
Epigenetic Silencing of MicroRNA-126 Promotes Cell Growth in Marek’s Disease

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. GaHV-2 Latent Infection in MSB-1 Cell Line and Reagents Used as Reactivation Stimuli
2.2. GaHV-2 Productive Infection in Chicken Embryo Fibroblasts
2.3. Ethics Statement of the In Vivo Experiment
2.4. Experimental Animals
2.5. Experimental Design
2.6. Cell Isolation
2.7. RNA Extraction
2.8. Specific Reverse Transcription (RT)
2.9. Non-Specific Reverse Transcription
2.10. Quantitative PCR
2.11. Reference Gene Selection
2.12. 3′ RACE
2.13. DNA Extraction
2.14. Bisulfite Genomic Sequencing Analysis (BGSA)
2.15. Polymerase Chain Reaction (PCR)
2.16. Cloning and Sequencing of PCR Amplicons
2.17. Western Blotting
2.18. Plasmid Constructs
2.19. Generation of MSB-1 Cells Stably Expressing Pre-miRs Regulated by T-REx™ System
2.20. MTS Cell Proliferation Assay
2.21. Statistical Analysis
3. Results
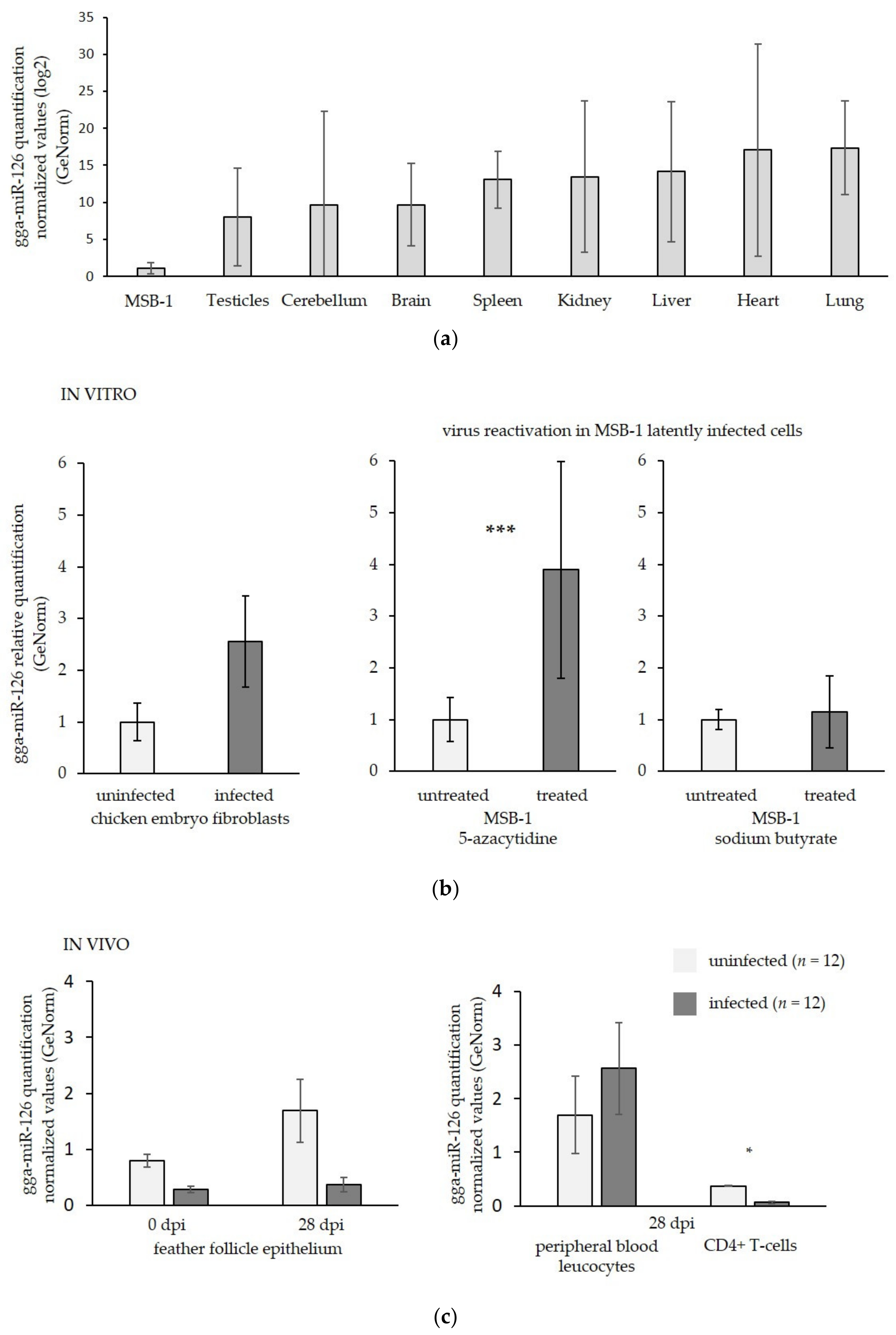
3.1. MiR-126 Is Repressed in the Target Cells of GaHV-2 Latency and Transformation
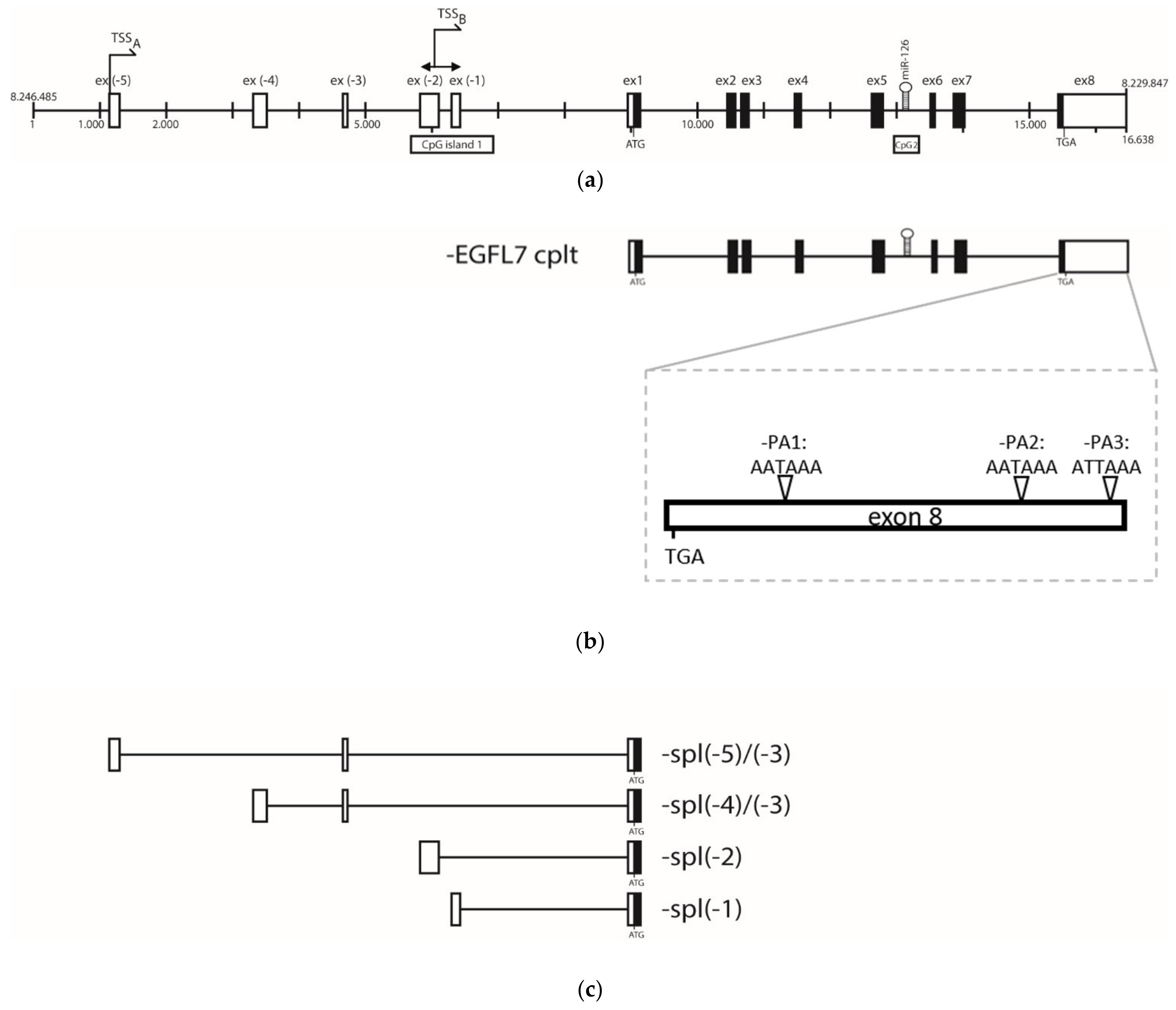
3.2. Epidermal Growth Factor Like 7 (EGFL-7) Transcripts Analysis
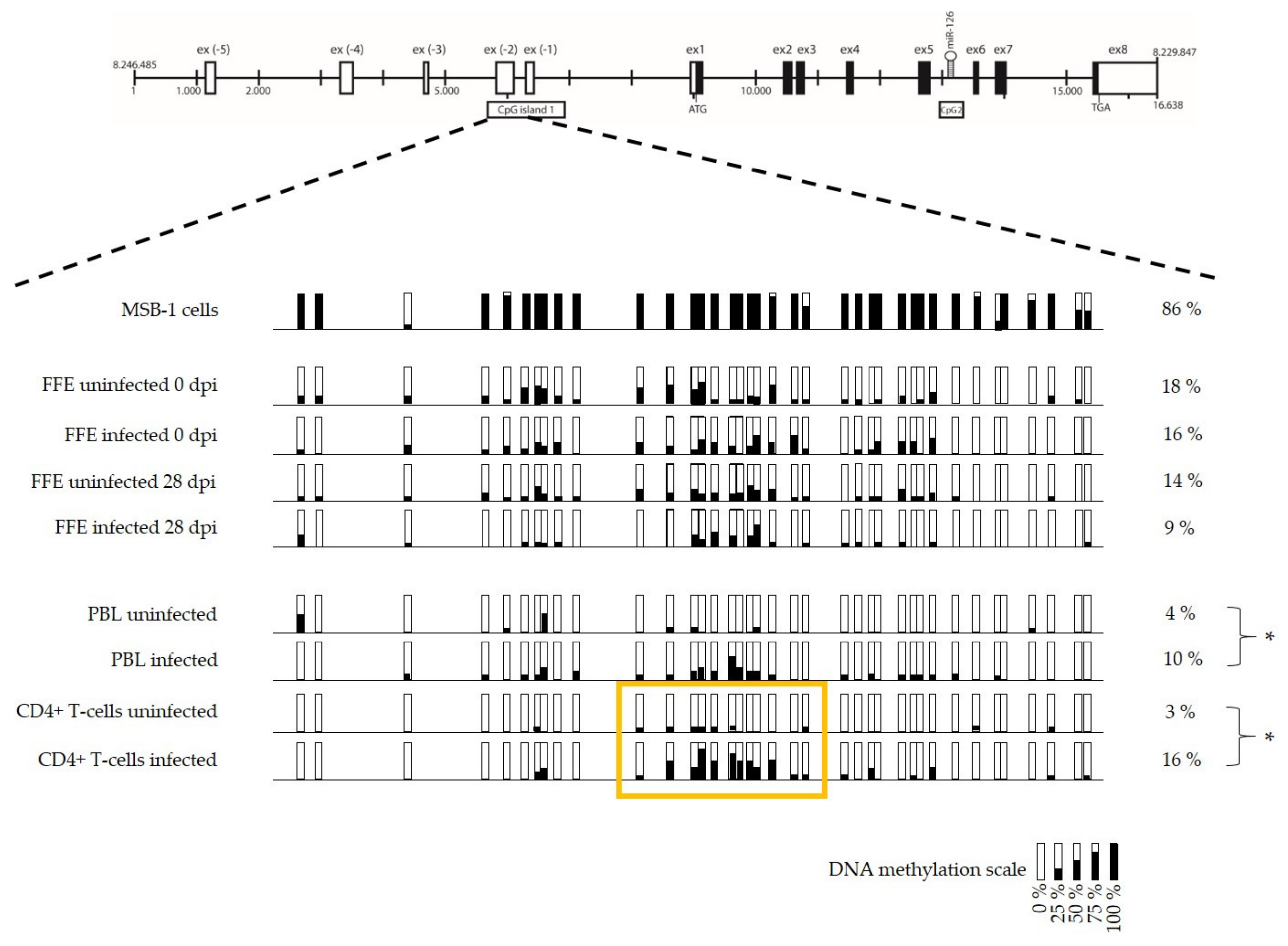
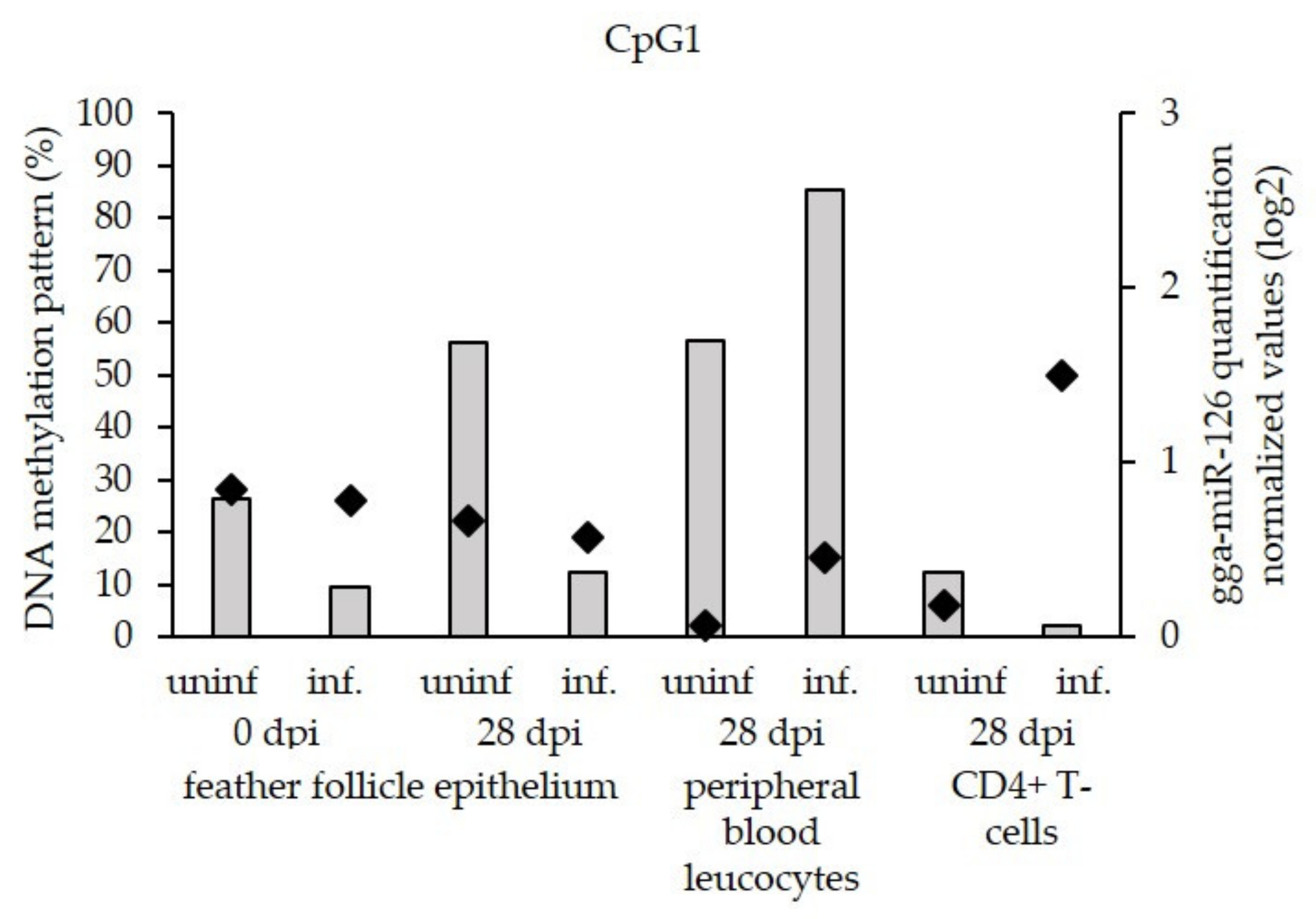
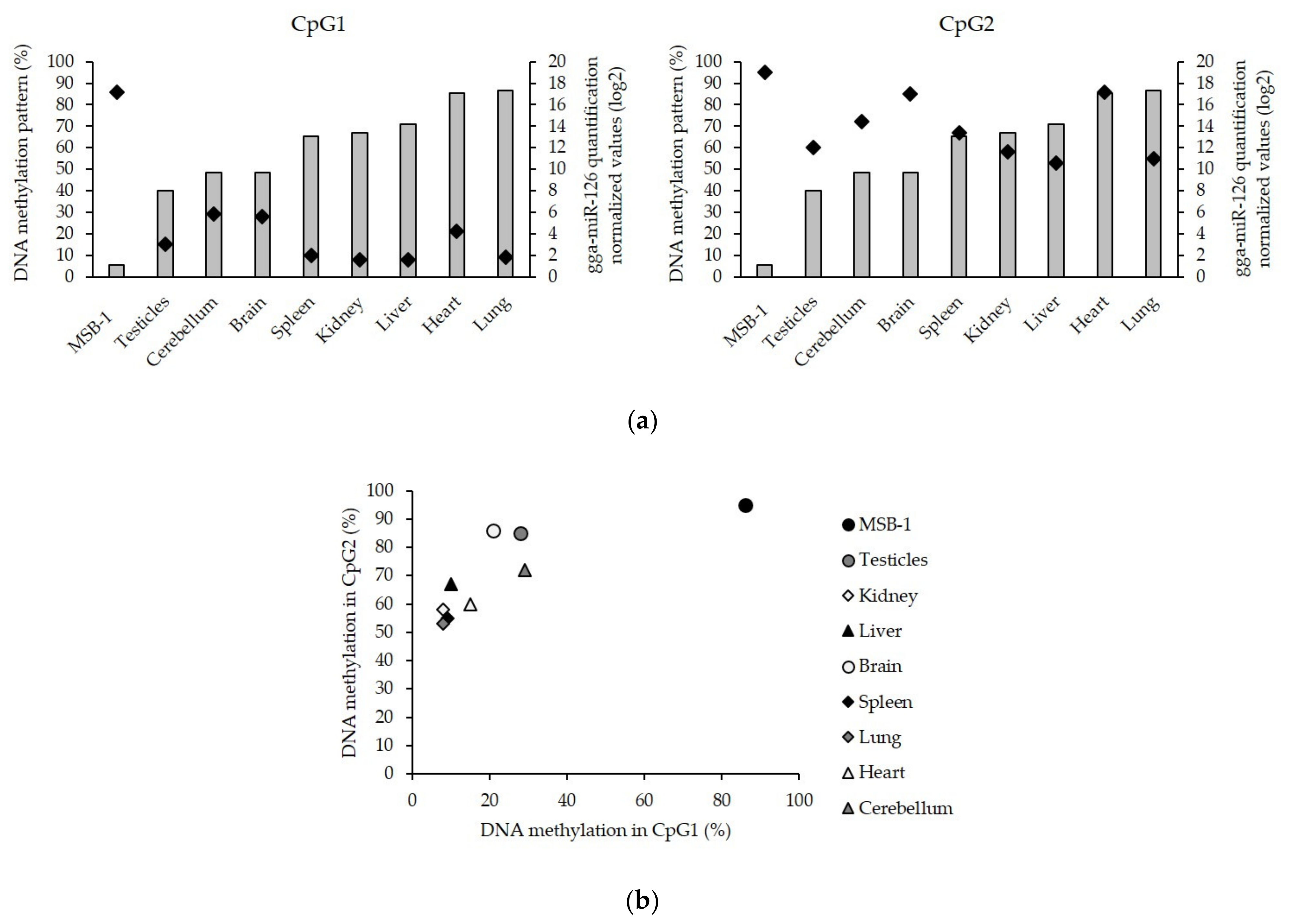
3.3. Low Level of miR-126 Expression Is Associated with DNA Hypermethylation of EGFL-7 CpG Islands in the Target Cells of Latent GaHV-2 Infection
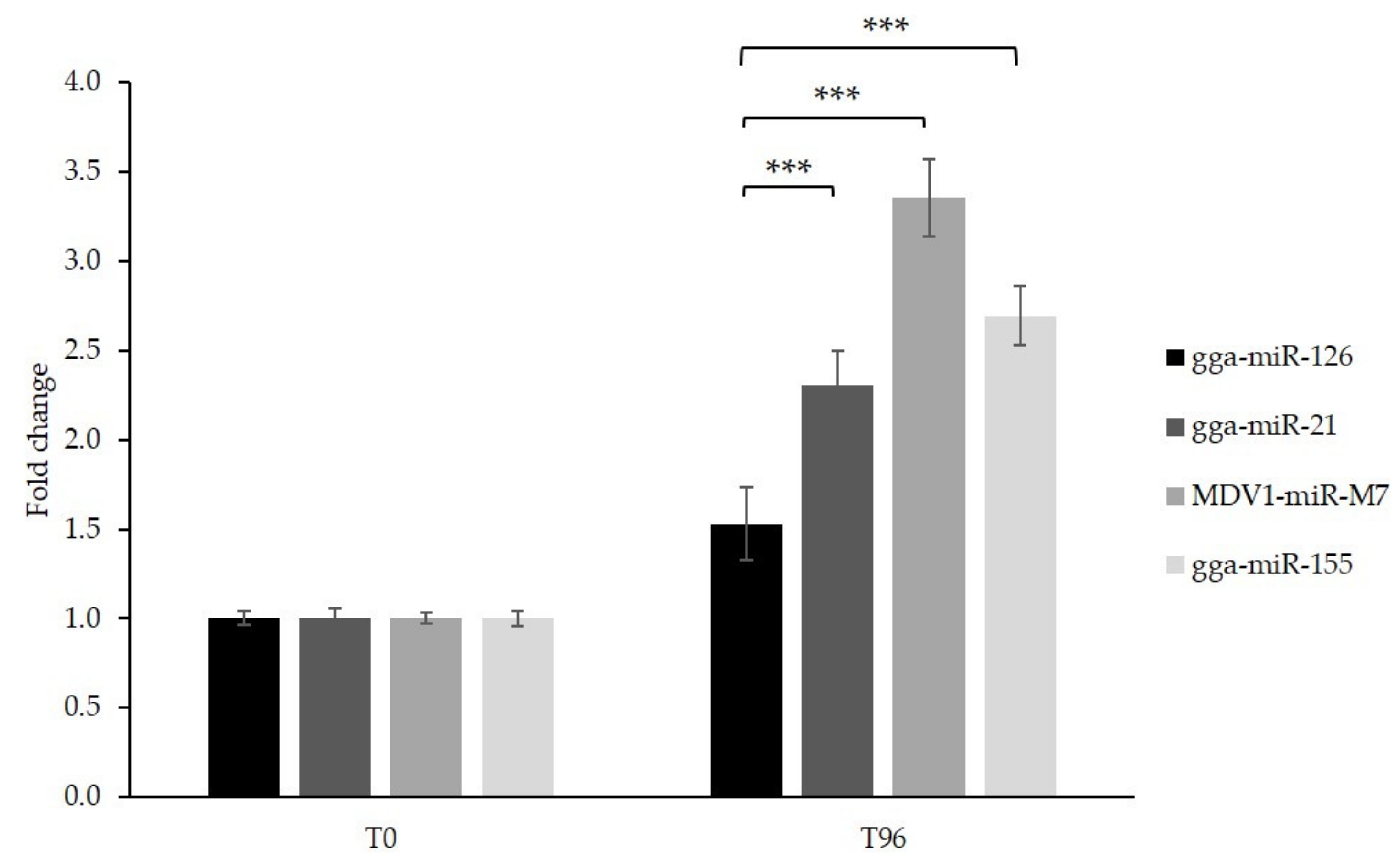
3.4. Generation of GaHV-2 Tumor Cell Lines with Inducible Overexpression of miR-126 and Control miRNAs
3.5. Cell Proliferation Is Impaired Following miR-126 Overexpression
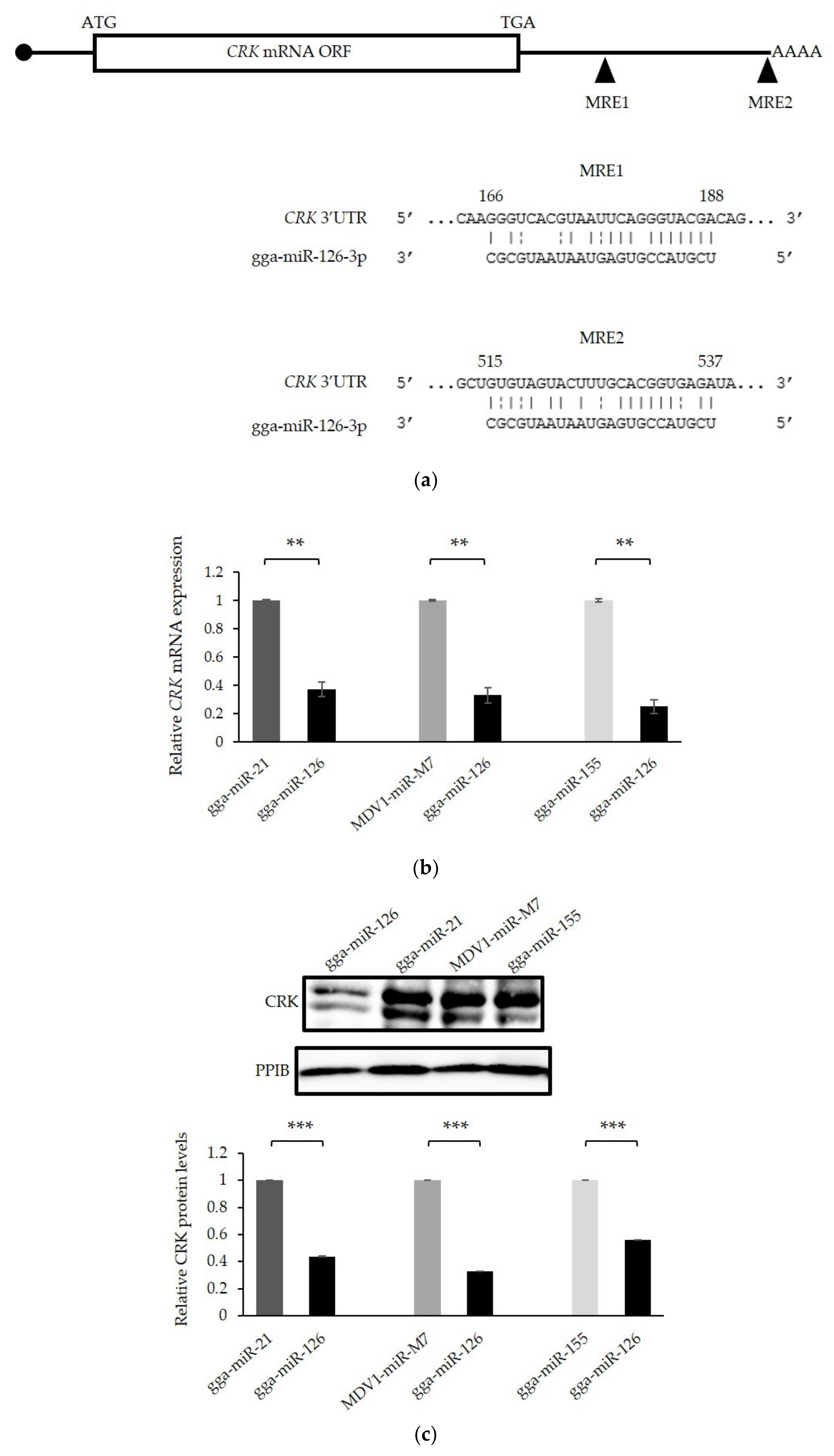
3.6. The Proto-Oncogene CRK Is Targeted and Down-Regulated by miR-126 in GaHV-2 Transformed Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers of qRT-PCR | ||
| Primers Name | Sequences (5′–3′) | GenBank Accession No. |
| gga-miR-126-3p | GSP: CATGATCAGCTGGGCCAAGAGCGCATTATT LNA: TCGTACCGTGAGTA UP: CATGATCAGCTGGGCCAAGA | |
| SDHA | Fwd: TTCCCGTTTTGCCTACGGTG Rev: CTGCCTCGCCACAAGCATAT | XM 419054.2 |
| U6 | Fwd: CTCGCTTCGGCAGCACATATAC Rev: TTTGCGTGTCATCCTTGCGC | (Zhao et al., 2015 [69]) |
| 18S rRNA | Fwd: GGCGGCTTTGGTGACTCTAG Rev: ATCGAACCCTGATTCCCCGT | AF173612.1 |
| HMBS | Fwd: GGCTGGGAGAATCGCATAGG Rev: TCCTGCAGGGCAGATACCAT | XM 417846.2 |
| GAPDH | Fwd: GTCCTCTCTGGCAAAGTCCAAG Rev: CCACAACATACTCAGCACCTGC | NM 204305.1 |
| ß-actin | Fwd: GACTCTGGTGATGGTGTTAC Rev: AGCACAGCTTCTCCTTGATG | NM 205518.1 |
| CRK | Fwd: CGGGTTATCCAGAAGCGAGTC Rev: TTCTCCTTCCCACTGACCACTC | |
| Primers Used in Bisulfite Genomic Sequencing Assay | ||
| Primers Name | Sequences (5′–3′) | GenBank Accession No. |
| gga-miR-126 CpGi-1 | Fwd-1: GTTGTTTGGTTAGTATAGAGAGAAATTTA Rev-1: ATTTCCTCCCCCCCAARCCA Fwd-2: TTYGGTTATTGTATYGGTGATGGGATTTTA Rev-2: ATCCCTCCRCCCCRACCCAATAAA | NR 031468.1 |
