
Characterization of a Novel Viral Interleukin 8 (vIL-8) Splice Variant Encoded by Marek’s Disease Virus

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Cells
2.3. Viruses
2.4. Western Blotting
2.5. Indirect Immunofluorescence
2.6. Plaque Size Assays
2.7. Multi-Step Growth Kinetics
2.8. In Vivo Experiment
2.9. Virus Quantification in Blood and Feather Follicles
2.10. Statistical Analyses
3. Results
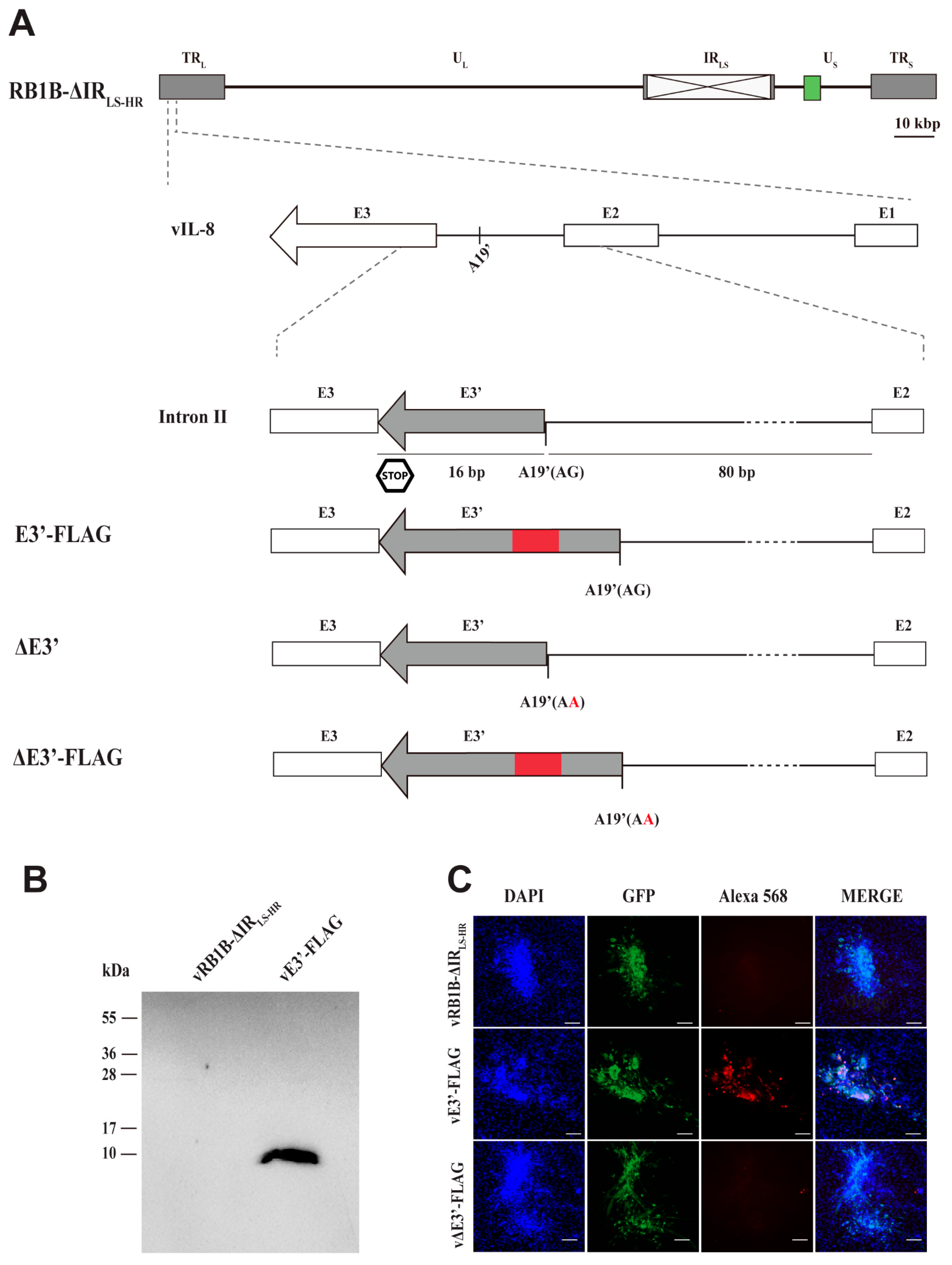
3.1. Detection of Protein Expression of the Novel vIL-8 Splice Variant
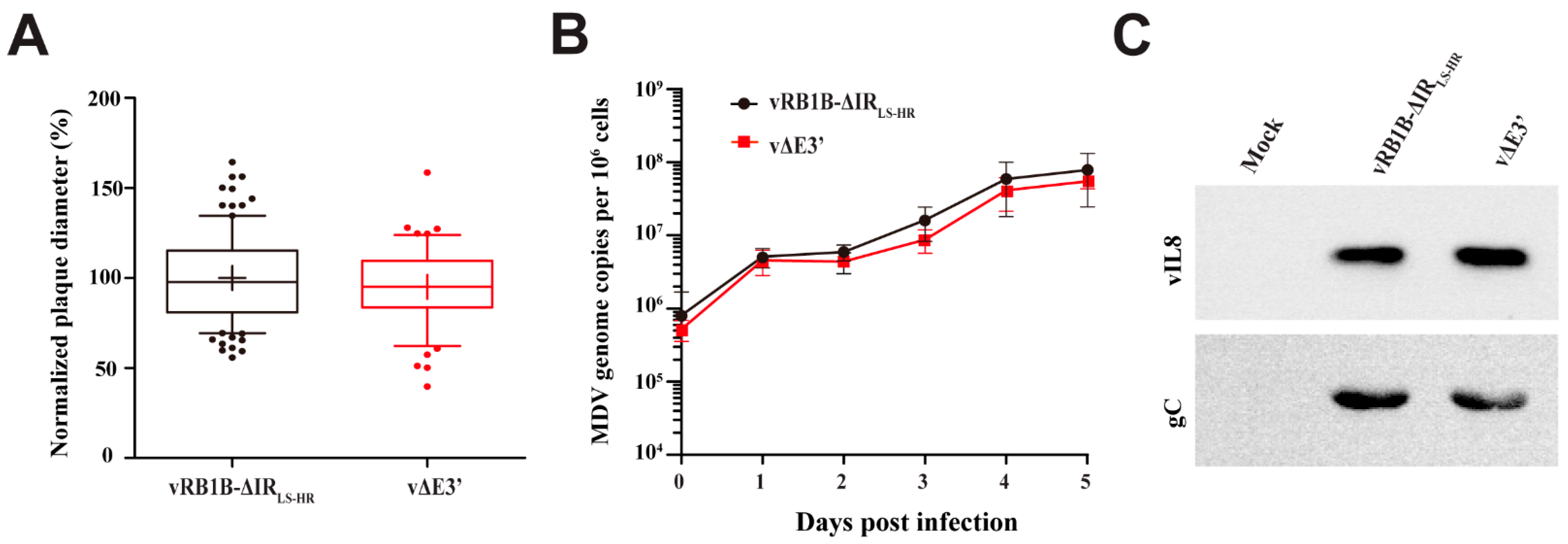
3.2. Abrogation of the Novel vIL-8 Splice Variant
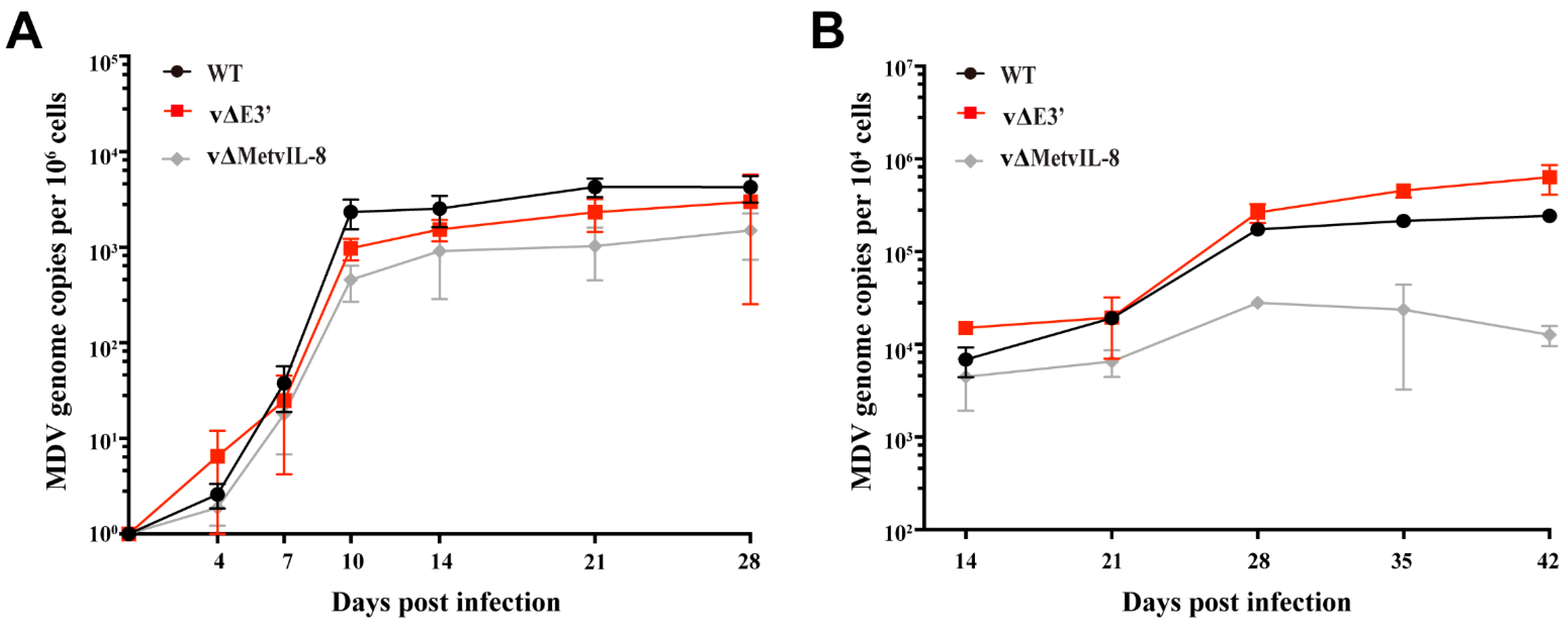
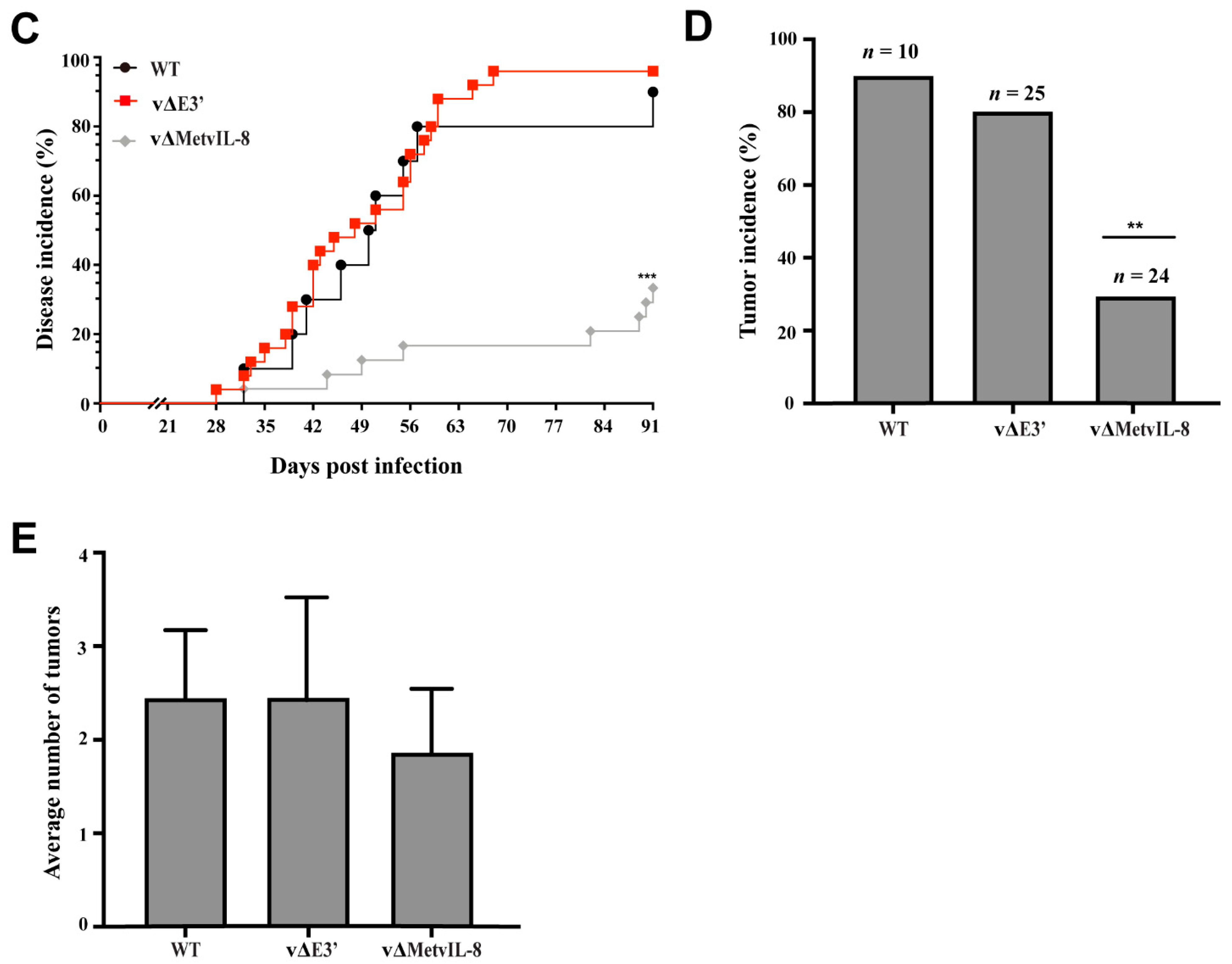
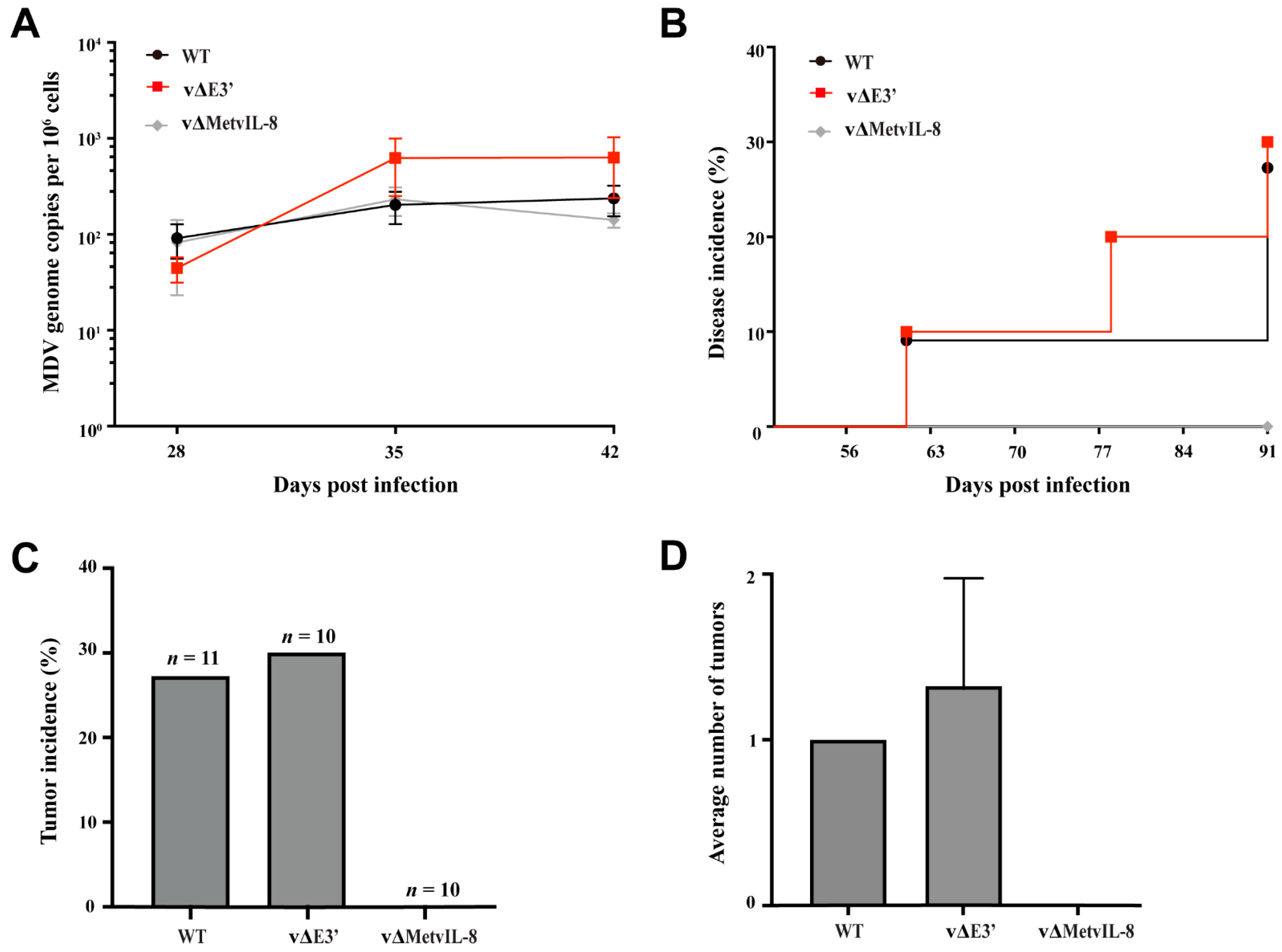
3.3. Role of the Novel vIL-8 Splice Variant in MDV Pathogenesis and Tumorigenesis
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Davison, A.J. Herpesvirus systematics. Vet. Microbiol. 2010, 143, 52–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osterrieder, N.; Kamil, J.P.; Schumacher, D.; Tischer, B.K.; Trapp, S. Marek’s disease virus: From miasma to model. Nat. Rev. Microbiol. 2006, 4, 283–294. [Google Scholar] [CrossRef]
- Rozins, C.; Day, T.; Greenhalgh, S. Managing Marek’s disease in the egg industry. Epidemics 2019, 27, 52–58. [Google Scholar] [CrossRef]
- Gimeno, I.; Schat, K. Virus-induced immunosuppression in chickens. Avian Dis. 2018, 62, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Bertzbach, L.D.; Conradie, A.M.; You, Y.; Kaufer, B.B. Latest insights into Marek’s disease virus pathogenesis and tumorigenesis. Cancers 2020, 12, 647. [Google Scholar] [CrossRef] [Green Version]
