef] [PubMed]
- Caradonna, T.; Marangi, M.; Del Chierico, F.; Ferrari, N.; Reddel, S.; Bracaglia, G.; Normanno, G.; Putignani, L.; Giangaspero, A. Detection and prevalence of protozoan parasites in ready-to-eat packaged salads on sale in Italy. Food Microbiol. 2017, 67, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.; Chye, T.; Karmacharya, B.; Govind, S. Blastocystis sp.: Waterborne zoonotic organism, a possibility? Parasit. Vectors 2012, 5, 130. [Google Scholar] [CrossRef] [Green Version]
- Leelayoova, S.; Siripattanapipong, S.; Thathaisong, U.; Naaglor, T.; Taamasri, P.; Piyaraj, P.; Mungthin, M. Drinking water: A possible source of Blastocystis spp. subtype 1 infection in schoolchildren of a rural community in central Thailand. Am. J. Trop. Med. Hyg. 2008, 79, 401–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ajjampur, S.S.R.; Tan, K.S.W. Pathogenic mechanisms in Blastocystis spp.—Interpreting results from in vitro and in vivo studies. Parasitol. Int. 2016, 65, 772–779. [Google Scholar] [CrossRef] [PubMed]
- Casero, R.D.; Mongi, F.; Sánchez, A.; Ramírez, J.D. Blastocystis and urticaria: Examination of subtypes and morphotypes in an unusual clinical manifestation. Acta Trop. 2015, 148, 156–161. [Google Scholar] [CrossRef]
- Rojas-Velázquez, L.; Maloney, J.G.; Molokin, A.; Morán, P.; Serrano-Vázquez, A.; González, E.; Pérez-Juárez, H.; Ximénez, C.; Santin, M. Use of next-generation amplicon sequencing to study Blastocystis genetic diversity in a rural human population from Mexico. Parasit. Vectors 2019, 12, 566. [Google Scholar] [CrossRef] [Green Version]
- Stensvold, C.R.; Clark, C.G. Pre-empting Pandora’s Box: Blastocystis subtypes revisited. Trends Parasitol. 2020, 36, 229–232. [Google Scholar] [CrossRef]
- Maloney, J.G.; Molokin, A.; da Cunha, M.J.R.; Cury, M.C.; Santin, M. Blastocystis subtype distribution in domestic and captive wild bird species from Brazil using next generation amplicon sequencing. Parasite Epidemiol. Control. 2020, 9, e00138. [Google Scholar] [CrossRef]
- Maloney, J.G.; da Cunha, M.J.R.; Molokin, A.; Cuty, M.C.; Santin, M. Next generation sequencing reveals wide genetic diversity of Blastocystis subtypes in chickens including potentially zoonotic subtypes. Parasitol. Res. 2021. [Google Scholar] [CrossRef]
- Maloney, J.G.; Molokin, A.; Santin, M. Next generation amplicon sequencing improves detection of Blastocystis mixed subtype infections. Infect. Genet. Evol. 2019, 73, 119–125. [Google Scholar] [CrossRef]