| gga-miR-126 CpGi-2 | Fwd-1: GTATAAGTTTAGGTTTTGTAGGG Rev-1: TTCRTACCTTTACTATCAACAAATA Fwd-2: TTGTAGGGGTGATAAAGTTTGGTTG Rev-2: TATCAACAAATAAATTAACACTCATATCTTCC | NR 031468.1 |
| M13 | Fwd: GTAAAACGACGGCCATG Rev: CAGGAAACAGCTATGAC | |
| Primers Used in EGFL-7 3′ RACE Analysis | ||
| Primers Name | Sequences (5′–3′) | Position on Chromosome 17 |
| ex8-1 | Fwd: ATCATCTCCAGGAAGGCACAAGGTA | 8230922-8230898 |
| ex8-2 | Fwd: AAAGAGAAGGCTGGTTTCCCTCATC | 8230896-8230872 |
| ex8-3 | Fwd: GTTTACACTGGCACAGCACCAGCT | 8230443-8230420 |
| ex8-4 | Fwd: CTCCCCAGCTCTGTTTGCTCAAG | 8230362-8230340 |
| Primers Used in EGFL-7 Transcript Analysis | ||
| Primers Name | Sequences (5′–3′) | Position on Chromosome 17 |
| ex(-5)-1 | Fwd: CCACGAGGCAGTGTGGCAGT | 8245224-8245205 |
| ex(-5)-2 | Fwd: ACGAGGCAGTGTGGCAGTGG | 8245222-8245203 |
| ex(-4)-1 | Fwd: AGCAAGTAGTCCCAGTTCTGAGC | 8243008-8242986 |
| ex(-3)-1 | Fwd: CAGTGAGAAACCAAGATGCTCCT | 8241801-8241779 |
| ex(-2)-1 | Fwd: GTGATGGGATTTCACGGTAGC | 8240504-8240484 |
| ex(-2)-2 | Fwd: CAGGAGAGCTGCTCTGCGAG | 8240422-8240403 |
| ex(-1)-1 | Fwd: AGGGACCGACTCGGCCTGG | 8240112-8240094 |
| ex(-1)-2 | Fwd: CGGCTCCGCCACCGCCAC | 8240049-8240032 |
| ex1-1 | Rev: GGCTCCTACTGCATGGCTTG | 8237474-8237493 |
| ex1-1 | Fwd: GAACAGCAAGCCATGCAGTAG | 8237499-8237479 |
| ex1-2 | Fwd: AAGCCATGCAGTAGGAGCCCAC | 8237492-8237471 |
| ex 8-1 | Rev: CAGCCTTCTCTTTCTACCTTGTG | 8230884-8230906 |
| ex8-2 | Rev:CCTTGTGCCTTCCTGGAGATGA | 8230900-8230921 |
| Primers for Plasmid Constructs | ||
| Primers Name | Sequences (5′–3′) | GenBank/miRBase Accession No. |
| gga-miR-126 PmeI | Fwd: GACTGTTTAAACGCACATCCATCCGAGCCACAAG Rev: GACTGTTTAAACGAGCATGTAGATGGCTCTCCCAG | NR 031468.1 |
| gga-miR-21 PmeI | Fwd: GACTGTTTAAACTGAATGTCCTCCTGTGTTGCCAG Rev: GACTGTTTAAACCTGGAGATGGGTGAGCAAACG | MI0004994 |
| MDV1-miR-M7 PmeI | Fwd: GACTGTTTAAACGATGCTCTCTAGCCAAGAGAG Rev: GACTGTTTAAACGCAGTTCTGAGGACACATTT | MI0005099 |
| gga-miR-155 PmeI | Fwd: GACTGTTTAAACCTAGAGTTCTTCTGTAGGCTGTATG Rev: GACTGTTTAAACGAGTTCTGATGAGAGGCATGGTAC | MI0001176 |
| Primers for tetR | ||
| Primers Name | Sequences (5′–3′) | |
| tetR | Fwd: GATGTTAGATAGGCACCATACTC Rev: GTAGTAGGTGTTTCCCTTTCTTC | |
References
- He, L.; Hannon, G.J. MicroRNAs: Small RNAs with a Big Role in Gene Regulation. Nat. Rev. Genet. 2004, 5, 522–531. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Croce, C.M. Causes and Consequences of MicroRNA Dysregulation in Cancer. Nat. Rev. Genet. 2009, 10, 704–714. [Google Scholar] [CrossRef]
- Shu, J.; Silva, B.V.R.E.; Gao, T.; Xu, Z.; Cui, J. Dynamic and Modularized MicroRNA Regulation and Its Implication in Human Cancers. Sci. Rep. 2017, 7, 13356. [Google Scholar] [CrossRef] [Green Version]
- Lambeth, L.S.; Yao, Y.; Smith, L.P.; Zhao, Y.; Nair, V. MicroRNAs 221 and 222 Target P27Kip1 in Marek’s Disease Virus-Transformed Tumour Cell Line MSB-1. J. Gen. Virol. 2009, 90, 1164–1171. [Google Scholar] [CrossRef]