- Bertzbach, L.D.; Pfaff, F.; Pauker, V.I.; Kheimar, A.M.; Höper, D.; Härtle, S.; Karger, A.; Kaufer, B.B. The Transcriptional Landscape of Marek’s Disease Virus in Primary Chicken B Cells Reveals Novel Splice Variants and Genes. Viruses 2019, 11, 264. [Google Scholar] [CrossRef] [Green Version]
- Haertle, S.; Alzuheir, I.; Busalt, F.; Waters, V.; Kaiser, P.; Kaufer, B.B. Identification of the Receptor and Cellular Ortholog of the Marek’s Disease Virus (MDV) CXC Chemokine. Front. Microbiol. 2017, 8, 2543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, X.; Lee, L.F.; Reed, W.M.; Kung, H.-J.; Reddy, S.M. Marek’s Disease Virus-Encoded vIL-8 Gene Is Involved in Early Cytolytic Infection but Dispensable for Establishment of Latency. J. Virol. 2004, 78, 4753–4760. [Google Scholar] [CrossRef] [Green Version]
- Engel, A.T.; Selvaraj, R.K.; Kamil, J.P.; Osterrieder, N.; Kaufer, B.B. Marek’s disease viral interleukin-8 promotes lymphoma formation through targeted recruitment of B cells and CD4+ CD25+ T cells. J. Virol. 2012, 86, 8536–8545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertzbach, L.D.; Laparidou, M.; Härtle, S.; Etches, R.J.; Kaspers, B.; Schusser, B.; Kaufer, B.B. Unraveling the role of B cells in the pathogenesis of an oncogenic avian herpesvirus. Proc. Natl. Acad. Sci. USA 2018, 115, 11603–11607. [Google Scholar] [CrossRef] [Green Version]
- Parcells, M.S.; Lin, S.-F.; Dienglewicz, R.L.; Majerciak, V.; Robinson, D.R.; Chen, H.-C.; Wu, Z.; Dubyak, G.R.; Brunovskis, P.; Hunt, H.D. Marek’s disease virus (MDV) encodes an interleukin-8 homolog (vIL-8): Characterization of the vIL-8 protein and a vIL-8 deletion mutant MDV. J. Virol. 2001, 75, 5159–5173. [Google Scholar] [CrossRef] [Green Version]