- Santín, M.; Gómez-Muñoz, M.T.; Solano-Aguilar, G.; Fayer, R. Development of a new PCR protocol to detect and subtype Blastocystis spp. from humans and animals. Parasitol. Res. 2011, 109, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Scicluna, S.M.; Tawari, B.; Clark, C.G. DNA barcoding of Blastocystis. Protist 2006, 157, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, H.; Koyama, Y.; Tsuchiya, E.; Takami, K. Blastocystis phylogeny among various isolates from humans to insects. Parasitol. Int. 2016, 65, 750–759. [Google Scholar] [CrossRef]
- Maloney, J.G.; Molokin, A.; Santin, M. Use of Oxford Nanopore MinION to generate full-length sequences of the Blastocystis small subunit (SSU) rRNA gene. Parasit. Vectors 2020, 13, 595. [Google Scholar] [CrossRef]
- Xiao, L.; Escalante, L.; Yang, C.; Sulaiman, I.; Escalante, A.A.; Montali, R.J.; Fayer, R.; Lal, A.A. Phylogenetic analysis of Cryptosporidium parasites based on the small-subunit rRNA gene locus. Appl. Environ. Microbiol. 1999, 65, 1578–1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, M. A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Maloney, J.G.; Lombard, J.E.; Shivley, C.B.; Urie, N.J.; Santin, M. Zoonotic and genetically diverse subtypes of Blastocystis in US pre-weaned dairy heifer calves. Parasitol. Res. 2019, 118, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.H.; Hu, X.F.; Liu, T.L.; Hu, R.S.; Yu, Z.Q.; Yang, W.B.; Wu, Y.L.; Yu, S.K.; Song, J.K. Molecular characterization of Blastocystis sp. in captive wild animals in Qinling Mountains. Parasitol. Res. 2017, 116, 2327–2333. [Google Scholar] [CrossRef] [PubMed]
- Alfellani, M.A.; Taner-Mulla, D.; Jacob, A.S.; Imeede, C.A.; Yoshikawa, H.; Stensvold, C.R.; Clark, C.G. Genetic diversity of Blastocystis in livestock and zoo animals. Protist 2013, 164, 497–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greige, S.; El Safadi, D.; Khaled, S.; Gantois, N.; Baydoun, M.; Chemaly, M.; Benamrouz-Vanneste, S.; Chabé, M.; Osman, M.; Certad, G.; et al. First report on the prevalence and subtype distribution of Blastocystis sp. in dairy cattle in Lebanon and assessment of zoonotic transmission. Acta Trop. 2019, 194, 23–29. [Google Scholar] [CrossRef]
- Greige, S.; El Safadi, D.; Bécu, N.; Gantois, N.; Pereira, B.; Chabé, M.; Benamrouz-Vanneste, S.; Certad, G.; El Hage, R.; Chemaly, M.; et al. Prevalence and subtype distribution of Blastocystis sp. isolates from poultry in Lebanon and evidence of zoonotic potential. Parasit. Vectors 2018, 11, 389. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Specimen ID/Isolate | Host | Location | Blastocystis Subtype (GenBank Accession Number) | Similarity to MiSeq Sequence (%) |
|---|---|---|---|---|
| 1 (Deer4) | White-tailed deer | Maryland, USA | ST24 (MW887928) | 100 |