- Stik, G.; Dambrine, G.; Pfeffer, S.; Rasschaert, D. The Oncogenic MicroRNA OncomiR-21 Overexpressed during Marek’s Disease Lymphomagenesis Is Transactivated by the Viral Oncoprotein Meq. J. Virol. 2013, 87, 80–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Yao, Y.; Smith, L.P.; Nair, V. MicroRNA-26a-Mediated Regulation of Interleukin-2 Expression in Transformed Avian Lymphocyte Lines. Cancer Cell Int. 2010, 10, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oduor, C.I.; Kaymaz, Y.; Chelimo, K.; Otieno, J.A.; Ong’echa, J.M.; Moormann, A.M.; Bailey, J.A. Integrative MicroRNA and MRNA Deep-Sequencing Expression Profiling in Endemic Burkitt Lymphoma. BMC Cancer 2017, 17, 761. [Google Scholar] [CrossRef]
- Han, Z.; Rosen, S.T.; Querfeld, C. Targeting MicroRNA in Hematologic Malignancies. Curr. Opin. Oncol. 2020, 32, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, S.; Tagawa, H. Dysregulation of MicroRNAs and Their Association in the Pathogenesis of T-Cell Lymphoma/Leukemias. Int. J. Hematol. 2014, 99, 542–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrie, C.H. MicroRNAs in Hematological Malignancies. Blood Rev. 2013, 27, 143–154. [Google Scholar] [CrossRef]
- Fiorucci, G.; Chiantore, M.V.; Mangino, G.; Romeo, G. MicroRNAs in Virus-Induced Tumorigenesis and IFN System. Cytokine Growth Factor Rev. 2015, 26, 183–194. [Google Scholar] [CrossRef]
- Osterrieder, N.; Kamil, J.P.; Schumacher, D.; Tischer, B.K.; Trapp, S. Marek’s Disease Virus: From Miasma to Model. Nat. Rev. Microbiol. 2006, 4, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Read, A.F.; Baigent, S.J.; Powers, C.; Kgosana, L.B.; Blackwell, L.; Smith, L.P.; Kennedy, D.A.; Walkden-Brown, S.W.; Nair, V.K. Imperfect Vaccination Can Enhance the Transmission of Highly Virulent Pathogens. PLoS Biol. 2015, 13, e1002198. [Google Scholar] [CrossRef]
- Burgess, S.C.; Young, J.R.; Baaten, B.J.G.; Hunt, L.; Ross, L.N.J.; Parcells, M.S.; Kumar, P.M.; Tregaskes, C.A.; Lee, L.F.; Davison, T.F. Marek’s Disease Is a Natural Model for Lymphomas Overexpressing Hodgkin’s Disease Antigen (CD30). Proc. Natl. Acad. Sci. USA 2004, 101, 13879–13884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gennart, I.; Coupeau, D.; Pejaković, S.; Laurent, S.; Rasschaert, D.; Muylkens, B. Marek’s Disease: Genetic Regulation of Gallid Herpesvirus 2 Infection and Latency. Vet. J. 2015, 205, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, H.; Chen, Y.; Gao, X.; Lu, Z.; Gao, L.; Wang, Y.; Gao, Y.; Gao, H.; Liu, C.; et al. The Down-Regulation of Casein Kinase 1 Alpha as a Host Defense Response against Infectious Bursal Disease Virus Infection. Virology 2017, 512, 211–221. [Google Scholar] [CrossRef]
- Catrina Ene, A.M.; Borze, I.; Guled, M.; Costache, M.; Leen, G.; Sajin, M.; Ionica, E.; Chitu, A.; Knuutila, S. MicroRNA Expression Profiles in Kaposi’s Sarcoma. Pathol. Oncol. Res. 2014, 20, 153–159. [Google Scholar] [CrossRef]