- Jarosinski, K.W.; Schat, K.A. Multiple alternative splicing to exons II and III of viral interleukin-8 (vIL-8) in the Marek’s disease virus genome: The importance of vIL-8 exon I. Virus Genes 2007, 34, 9–22. [Google Scholar] [CrossRef]
- Sadigh, Y.; Tahiri-Alaoui, A.; Spatz, S.; Nair, V.; Ribeca, P. Pervasive Differential Splicing in Marek’s Disease Virus can Discriminate CVI-988 Vaccine Strain from RB-1B Very Virulent Strain in Chicken Embryonic Fibroblasts. Viruses 2020, 12, 329. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, D.; Tischer, B.K.; Fuchs, W.; Osterrieder, N. Reconstitution of Marek’s disease virus serotype 1 (MDV-1) from DNA cloned as a bacterial artificial chromosome and characterization of a glycoprotein B-negative MDV-1 mutant. J. Virol. 2000, 74, 11088–11098. [Google Scholar] [CrossRef] [Green Version]
- Vychodil, T.; Conradie, A.M.; Trimpert, J.; Aswad, A.; Bertzbach, L.D.; Kaufer, B.B. Marek’s disease virus requires both copies of the inverted repeat regions for efficient in vivo replication and pathogenesis. J. Virol. 2020, 95, e01256-20. [Google Scholar] [CrossRef]
- Tischer, B.K.; von Einem, J.; Kaufer, B.; Osterrieder, N. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 2006, 40, 191–197. [Google Scholar]
- Tischer, B.K.; Kaufer, B.B. Viral Bacterial Artificial Chromosomes: Generation, Mutagenesis, and Removal of Mini-F Sequences. J. Biomed. Biotechnol. 2012, 2012, 472537. [Google Scholar] [CrossRef] [Green Version]