| 2 (Deer 79) | White-tailed deer | Maryland, USA | ST21 (MW887929) ST24 (MW887930) | 100 100 |
| 3 (2817.14 m) | Cattle | Maryland, USA | ST23 (MW887931) ST26 (MW887932) | 100 100 |
| 4 (2813.20 m) | Cattle | Maryland, USA | ST25 (MW887933) | 100 |
| 5 (Bird36) | Indian peafowl | Uberlândia, Brazil | ST27 (MW887934) ST28 (MW887935) | 99.6 99.8 |
| Subtype | Host | Country | Isolate/Strain | Sequence Length (bp) | GenBank Accession No. |
|---|---|---|---|---|---|
| ST1 | Human | USA | Nand a | 1770 | U51151 |
| Human | Thailand | 989 | 1781 | AY618266 | |
| Human | Japan | PJ99-172 | 1770 | AB107962 | |
| Human | N/A | HJ96A-29 | 1770 | AB070989 | |
| Human | USA | 1 a | 1766 | MT898451 | |
| ST2 | Human | Japan | HJ96-1 | 1768 | AB070987 |
| NHP | Japan | JM92-2 | 1768 | AB070997 | |
| NHP | Philippines | M24 | 1721 | EU445491 | |
| ST3 | Human | N/A | HJ96A-26 | 1719 | AB091234 |
| Human | Senegal | N/A | 1755 | JX132219 | |
| Human | Singapore | N/A | 1769 | KX618192 | |
| Cattle | Japan | CJ99-363 | 1769 | AB107965 | |
| ST4 | Human | Germany | HG00-12 | 1778 | AY244620 |
| Human | Spain | H-1/3 b | 1773 | MT898453 | |
| Human | USA | 2 c | 1772 | MT898452 | |
| Guinea pig | USA | NIH:1295:1 d | 1778 | U51152 | |
| Rat | Japan | RN94-9 | 1778 | AB071000 | |
| ST5 | Cattle | Japan | CJ99-284 | 1784 | AB107966 |
| Pig | Austria | L01064_Blast_ST5_99 | 1749 | MK801415 | |
| Pig | Germany | L00855_Blast_ST5_97 | 1738 | MK801369 | |
| Pig | Germany | L00926_Blast_ST5_99 | 1738 | MK801378 | |
| Pig | Japan | SY94-3 | 1784 | AB070998 | |
| ST6 | Human | N/A | HJ96AS-1 | 1691 | AB091236 |
| Human | N/A | HJ96AS-1 | 1741 | AB070990 | |
| Chicken | Brazil | 96.7 | 1738 | MW538478 | |
| Chicken | N/A | CK92-4 | 1692 | AB091242 | |
| Japanese quail | Japan | QQ93-3 | 1741 | AB070995 | |
| Partridge | Japan | BJ99-310 | 1741 | AB107972 | |
| ST7 | Human | China | HC05-10 | 1797 | DQ366343 |
| Human | China | HC06-08 | 1795 | EF079872 | |
| Human | Singapore | B | 1819 | KF447161 | |
| Chicken | Brazil | 96.9 | 1813 | MW538475 | |
| Chicken | Brazil | 96.10 | 1786 | MW538476 | |
| Goose | Japan | BJ99-569 | 1796 | AB107973 | |
| Quail | N/A | QQ98-4 | 1756 | AB091244 | |
| ST8 | NHP | Japan | MJ99-132 | 1779 | AB107970 |
| Pheasant | Japan | BJ99-319 | 1779 | AB107971 | |
| ST9 | Human | Japan | HJ00-4 | 1736 | AF408425 |
| Human | Japan | HJ05-4 | 1741 | KT438703 | |
| Human | Denmark | N/A | 1668 | KC138681 | |
| Human | Japan | HJ00-5 | 1737 | AF408426 | |
| ST10 | Cattle | USA | 5 | 1770 | MT898456 |
| Cattle | USA | 6a | 1770 | MT898457 | |
| Dromedary | Libya | CA6 | 1728 | KC148207 | |
| ST11 | Elephant | USA | 4a | 1762 | MT898454 |
| Elephant | USA | 4b | 1763 | MT898455 | |