- Ebrahimi, F.; Gopalan, V.; Smith, R.A.; Lam, A.K.-Y. MiR-126 in Human Cancers: Clinical Roles and Current Perspectives. Exp. Mol. Pathol. 2014, 96, 98–107. [Google Scholar] [CrossRef]
- Nikolic, I.; Plate, K.-H.; Schmidt, M.H.H. EGFL7 Meets MiRNA-126: An Angiogenesis Alliance. J. Angiogenes. Res. 2010, 2, 9. [Google Scholar] [CrossRef] [Green Version]
- Fish, J.E.; Santoro, M.M.; Morton, S.U.; Yu, S.; Yeh, R.-F.; Wythe, J.D.; Ivey, K.N.; Bruneau, B.G.; Stainier, D.Y.R.; Srivastava, D. MiR-126 Regulates Angiogenic Signaling and Vascular Integrity. Dev. Cell 2008, 15, 272–284. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Aurora, A.B.; Johnson, B.A.; Qi, X.; McAnally, J.; Hill, J.A.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. The Endothelial-Specific MicroRNA MiR-126 Governs Vascular Integrity and Angiogenesis. Dev. Cell 2008, 15, 261–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miko, E.; Margitai, Z.; Czimmerer, Z.; Várkonyi, I.; Dezso, B.; Lányi, A.; Bacsó, Z.; Scholtz, B. MiR-126 Inhibits Proliferation of Small Cell Lung Cancer Cells by Targeting SLC7A5. FEBS Lett. 2011, 585, 1191–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamada, S.; Satoh, K.; Fujibuchi, W.; Hirota, M.; Kanno, A.; Unno, J.; Masamune, A.; Kikuta, K.; Kume, K.; Shimosegawa, T. MiR-126 Acts as a Tumor Suppressor in Pancreatic Cancer Cells via the Regulation of ADAM9. Mol. Cancer Res. 2012, 10, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Crawford, M.; Brawner, E.; Batte, K.; Yu, L.; Hunter, M.G.; Otterson, G.A.; Nuovo, G.; Marsh, C.B.; Nana-Sinkam, S.P. MicroRNA-126 Inhibits Invasion in Non-Small Cell Lung Carcinoma Cell Lines. Biochem. Biophys. Res. Commun. 2008, 373, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Friedman, J.M.; Chihara, Y.; Egger, G.; Chuang, J.C.; Liang, G. Epigenetic Therapy Upregulates the Tumor Suppressor MicroRNA-126 and Its Host Gene EGFL7 in Human Cancer Cells. Biochem. Biophys. Res. Commun. 2009, 379, 726–731. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, X.; Xu, B.; Wang, B.; Wang, Z.; Liang, Y.; Zhou, J.; Hu, J.; Jiang, B. Epigenetic Silencing of MiR-126 Contributes to Tumor Invasion and Angiogenesis in Colorectal Cancer. Oncol. Rep. 2013, 30, 1976–1984. [Google Scholar] [CrossRef] [Green Version]
- Cui, H.; Mu, Y.; Yu, L.; Xi, Y.; Matthiesen, R.; Su, X.; Sun, W. Methylation of the MiR-126 Gene Associated with Glioma Progression. Fam. Cancer 2016, 15, 317–324. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, K.; Emoto, N.; Hamano, E.; Sunohara, M.; Kawakami, M.; Kage, H.; Kitano, K.; Nakajima, J.; Goto, A.; Fukayama, M.; et al. Genome Structure-Based Screening Identified Epigenetically Silenced MicroRNA Associated with Invasiveness in Non-Small-Cell Lung Cancer. Int. J. Cancer 2012, 130, 2580–2590. [Google Scholar] [CrossRef]