- Bertzbach, L.D.; van Haarlem, D.A.; Härtle, S.; Kaufer, B.B.; Jansen, C.A. Marek’s Disease Virus Infection of Natural Killer Cells. Microorganisms 2019, 7, 588. [Google Scholar] [CrossRef] [Green Version]
- Conradie, A.M.; Bertzbach, L.D.; Trimpert, J.; Patria, J.N.; Murata, S.; Parcells, M.S.; Kaufer, B.B. Distinct polymorphisms in a single herpesvirus gene are capable of enhancing virulence and mediating vaccinal resistance. PLoS Pathog. 2020, 16, e1009104. [Google Scholar] [CrossRef]
- Schippers, T.; Jarosinski, K.; Osterrieder, N. The ORF012 gene of Marek’s disease virus type 1 produces a spliced transcript and encodes a novel nuclear phosphoprotein essential for virus growth. J. Virol. 2015, 89, 1348–1363. [Google Scholar] [CrossRef] [Green Version]
- Tischer, B.K.; Schumacher, D.; Chabanne-Vautherot, D.; Zelnik, V.; Vautherot, J.-F.; Osterrieder, N. High-level expression of Marek’s disease virus glycoprotein C is detrimental to virus growth in vitro. J. Virol. 2005, 79, 5889–5899. [Google Scholar] [CrossRef] [Green Version]
- Previdelli, R.L.; Bertzbach, L.D.; Wight, D.J.; Vychodil, T.; You, Y.; Arndt, S.; Kaufer, B.B. The Role of Marek’s Disease Virus UL12 and UL29 in DNA Recombination and the Virus Lifecycle. Viruses 2019, 11, 111. [Google Scholar] [CrossRef] [Green Version]
- Conradie, A.M.; Bertzbach, L.D.; Bhandari, N.; Parcells, M.; Kaufer, B.B. A Common Live-Attenuated Avian Herpesvirus Vaccine Expresses a Very Potent Oncogene. mSphere 2019, 4, e00658-19. [Google Scholar] [CrossRef] [Green Version]