| ST12 | Marsupial | N/A | MWJ04-41 | 1772 | EU427515 |
| ST13 | Mouse deer | UK | Mousedeer | 1765 | KC148209 |
| ST14 | Cattle | USA | 6b | 1771 | MT898458 |
| Cattle | USA | 7 | 1771 | MT898459 | |
| ST15 | NHP | UK | MA7 | 1898 | KC148211 |
| ST16 | Marsupial | N/A | MKJ04-10 | 1748 | EU427512 |
| ST16 | Marsupial | N/A | MKJ04-30 | 1748 | EU427514 |
| ST17 | Gundi | Libya | Gundi | 1983 | KC148208 |
| ST21 | White-tailed deer | USA | Deer79 | 1776 | MW887929 |
| ST23 | Cattle | USA | 2817.14 m | 1777 | MW887931 |
| ST24 | White-tailed deer | USA | Deer4 | 1769 | MW887928 |
| ST24 | White-tailed deer | USA | Deer79 | 1770 | MW887930 |
| ST25 | Cattle | USA | 2813.20 m | 1782 | MW887933 |
| ST26 | Cattle | USA | 2817.14 m | 1785 | MW887932 |
| ST27 | Indian peafowl | Brazil | Bird36 | 1729 | MW887934 |
| ST28 | Indian peafowl | Brazil | Bird36 | 1808 | MW887935 |
| ST29 | Chicken | Brazil | 96.6 | 1776 | MW538473 |
| Subtypes | ||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 21 | 23 | 24 | 25 | 26 | 27 | 28 | 29 | ||
| Subtypes | 1 | |||||||||||||||||||||||||
| 2 | 0.07 | |||||||||||||||||||||||||
| 3 | 0.14 | 0.13 | ||||||||||||||||||||||||
| 4 | 0.12 | 0.13 | 0.09 | |||||||||||||||||||||||
| 5 | 0.11 | 0.11 | 0.13 | 0.12 | ||||||||||||||||||||||
| 6 | 0.13 | 0.14 | 0.15 | 0.14 | 0.13 | |||||||||||||||||||||
| 7 | 0.17 | 0.17 | 0.18 | 0.17 | 0.15 | 0.12 | ||||||||||||||||||||
| 8 | 0.11 | 0.12 | 0.10 | 0.06 | 0.12 | 0.14 | 0.17 | |||||||||||||||||||
| 9 | 0.13 | 0.13 | 0.15 | 0.14 | 0.13 | 0.03 | 0.11 | 0.14 | ||||||||||||||||||
| 10 | 0.13 | 0.13 | 0.11 | 0.08 | 0.11 | 0.13 | 0.17 | 0.08 | 0.13 | |||||||||||||||||
| 11 | 0.06 | 0.08 | 0.14 | 0.12 | 0.11 | 0.13 | 0.16 | 0.12 | 0.13 | 0.12 | ||||||||||||||||
| 12 | 0.10 | 0.11 | 0.13 | 0.13 | 0.06 | 0.12 | 0.15 | 0.12 | 0.12 | 0.12 | 0.11 | |||||||||||||||
| 13 | 0.11 | 0.11 | 0.14 | 0.13 | 0.07 | 0.13 | 0.16 | 0.12 | 0.12 | 0.12 | 0.11 | 0.06 | ||||||||||||||
| 14 | 0.10 | 0.11 | 0.13 | 0.12 | 0.06 | 0.13 | 0.16 | 0.12 | 0.12 | 0.11 | 0.10 | 0.05 | 0.05 | |||||||||||||
| 15 | 0.20 | 0.20 | 0.18 | 0.18 | 0.17 | 0.17 | 0.20 | 0.18 | 0.16 | 0.17 | 0.19 | 0.17 | 0.18 | 0.17 | ||||||||||||
| 16 | 0.15 | 0.15 | 0.12 | 0.12 | 0.14 | 0.15 | 0.18 | 0.13 | 0.15 | 0.13 | 0.15 | 0.14 | 0.15 | 0.15 | 0.18 | |||||||||||
| 17 | 0.17 | 0.18 | 0.17 | 0.18 | 0.16 | 0.16 | 0.20 | 0.18 | 0.16 | 0.18 | 0.17 | 0.16 | 0.17 | 0.17 | 0.19 | 0.18 | ||||||||||
| 21 | 0.11 | 0.11 | 0.13 | 0.13 | 0.07 | 0.12 | 0.15 | 0.12 | 0.12 | 0.12 | 0.11 | 0.06 | 0.07 | 0.06 | 0.18 | 0.14 | 0.17 | |||||||||