- Widschwendter, M.; Jones, A.; Evans, I.; Reisel, D.; Dillner, J.; Sundström, K.; Steyerberg, E.W.; Vergouwe, Y.; Wegwarth, O.; Rebitschek, F.G.; et al. Epigenome-Based Cancer Risk Prediction: Rationale, Opportunities and Challenges. Nat. Rev. Clin. Oncol. 2018, 15, 292–309. [Google Scholar] [CrossRef]
- Kim, Y.-K.; Kim, V.N. Processing of Intronic MicroRNAs. EMBO J. 2007, 26, 775–783. [Google Scholar] [CrossRef] [Green Version]
- Hirai, K.; Yamada, M.; Arao, Y.; Kato, S.; Nii, S. Replicating Marek’s Disease Virus (MDV) Serotype 2 DNA with Inserted MDV Serotype 1 DNA Sequences in a Marek’s Disease Lymphoblastoid Cell Line MSB1-41C. Arch. Virol. 1990, 114, 153–165. [Google Scholar] [CrossRef]
- Akiyama, Y.; Kato, S.; Iwa, N. Continuous Cell Culture from Lymphoma of Marek’s Disease. Biken J. 1973, 16, 177–179. [Google Scholar]
- Parcells, M.S.; Arumugaswami, V.; Prigge, J.T.; Pandya, K.; Dienglewicz, R.L. Marek’s Disease Virus Reactivation from Latency: Changes in Gene Expression at the Origin of Replication. Poult. Sci. 2003, 82, 893–898. [Google Scholar] [CrossRef]
- Petherbridge, L.; Brown, A.C.; Baigent, S.J.; Howes, K.; Sacco, M.A.; Osterrieder, N.; Nair, V.K. Oncogenicity of Virulent Marek’s Disease Virus Cloned as Bacterial Artificial Chromosomes. J. Virol. 2004, 78, 13376–13380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muylkens, B.; Farnir, F.; Meurens, F.; Schynts, F.; Vanderplasschen, A.; Georges, M.; Thiry, E. Coinfection with Two Closely Related Alphaherpesviruses Results in a Highly Diversified Recombination Mosaic Displaying Negative Genetic Interference. J. Virol. 2009, 83, 3127–3137. [Google Scholar] [CrossRef] [Green Version]
- Stik, G.; Muylkens, B.; Coupeau, D.; Laurent, S.; Dambrine, G.; Messmer, M.; Chane-Woon-Ming, B.; Pfeffer, S.; Rasschaert, D. Small RNA Cloning and Sequencing Strategy Affects Host and Viral MicroRNA Expression Signatures. J. Biotechnol. 2014, 181, 35–44. [Google Scholar] [CrossRef]
- Debba-Pavard, M.; Ait-Lounis, A.; Soubieux, D.; Rasschaert, D.; Dambrine, G. Vaccination against Marek’s Disease Reduces Telomerase Activity and Viral Gene Transcription in Peripheral Blood Leukocytes from Challenged Chickens. Vaccine 2008, 26, 4904–4912. [Google Scholar] [CrossRef] [PubMed]
- Raymond, C.K.; Roberts, B.S.; Garrett-Engele, P.; Lim, L.P.; Johnson, J.M. Simple, Quantitative Primer-Extension PCR Assay for Direct Monitoring of MicroRNAs and Short-Interfering RNAs. RNA 2005, 11, 1737–1744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate Normalization of Real-Time Quantitative RT-PCR Data by Geometric Averaging of Multiple Internal Control Genes. Genome Biol. 2002, 3, RESEARCH0034. [Google Scholar] [CrossRef] [Green Version]
- Hillen, W.; Berens, C. Mechanisms Underlying Expression of Tn10 Encoded Tetracycline Resistance. Annu. Rev. Microbiol. 1994, 48, 345–369. [Google Scholar] [CrossRef]