- Bello, N.; Francino, O.; Sánchez, A. Isolation of genomic DNA from feathers. J. Vet. Diagn. Invest. 2001, 13, 162–164. [Google Scholar] [CrossRef] [Green Version]
- Schommartz, T.; Loroch, S.; Alawi, M.; Grundhoff, A.; Sickmann, A.; Brune, W. Functional dissection of an alternatively spliced herpesvirus gene by splice site mutagenesis. J. Virol. 2016, 90, 4626–4636. [Google Scholar] [CrossRef] [Green Version]
- Anna, A.; Monika, G. Splicing mutations in human genetic disorders: Examples, detection, and confirmation. J. Appl. Genet. 2018, 59, 253–268. [Google Scholar] [CrossRef] [Green Version]
- Tombácz, D.; Csabai, Z.; Szűcs, A.; Balázs, Z.; Moldován, N.; Sharon, D.; Snyder, M.; Boldogkői, Z. Long-read isoform sequencing reveals a hidden complexity of the transcriptional landscape of herpes simplex virus type 1. Front. Microbiol. 2017, 8, 1079. [Google Scholar] [CrossRef]
- O’Grady, T.; Wang, X.; Höner zu Bentrup, K.; Baddoo, M.; Concha, M.; Flemington, E.K. Global transcript structure resolution of high gene density genomes through multi-platform data integration. Nucleic Acids Res. 2016, 44, e145. [Google Scholar] [CrossRef]
- Gatherer, D.; Seirafian, S.; Cunningham, C.; Holton, M.; Dargan, D.J.; Baluchova, K.; Hector, R.D.; Galbraith, J.; Herzyk, P.; Wilkinson, G.W. High-resolution human cytomegalovirus transcriptome. Proc. Natl. Acad. Sci. USA 2011, 108, 19755–19760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anobile, J.M.; Arumugaswami, V.; Downs, D.; Czymmek, K.; Parcells, M.; Schmidt, C.J. Nuclear localization and dynamic properties of the Marek’s disease virus oncogene products Meq and Meq/vIL8. J. Virol. 