| 23 | 0.13 | 0.13 | 0.11 | 0.08 | 0.11 | 0.13 | 0.17 | 0.08 | 0.13 | 0.02 | 0.12 | 0.11 | 0.12 | 0.11 | 0.18 | 0.14 | 0.17 | 0.12 | ||||||||
| 24 | 0.11 | 0.11 | 0.13 | 0.12 | 0.06 | 0.13 | 0.15 | 0.12 | 0.12 | 0.11 | 0.10 | 0.05 | 0.05 | 0.02 | 0.16 | 0.14 | 0.17 | 0.06 | 0.11 | |||||||
| 25 | 0.10 | 0.10 | 0.13 | 0.12 | 0.06 | 0.12 | 0.15 | 0.12 | 0.12 | 0.11 | 0.10 | 0.05 | 0.05 | 0.02 | 0.17 | 0.14 | 0.16 | 0.06 | 0.11 | 0.02 | ||||||
| 26 | 0.11 | 0.10 | 0.13 | 0.13 | 0.06 | 0.13 | 0.15 | 0.12 | 0.12 | 0.12 | 0.11 | 0.06 | 0.06 | 0.05 | 0.17 | 0.14 | 0.16 | 0.04 | 0.11 | 0.05 | 0.05 | |||||
| 27 | 0.13 | 0.14 | 0.16 | 0.15 | 0.13 | 0.05 | 0.12 | 0.15 | 0.05 | 0.14 | 0.13 | 0.13 | 0.13 | 0.12 | 0.18 | 0.16 | 0.18 | 0.12 | 0.15 | 0.13 | 0.12 | 0.13 | ||||
| 28 | 0.20 | 0.20 | 0.17 | 0.18 | 0.16 | 0.18 | 0.20 | 0.19 | 0.18 | 0.18 | 0.18 | 0.17 | 0.19 | 0.17 | 0.07 | 0.18 | 0.18 | 0.17 | 0.18 | 0.17 | 0.17 | 0.17 | 0.19 | |||
| 29 | 0.16 | 0.16 | 0.17 | 0.16 | 0.17 | 0.17 | 0.18 | 0.16 | 0.16 | 0.17 | 0.16 | 0.17 | 0.16 | 0.15 | 0.22 | 0.18 | 0.20 | 0.16 | 0.17 | 0.15 | 0.15 | 0.16 | 0.16 | 0.20 | ||
| Subtypes | ||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 21 | 23 | 24 | 25 | 26 | 27 | 28 | 29 | ||
| Subtypes | 1 | |||||||||||||||||||||||||
| 2 | 0.05 | |||||||||||||||||||||||||
| 3 | 0.09 | 0.08 | ||||||||||||||||||||||||
| 4 | 0.09 | 0.10 | 0.06 | |||||||||||||||||||||||
| 5 | 0.09 | 0.09 | 0.10 | 0.08 | ||||||||||||||||||||||
| 6 | 0.14 | 0.14 | 0.14 | 0.14 | 0.14 | |||||||||||||||||||||
| 7 | 0.18 | 0.17 | 0.15 | 0.17 | 0.18 | 0.09 | ||||||||||||||||||||
| 8 | 0.10 | 0.11 | 0.05 | 0.06 | 0.10 | 0.13 | 0.16 | |||||||||||||||||||
| 9 | 0.14 | 0.14 | 0.13 | 0.11 | 0.14 | 0.03 | 0.09 | 0.12 | ||||||||||||||||||
| 10 | 0.10 | 0.11 | 0.08 | 0.06 | 0.11 | 0.13 | 0.16 | 0.05 | 0.12 | |||||||||||||||||
| 11 | 0.05 | 0.07 | 0.10 | 0.09 | 0.09 | 0.14 | 0.17 | 0.11 | 0.14 | 0.09 | ||||||||||||||||
| 12 | 0.10 | 0.10 | 0.09 | 0.09 | 0.04 | 0.14 | 0.17 | 0.09 | 0.15 | 0.09 | 0.09 | |||||||||||||||
| 13 | 0.10 | 0.10 | 0.10 | 0.09 | 0.05 | 0.13 | 0.18 | 0.10 | 0.14 | 0.09 | 0.09 | 0.04 | ||||||||||||||
| 14 | 0.08 | 0.09 | 0.10 | 0.07 | 0.04 | 0.14 | 0.18 | 0.10 | 0.14 | 0.09 | 0.08 | 0.03 | 0.03 | |||||||||||||
| 15 | 0.19 | 0.18 | 0.15 | 0.16 | 0.17 | 0.15 | 0.15 | 0.16 | 0.16 | 0.15 | 0.17 | 0.15 | 0.17 | 0.16 | ||||||||||||
| 16 | 0.12 | 0.13 | 0.09 | 0.09 | 0.12 | 0.14 | 0.18 | 0.10 | 0.13 | 0.10 | 0.13 | 0.11 | 0.11 | 0.11 | 0.14 | |||||||||||
| 17 | 0.18 | 0.17 | 0.13 | 0.14 | 0.17 | 0.15 | 0.16 | 0.15 | 0.16 | 0.15 | 0.17 | 0.16 | 0.16 | 0.16 | 0.15 | 0.14 | ||||||||||
| 21 | 0.09 | 0.10 | 0.10 | 0.08 | 0.05 | 0.12 | 0.16 | 0.09 | 0.14 | 0.08 | 0.09 | 0.03 | 0.04 | 0.03 | 0.16 | 0.11 | 0.15 | |||||||||