- Lizio, M.; Deviatiiarov, R.; Nagai, H.; Galan, L.; Arner, E.; Itoh, M.; Lassmann, T.; Kasukawa, T.; Hasegawa, A.; Ros, M.A.; et al. Systematic Analysis of Transcription Start Sites in Avian Development. PLoS Biol. 2017, 15, e2002887. [Google Scholar] [CrossRef] [Green Version]
- Gardiner-Garden, M.; Frommer, M. CpG Islands in Vertebrate Genomes. J. Mol. Biol. 1987, 196, 261–282. [Google Scholar] [CrossRef]
- Rasschaert, P.; Figueroa, T.; Dambrine, G.; Rasschaert, D.; Laurent, S. Alternative Splicing of a Viral Mirtron Differentially Affects the Expression of Other MicroRNAs from Its Cluster and of the Host Transcript. RNA Biol. 2016, 13, 1310–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muylkens, B.; Coupeau, D.; Dambrine, G.; Trapp, S.; Rasschaert, D. Marek’s Disease Virus MicroRNA Designated Mdv1-Pre-MiR-M4 Targets Both Cellular and Viral Genes. Arch. Virol. 2010, 155, 1823–1837. [Google Scholar] [CrossRef]
- Feng, R.; Chen, X.; Yu, Y.; Su, L.; Yu, B.; Li, J.; Cai, Q.; Yan, M.; Liu, B.; Zhu, Z. MiR-126 Functions as a Tumour Suppressor in Human Gastric Cancer. Cancer Lett. 2010, 298, 50–63. [Google Scholar] [CrossRef]
- Lu, W.; Tao, X.; Fan, Y.; Tang, Y.; Xu, X.; Fan, S.; Huang, Y.; Yu, Y.; Luo, D. LINC00888 Promoted Tumorigenicity of Melanoma via MiR-126/CRK Signaling Axis. OncoTargets Ther. 2018, 11, 4431–4442. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.Y.; Wang, W.; Zhao, L.Y.; Guo, B.; Yang, J.; Zhao, X.G.; Hou, N.; Ni, L.; Wang, A.Y.; Song, T.S.; et al. Mir-126 Inhibits Growth of SGC-7901 Cells by Synergistically Targeting the Oncogenes PI3KR2 and Crk, and the Tumor Suppressor PLK2. Int. J. Oncol. 2014, 45, 1257–1265. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Shen, Y.; Ichikawa, H.; Antes, T.; Goldberg, G.S. Regulation of MiRNA Expression by Src and Contact Normalization: Effects on Nonanchored Cell Growth and Migration. Oncogene 2009, 28, 4272–4283. [Google Scholar] [CrossRef] [Green Version]
- Miller, C.T.; Chen, G.; Gharib, T.G.; Wang, H.; Thomas, D.G.; Misek, D.E.; Giordano, T.J.; Yee, J.; Orringer, M.B.; Hanash, S.M.; et al. Increased C-CRK Proto-Oncogene Expression Is Associated with an Aggressive Phenotype in Lung Adenocarcinomas. Oncogene 2003, 22, 7950–7957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, S.; Shi, H.; Han, J.; Zhang, T.; Zhu, W.; Zhang, D. Prognostic Value of MicroRNA-126 and CRK Expression in Gastric Cancer. OncoTargets Ther. 2016, 9, 6127–6135. [Google Scholar] [CrossRef] [Green Version]
- Frampton, A.E.; Krell, J.; Jacob, J.; Stebbing, J.; Castellano, L.; Jiao, L.R. Loss of MiR-126 Is Crucial to Pancreatic Cancer Progression. Expert Rev. Anticancer Ther. 2012, 12, 881–884. [Google Scholar] [CrossRef]