2006, 80, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Jarosinski, K.W.; Osterrieder, N. Marek’s disease virus expresses multiple UL44 (gC) variants through mRNA splicing that are all required for efficient horizontal transmission. J. Virol. 2012, 86, 7896–7906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Bajwa, K.; Al-Mahmood, M.; Gimeno, I.M.; Reddy, S.M.; Lupiani, B. The role of Meq-vIL8 in regulating Marek’s disease virus pathogenesis. J. Gen. Virol. 2021, 102, 001528. [Google Scholar] [CrossRef] [PubMed]
- Arumugaswami, V.; Kumar, P.M.; Konjufca, V.; Dienglewicz, R.L.; Reddy, S.M.; Parcells, M.S. Latency of Marek’s Disease Virus (MDV) in a Reticuloendotheliosis Virus–Transformed T-Cell Line. II: Expression of the Latent MDV Genome. Avian Dis. 2009, 53, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Kelemen, O.; Convertini, P.; Zhang, Z.; Wen, Y.; Shen, M.; Falaleeva, M.; Stamm, S. Function of alternative splicing. Gene 2013, 514, 1–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, F.; Li, Y.; Yang, X.; Wu, Y.-P.; Lin, L.-J.; Liu, X.-M. Significance of alternative splicing in cancer cells. Chin. Med. J. 2020, 133, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Francies, F.Z.; Dlamini, Z. Aberrant Splicing Events and Epigenetics in Viral Oncogenomics: Current Therapeutic Strategies. Cells 2021, 10, 239. [Google Scholar] [CrossRef]
- Bryant, H.; Wadd, S.; Lamond, A.; Silverstein, S.; Clements, J. Herpes simplex virus IE63 (ICP27) protein interacts with spliceosome-associated protein 145 and inhibits splicing prior to the first catalytic step. J. Virol. 2001, 75, 4376–4385. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.; Patel, A.; Krause, P.R. Hidden regulation of herpes simplex virus 1 pre-mRNA splicing and polyadenylation by virally encoded immediate early gene ICP27. PLoS Pathog. 2019, 15, e1007884. [Google Scholar] [CrossRef]