| 23 | 0.11 | 0.12 | 0.07 | 0.06 | 0.11 | 0.12 | 0.18 | 0.06 | 0.12 | 0.01 | 0.10 | 0.10 | 0.09 | 0.09 | 0.15 | 0.10 | 0.15 | 0.08 | ||||||||
| 24 | 0.09 | 0.10 | 0.10 | 0.08 | 0.04 | 0.14 | 0.17 | 0.10 | 0.14 | 0.08 | 0.08 | 0.03 | 0.04 | 0.01 | 0.16 | 0.10 | 0.16 | 0.03 | 0.09 | |||||||
| 25 | 0.08 | 0.09 | 0.09 | 0.07 | 0.04 | 0.13 | 0.16 | 0.09 | 0.13 | 0.08 | 0.07 | 0.03 | 0.04 | 0.01 | 0.16 | 0.10 | 0.16 | 0.03 | 0.09 | 0.01 | ||||||
| 26 | 0.09 | 0.10 | 0.10 | 0.08 | 0.05 | 0.14 | 0.16 | 0.09 | 0.14 | 0.08 | 0.08 | 0.03 | 0.04 | 0.03 | 0.17 | 0.10 | 0.16 | 0.01 | 0.08 | 0.02 | 0.02 | |||||
| 27 | 0.15 | 0.16 | 0.14 | 0.13 | 0.15 | 0.05 | 0.09 | 0.14 | 0.05 | 0.14 | 0.15 | 0.16 | 0.14 | 0.15 | 0.17 | 0.15 | 0.16 | 0.15 | 0.13 | 0.16 | 0.15 | 0.15 | ||||
| 28 | 0.19 | 0.17 | 0.15 | 0.16 | 0.16 | 0.16 | 0.18 | 0.16 | 0.18 | 0.16 | 0.17 | 0.15 | 0.17 | 0.16 | 0.06 | 0.15 | 0.15 | 0.15 | 0.16 | 0.16 | 0.15 | 0.16 | 0.19 | |||
| 29 | 0.16 | 0.16 | 0.15 | 0.17 | 0.18 | 0.16 | 0.17 | 0.16 | 0.16 | 0.15 | 0.17 | 0.16 | 0.16 | 0.16 | 0.20 | 0.15 | 0.20 | 0.16 | 0.15 | 0.16 | 0.16 | 0.16 | 0.16 | 0.18 | ||
| Subtypes | ||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 21 | 23 | 24 | 25 | 26 | 27 | 28 | 29 | ||
| Subtypes | 1 | |||||||||||||||||||||||||
| 2 | 0.11 | |||||||||||||||||||||||||
| 3 | 0.18 | 0.22 | ||||||||||||||||||||||||
| 4 | 0.16 | 0.18 | 0.15 | |||||||||||||||||||||||
| 5 | 0.17 | 0.16 | 0.19 | 0.17 | ||||||||||||||||||||||
| 6 | 0.21 | 0.21 | 0.22 | 0.20 | 0.20 | |||||||||||||||||||||
| 7 | 0.25 | 0.27 | 0.24 | 0.24 | 0.22 | 0.24 | ||||||||||||||||||||
| 8 | 0.18 | 0.19 | 0.15 | 0.08 | 0.19 | 0.20 | 0.25 | |||||||||||||||||||
| 9 | 0.20 | 0.21 | 0.22 | 0.20 | 0.19 | 0.07 | 0.23 | 0.20 | ||||||||||||||||||
| 10 | 0.19 | 0.18 | 0.17 | 0.13 | 0.17 | 0.19 | 0.25 | 0.13 | 0.19 | |||||||||||||||||
| 11 | 0.12 | 0.14 | 0.21 | 0.17 | 0.18 | 0.21 | 0.25 | 0.19 | 0.22 | 0.19 | ||||||||||||||||
| 12 | 0.18 | 0.17 | 0.18 | 0.15 | 0.11 | 0.20 | 0.24 | 0.15 | 0.19 | 0.14 | 0.18 | |||||||||||||||
| 13 | 0.18 | 0.17 | 0.18 | 0.18 | 0.12 | 0.23 | 0.22 | 0.17 | 0.21 | 0.16 | 0.18 | 0.10 | ||||||||||||||
| 14 | 0.15 | 0.16 | 0.19 | 0.16 | 0.10 | 0.21 | 0.24 | 0.17 | 0.20 | 0.16 | 0.15 | 0.09 | 0.08 | |||||||||||||
| 15 | 0.31 | 0.33 | 0.27 | 0.29 | 0.24 | 0.28 | 0.32 | 0.29 | 0.27 | 0.29 | 0.32 | 0.27 | 0.28 | 0.29 | ||||||||||||
| 16 | 0.28 | 0.28 | 0.24 | 0.24 | 0.24 | 0.26 | 0.30 | 0.26 | 0.24 | 0.27 | 0.27 | 0.24 | 0.27 | 0.26 | 0.22 | |||||||||||
| 17 | 0.32 | 0.32 | 0.28 | 0.29 | 0.24 | 0.28 | 0.31 | 0.29 | 0.26 | 0.28 | 0.30 | 0.29 | 0.30 | 0.28 | 0.25 | 0.23 | ||||||||||