- Gong, X.-H.; Chen, C.; Hou, P.; Zhu, S.-C.; Wu, C.-Q.; Song, C.-L.; Ni, W.; Hu, J.-F.; Yao, D.-K.; Kang, J.-H.; et al. Overexpression of MiR-126 Inhibits the Activation and Migration of HSCs through Targeting CRK. Cell Physiol. Biochem. 2014, 33, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Zhao, Y.; Smith, L.P.; Lawrie, C.H.; Saunders, N.J.; Watson, M.; Nair, V. Differential Expression of MicroRNAs in Marek’s Disease Virus-Transformed T-Lymphoma Cell Lines. J. Gen. Virol. 2009, 90, 1551–1559. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Luo, J.; Zhang, H.; Chang, S.; Song, J. MiRNA Expression Signatures Induced by Marek’s Disease Virus Infection in Chickens. Genomics 2012, 99, 152–159. [Google Scholar] [CrossRef] [Green Version]
- Landgraf, P.; Rusu, M.; Sheridan, R.; Sewer, A.; Iovino, N.; Aravin, A.; Pfeffer, S.; Rice, A.; Kamphorst, A.O.; Landthaler, M.; et al. A Mammalian MicroRNA Expression Atlas Based on Small RNA Library Sequencing. Cell 2007, 129, 1401–1414. [Google Scholar] [CrossRef] [Green Version]
- Chu, F.; Hu, Y.; Zhou, Y.; Guo, M.; Lu, J.; Zheng, W.; Xu, H.; Zhao, J.; Xu, L. MicroRNA-126 Deficiency Enhanced the Activation and Function of CD4+ T Cells by Elevating IRS-1 Pathway. Clin. Exp. Immunol. 2018, 191, 166–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lian, L.; Qu, L.; Chen, Y.; Lamont, S.J.; Yang, N. A Systematic Analysis of MiRNA Transcriptome in Marek’s Disease Virus-Induced Lymphoma Reveals Novel and Differentially Expressed MiRNAs. PLoS ONE 2012, 7, e51003. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, A.; Griffiths-Jones, S.; Ashurst, J.L.; Bradley, A. Identification of Mammalian MicroRNA Host Genes and Transcription Units. Genome Res. 2004, 14, 1902–1910. [Google Scholar] [CrossRef] [Green Version]
- Baskerville, S.; Bartel, D.P. Microarray Profiling of MicroRNAs Reveals Frequent Coexpression with Neighboring MiRNAs and Host Genes. RNA 2005, 11, 241–247. [Google Scholar] [CrossRef] [Green Version]
- Balakrishnan, L.; Milavetz, B. Epigenetic Regulation of Viral Biological Processes. Viruses 2017, 9, 346. [Google Scholar] [CrossRef] [Green Version]
- Flanagan, J.M. Host Epigenetic Modifications by Oncogenic Viruses. Br. J. Cancer 2007, 96, 183–188. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.-N.; Tsai, C.-L.; Tse, K.-P.; Chang, H.-Y.; Chang, Y.-S. The Epstein-Barr Virus Oncogene Product, Latent Membrane Protein 1, Induces the Downregulation of E-Cadherin Gene Expression via Activation of DNA Methyltransferases. Proc. Natl. Acad. Sci. USA 2002, 99, 10084–10089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platt, G.; Carbone, A.; Mittnacht, S. P16INK4a Loss and Sensitivity in KSHV Associated Primary Effusion Lymphoma. Oncogene 2002, 21, 1823–1831. [Google Scholar] [CrossRef] [Green Version]
- Shamay, M.; Krithivas, A.; Zhang, J.; Hayward, S.D. Recruitment of the de Novo DNA Methyltransferase Dnmt3a by Kaposi’s Sarcoma-Associated Herpesvirus LANA. Proc. Natl. Acad. Sci. USA 2006, 103, 14554–14559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Gu, J.; Jiang, P.; Zheng, Y.; Liu, X.; Jiang, X.; Huang, E.; Xiong, S.; Xu, F.; Liu, G.; et al. DNMT1-MicroRNA126 Epigenetic Circuit Contributes to Esophageal Squamous Cell Carcinoma Growth via ADAM9-EGFR-AKT Signaling. Clin. Cancer Res. 2015, 21, 854–863. [Google Scholar] [CrossRef] [Green Version]