- Sedlackova, L.; Perkins, K.D.; Lengyel, J.; Strain, A.K.; Van Santen, V.L.; Rice, S.A. Herpes simplex virus type 1 ICP27 regulates expression of a variant, secreted form of glycoprotein C by an intron retention mechanism. J. Virol. 2008, 82, 7443–7455. [Google Scholar] [CrossRef] [Green Version]
- Perkins, K.D.; Gregonis, J.; Borge, S.; Rice, S.A. Transactivation of a viral target gene by herpes simplex virus ICP27 is posttranscriptional and does not require the endogenous promoter or polyadenylation site. J. Virol. 2003, 77, 9872–9884. [Google Scholar] [CrossRef] [Green Version]
- Amor, S.; Strassheim, S.; Dambrine, G.; Remy, S.; Rasschaert, D.; Laurent, S. ICP27 protein of Marek’s disease virus interacts with SR proteins and inhibits the splicing of cellular telomerase chTERT and viral vIL8 transcripts. J. Gen. Virol. 2011, 92, 1273–1278. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Construct | Primer or Probe Sequence (5′–3′) | |
|---|---|---|
| E3′-FLAG | For | GTAGTGTCTGGCTGTAAAGCTAATTTGGTTAAGGTTTTCCGGCAGC GATTACAAGGATGACGACGATAAGTAGGGATAACAGGGTAATCGATTT |
| Rev | ACATACCTTCCTGTTCTTCTTGAGAGCAAAGCTACAAAAGCTTAT CGTCGTCATCCTTGTAATCGCTGCCGCCAGTGTTACAACCAATTAACC | |
| vΔE3′-FLAG | For | CTTCCTGTTCTTCTTGAGAGCAAAGCTACAAAAGGGAAAACTTTA ACCAAATTAGCTTTACAGCCAGTAGGGATAACAGGGTAATCGATTT |
| Rev | CTTAGGTGTAGTGTCTGGCTGTAAAGCTAATTTGGTTAAAGTTTTCCGCCAGTGTTACAACCAATTAACC | |
