
Hepatitis B Virus DNA Integration, Chronic Infections and Hepatocellular Carcinoma

 ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Evolution of HBV and Co-Existence with Human Populations
3. Geographical Distribution of Human HBV Genotypes and Their Role in the Natural History of the Infection
4. Natural History of Chronic HBV Infection
5. Genome Organization and Types of Genomic Material within the HBV Life Cycle
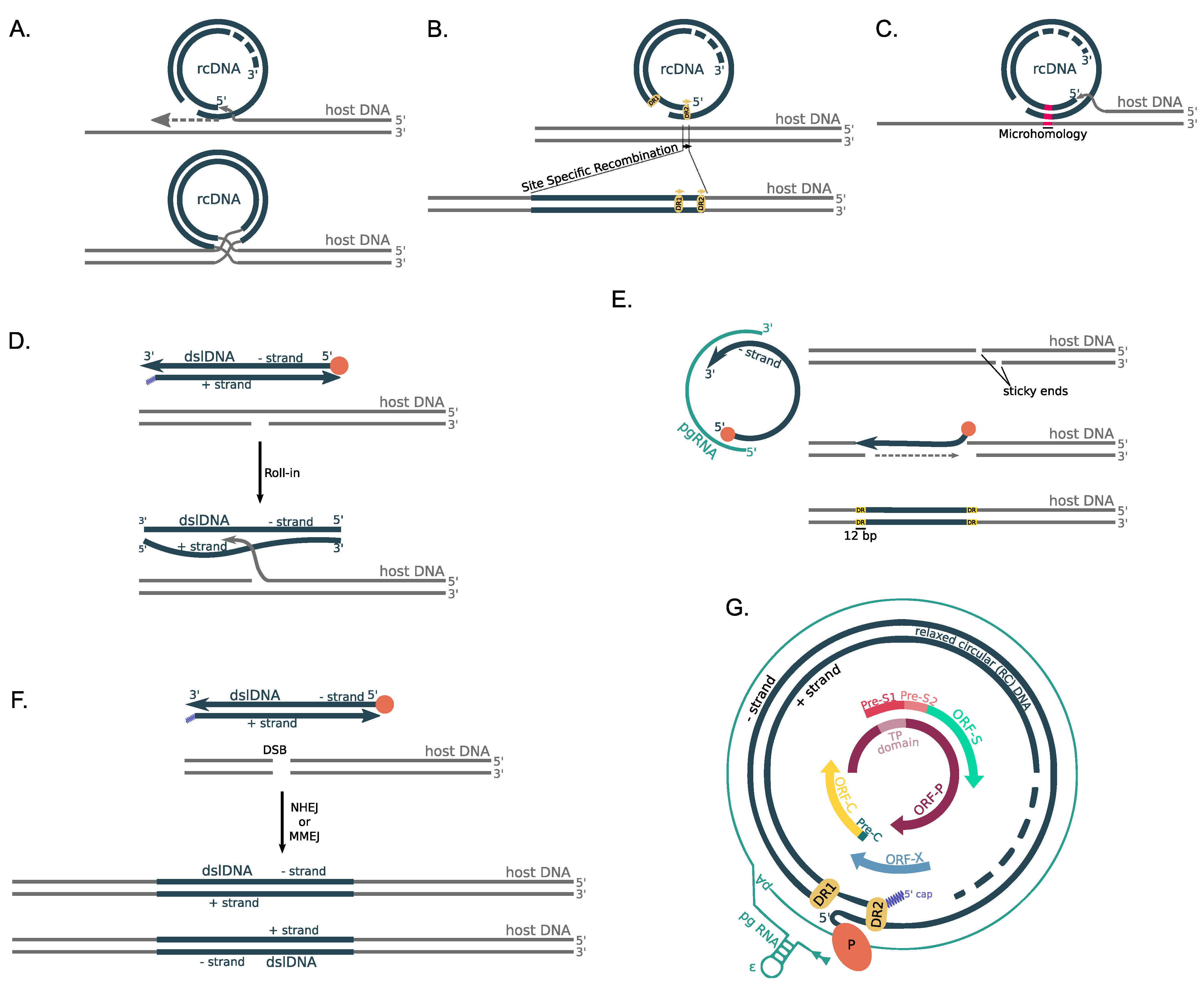
6. Proposed Molecular Mechanisms of HBV DNA Integration
7. Hepatocellular Carcinoma (HCC)
8. HBV Integration Sites into the Human Genome and Target Genes
8.1. Genes Involved in the Cell Cycle G1/S Transition (Gene Ontology (GO) ID:0044843)
8.2. Genes Involved in the DNA Replication (GO:0006260)
8.3. Genes Involved in the Histone Modification (GO:0016570)
8.4. Genes Involved in the Stem Cell Differentiation (GO:0048863)
8.5. Genes Involved in the Wnt Signaling Pathway (GO:0016055, GO:0198738)
8.6. Genes Involved in Angiogenesis (GO:0001525)
8.7. Genes Involved in Blood Circulation (GO:0008015)
8.8. Other Protein-Coding Affected Genes
8.9. Long Non-Coding RNA Genes (lincRNAs)
9. Molecular Mechanisms of Hepatocarcinogenesis Caused by HBV Integration
10. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| HBV | Hepatitis B Virus |
| CHB | Chronic HBV infection |
| HCC | Hepatocellular Carcinoma |
| HBeAg | Hepatitis B e antigen |
| HBcAg | Hepatitis B core antigen |
| HBsAg | Hepatitis B surface antigen |
| anti-HBs | Hepatitis B surface antibody |
| anti-HBc | Total hepatitis B core antibody |
| IgM anti-HBc | IgM antibody to hepatitis B core antigen |
| rcDNA | relaxed circular DNA |
| dslDNA | double-stranded linear DNA |
| cccDNA | covalently closed circular DNA |
| pgRNA | pre-genomic RNA |
| NHEJ | Non-Homologous End Joining |
| MMEJ | Microhomology-Mediated End Joining |
References
- Blumberg, B.S.; Alter, H.J.; Visnich, S. A “New” Antigen in Leukemia Sera. JAMA J. Am. Med. Assoc. 1984, 252, 252–257. [Google Scholar] [CrossRef]
- Block, T.M.; Alter, H.J.; London, W.T.; Bray, M. A historical perspective on the discovery and elucidation of the hepatitis B virus. Antivir. Res. 2016, 131, 109–123. [Google Scholar] [CrossRef]
- Gruber, W.; Virchow, R. Ueber das Vorkommen und den Nachweis des hepatogenen, insbesondere des katarrhalischen Icterus. Arch. Pathol. Anat. Physiol. Klin. Med. 1865, 32, 117–125. [Google Scholar] [CrossRef] [Green Version]
- Papavramidou, N.; Fee, E.; Christopoulou-Aletra, H. Jaundice in the hippocratic corpus. J. Gastrointest. Surg. 2007, 11, 1728–1731. [Google Scholar] [CrossRef]
- Neefe, J.R.; Gellis, S.S.; Stokes, J. Homologous serum hepatitis and infectious (epidemic) hepatitis: Studies in volunteers bearing on immunological and other characteristics of the etiological agents. Am. J. Med. 1946, 1, 3–22. [Google Scholar] [CrossRef]
- Ratnoff, O.D.; Patek, A.J.J.R. The natural history of laennec’s cirrhosis of the liver an Analysis of 386 CASES. Medicine 1942, 21, 207–268. [Google Scholar] [CrossRef]