- Lutz, G.; Jurak, I.; Kim, E.T.; Kim, J.Y.; Hackenberg, M.; Leader, A.; Stoller, M.L.; Fekete, D.M.; Weitzman, M.D.; Coen, D.M.; et al. Viral Ubiquitin Ligase Stimulates Selective Host MicroRNA Expression by Targeting ZEB Transcriptional Repressors. Viruses 2017, 9, 210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.-C.; Kao, C.-Y.; Lee, H.-J.; Creighton, C.J.; Ittmann, M.M.; Tsai, S.-J.; Tsai, S.Y.; Tsai, M.-J. Dysregulation of MiRNAs-COUP-TFII-FOXM1-CENPF Axis Contributes to the Metastasis of Prostate Cancer. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.; Li, Y.; Zhang, M.; Yang, Y.; Chang, L. MiR-126 Inhibits Cell Proliferation and Induces Cell Apoptosis of Hepatocellular Carcinoma Cells Partially by Targeting Sox2. Hum. Cell 2015, 28, 91–99. [Google Scholar] [CrossRef]
- Otsubo, T.; Akiyama, Y.; Hashimoto, Y.; Shimada, S.; Goto, K.; Yuasa, Y. MicroRNA-126 Inhibits SOX2 Expression and Contributes to Gastric Carcinogenesis. PLoS ONE 2011, 6, e16617. [Google Scholar] [CrossRef] [Green Version]
- Bell, E.S.; Park, M. Models of Crk Adaptor Proteins in Cancer. Genes Cancer 2012, 3, 341–352. [Google Scholar] [CrossRef]
- Schmidpeter, P.A.M.; Schmid, F.X. Molecular Determinants of a Regulatory Prolyl Isomerization in the Signal Adapter Protein C-CrkII. ACS Chem. Biol. 2014, 9, 1145–1152. [Google Scholar] [CrossRef]
- Huang, V.; Place, R.F.; Portnoy, V.; Wang, J.; Qi, Z.; Jia, Z.; Yu, A.; Shuman, M.; Yu, J.; Li, L.-C. Upregulation of Cyclin B1 by MiRNA and Its Implications in Cancer. Nucleic Acids Res. 2012, 40, 1695–1707. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gennart, I.; Petit, A.; Wiggers, L.; Pejaković, S.; Dauchot, N.; Laurent, S.; Coupeau, D.; Muylkens, B. Epigenetic Silencing of MicroRNA-126 Promotes Cell Growth in Marek’s Disease. Microorganisms 2021, 9, 1339. https://doi.org/10.3390/microorganisms9061339
Gennart I, Petit A, Wiggers L, Pejaković S, Dauchot N, Laurent S, Coupeau D, Muylkens B. Epigenetic Silencing of MicroRNA-126 Promotes Cell Growth in Marek’s Disease. Microorganisms. 2021; 9(6):1339. https://doi.org/10.3390/microorganisms9061339
Chicago/Turabian StyleGennart, Isabelle, Astrid Petit, Laetitia Wiggers, Srđan Pejaković, Nicolas Dauchot, Sylvie Laurent, Damien Coupeau, and Benoît Muylkens. 2021. "Epigenetic Silencing of MicroRNA-126 Promotes Cell Growth in Marek’s Disease" Microorganisms 9, no. 6: 1339. https://doi.org/10.3390/microorganisms9061339
APA StyleGennart, I., Petit, A., Wiggers, L., Pejaković, S., Dauchot, N., Laurent, S., Coupeau, D., & Muylkens, B. (2021). Epigenetic Silencing of MicroRNA-126 Promotes Cell Growth in Marek’s Disease. Microorganisms, 9(6), 1339. https://doi.org/10.3390/microorganisms9061339