| vΔE3′ | For | GCTACAAAAGCTTATCGTCGTCATCCTTGTAATCGGAAAACTTTA ACCAAATTAGCTTTACAGCCAGTAGGGATAACAGGGTAATCGATTT |
| Rev | CTTAGGTGTAGTGTCTGGCTGTAAAGCTAATTTGGTTAAAG TTTTCCGCCAGTGTTACAACCAATTAACC | |
| vIL-8 sequencing | For | CCGTATCCCTGCTCCATCCAATAGC |
| Rev | GGTCTCCAATATCACGTGTTGGTGG | |
| ICP4 | For | CGTGTTTTCCGGCATGTG |
| Rev | TCCCATACCAATCCTCATCCA | |
| Probe | FAM-CCCCCACCAGGTGCAGGCA-TAM | |
| iNOS | For | GAGTGGTTTAAGGAGTTGGATCTGA |
| Rev | TTCCAGACCTCCCACCTCAA | |
| Probe | FAM-CTCTGCCTGCTGTTGCCAACATGC-TAM | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
You, Y.; Hagag, I.T.; Kheimar, A.; Bertzbach, L.D.; Kaufer, B.B. Characterization of a Novel Viral Interleukin 8 (vIL-8) Splice Variant Encoded by Marek’s Disease Virus. Microorganisms 2021, 9, 1475. https://doi.org/10.3390/microorganisms9071475
You Y, Hagag IT, Kheimar A, Bertzbach LD, Kaufer BB. Characterization of a Novel Viral Interleukin 8 (vIL-8) Splice Variant Encoded by Marek’s Disease Virus. Microorganisms. 2021; 9(7):1475. https://doi.org/10.3390/microorganisms9071475
Chicago/Turabian StyleYou, Yu, Ibrahim T. Hagag, Ahmed Kheimar, Luca D. Bertzbach, and Benedikt B. Kaufer. 2021. "Characterization of a Novel Viral Interleukin 8 (vIL-8) Splice Variant Encoded by Marek’s Disease Virus" Microorganisms 9, no. 7: 1475. https://doi.org/10.3390/microorganisms9071475
APA StyleYou, Y., Hagag, I. T., Kheimar, A., Bertzbach, L. D., & Kaufer, B. B. (2021). Characterization of a Novel Viral Interleukin 8 (vIL-8) Splice Variant Encoded by Marek’s Disease Virus. Microorganisms, 9(7), 1475. https://doi.org/10.3390/microorganisms9071475