| 21 | 0.16 | 0.17 | 0.18 | 0.17 | 0.12 | 0.22 | 0.21 | 0.20 | 0.20 | 0.20 | 0.19 | 0.12 | 0.13 | 0.12 | 0.28 | 0.23 | 0.28 | |||||||||
| 23 | 0.20 | 0.17 | 0.17 | 0.13 | 0.15 | 0.19 | 0.23 | 0.13 | 0.19 | 0.05 | 0.18 | 0.15 | 0.16 | 0.17 | 0.28 | 0.25 | 0.29 | 0.18 | ||||||||
| 24 | 0.17 | 0.18 | 0.19 | 0.18 | 0.09 | 0.20 | 0.23 | 0.18 | 0.20 | 0.17 | 0.16 | 0.08 | 0.09 | 0.04 | 0.28 | 0.24 | 0.27 | 0.11 | 0.17 | |||||||
| 25 | 0.16 | 0.17 | 0.18 | 0.16 | 0.09 | 0.20 | 0.22 | 0.16 | 0.18 | 0.16 | 0.15 | 0.08 | 0.09 | 0.03 | 0.28 | 0.26 | 0.27 | 0.11 | 0.16 | 0.04 | ||||||
| 26 | 0.15 | 0.18 | 0.18 | 0.17 | 0.09 | 0.21 | 0.21 | 0.18 | 0.20 | 0.17 | 0.17 | 0.11 | 0.11 | 0.10 | 0.26 | 0.24 | 0.28 | 0.08 | 0.15 | 0.09 | 0.09 | |||||
| 27 | 0.21 | 0.24 | 0.23 | 0.20 | 0.22 | 0.09 | 0.25 | 0.21 | 0.10 | 0.21 | 0.23 | 0.22 | 0.22 | 0.21 | 0.29 | 0.27 | 0.30 | 0.23 | 0.23 | 0.22 | 0.20 | 0.23 | ||||
| 28 | 0.29 | 0.31 | 0.24 | 0.27 | 0.25 | 0.26 | 0.29 | 0.27 | 0.26 | 0.29 | 0.29 | 0.25 | 0.28 | 0.28 | 0.11 | 0.23 | 0.26 | 0.25 | 0.28 | 0.26 | 0.26 | 0.25 | 0.27 | |||
| 29 | 0.26 | 0.26 | 0.25 | 0.22 | 0.26 | 0.26 | 0.30 | 0.23 | 0.26 | 0.25 | 0.26 | 0.26 | 0.26 | 0.25 | 0.29 | 0.25 | 0.34 | 0.25 | 0.25 | 0.25 | 0.26 | 0.26 | 0.26 | 0.27 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maloney, J.G.; Santin, M. Mind the Gap: New Full-Length Sequences of Blastocystis Subtypes Generated via Oxford Nanopore Minion Sequencing Allow for Comparisons between Full-Length and Partial Sequences of the Small Subunit of the Ribosomal RNA Gene. Microorganisms 2021, 9, 997. https://doi.org/10.3390/microorganisms9050997
Maloney JG, Santin M. Mind the Gap: New Full-Length Sequences of Blastocystis Subtypes Generated via Oxford Nanopore Minion Sequencing Allow for Comparisons between Full-Length and Partial Sequences of the Small Subunit of the Ribosomal RNA Gene. Microorganisms. 2021; 9(5):997. https://doi.org/10.3390/microorganisms9050997
Chicago/Turabian StyleMaloney, Jenny G., and Monica Santin. 2021. "Mind the Gap: New Full-Length Sequences of Blastocystis Subtypes Generated via Oxford Nanopore Minion Sequencing Allow for Comparisons between Full-Length and Partial Sequences of the Small Subunit of the Ribosomal RNA Gene" Microorganisms 9, no. 5: 997. https://doi.org/10.3390/microorganisms9050997
APA StyleMaloney, J. G., & Santin, M. (2021). Mind the Gap: New Full-Length Sequences of Blastocystis Subtypes Generated via Oxford Nanopore Minion Sequencing Allow for Comparisons between Full-Length and Partial Sequences of the Small Subunit of the Ribosomal RNA Gene. Microorganisms, 9(5), 997. https://doi.org/10.3390/microorganisms9050997