- Millman, I.; Eisenstein, T.; Blumberg, B.S. The Development of the Hepatitis B Vaccine; Springer: Jersey City, NJ, USA, 1984; pp. 137–147. [Google Scholar] [CrossRef]
- Hoofnagle, J.H.; Mullen, K.D.; Jones, D.B.; Rustgi, V.; Di Bisceglie, A.; Peters, M.; Waggoner, J.G.; Park, Y.; Jones, E.A. Treatment of Chronic Non-A, Non-B Hepatitis with Recombinant Human Alpha Interferon. N. Engl. J. Med. 1986, 315, 1575–1578. [Google Scholar] [CrossRef] [Green Version]
- Lai, C.L.; Chien, R.N.; Leung, N.W.; Chang, T.T.; Guan, R.; Tai, D.I.; Ng, K.Y.; Wu, P.C.; Dent, J.C.; Barber, J.; et al. A One-Year Trial of Lamivudine for Chronic Hepatitis B. N. Engl. J. Med. 1998, 339, 61–68. [Google Scholar] [CrossRef] [Green Version]
- Shafritz, D.A.; Shouval, D.; Sherman, H.I.; Hadziyannis, S.J.; Kew, M.C. Integration of Hepatitis B Virus DNA into the Genome of Liver Cells in Chronic Liver Disease and Hepatocellular Carcinoma. N. Engl. J. Med. 1981, 305, 1067–1073. [Google Scholar] [CrossRef]
- Koshy, R.; Koch, S.; Freytag von Loringhoven, A.; Kahmann, R.; Murray, K.; Hofschneider, P.H. Integration of hepatitis B virus DNA: Evidence for integration in the single-stranded gap. Cell 1983, 34, 215–223. [Google Scholar] [CrossRef]
- Brechot, C.; Pourcel, C.; Louise, A.; Rain, B.; Tiollais, P. Presence of integrated hepatitis B virus DNA sequences in cellular DNA of human hepatocellular carcinoma. Nature 1980, 286, 533–535. [Google Scholar] [CrossRef]
- Chakraborty, P.R.; Ruiz-Opazo, N.; Shouval, D.; Shafritz, D.A. Identification of integrated hepatitis B virus DNA and expression of viral RNA in an HBsAg-producing human hepatocellular carcinoma cell line. Nature 1980, 286, 531–533. [Google Scholar] [CrossRef]
- Guerrero, R.B.; Roberts, L.R. The role of hepatitis B virus integrations in the pathogenesis of human hepatocellular carcinoma. J. Hepatol. 2005, 42, 760–777. [Google Scholar] [CrossRef]
- Murakami, Y.; Saigo, K.; Takashima, H.; Minami, M.; Okanoue, T.; Bréchet, C.; Paterlini-Bréchot, P. Large scaled analysis of hepatitis B virus (HBV) DNA integration in HBV related hepatocellular carcinomas. Gut 2005, 54, 1162–1168. [Google Scholar] [CrossRef]
- Saigo, K.; Yoshida, K.; Ikeda, R.; Sakamoto, Y.; Murakami, Y.; Urashima, T.; Asano, T.; Kenmochi, T.; Inoue, I. Integration of hepatitis B virus DNA into the myeloid/lymphoid or mixed-lineage leukemia (MLL4) gene and rearrangements of MLL4 in human hepatocellular carcinoma. Hum. Mutat. 2008, 29, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Ferber, M.J.; Montoya, D.P.; Yu, C.; Aderca, I.; McGee, A.; Thorland, E.C.; Nagorney, D.M.; Gostout, B.S.; Burgart, L.J.; Boix, L.; et al. Integrations of the hepatitis B virus (HBV) and human papillomavirus (HPV) into the human telomerase reverse transcriptase (hTERT) gene in liver and cervical cancers. Oncogene 2003, 22, 3813–3820. [Google Scholar] [CrossRef] [Green Version]
- Paterlini-Bréchot, P.; Saigo, K.; Murakami, Y.; Chami, M.; Gozuacik, D.; Mugnier, C.; Lagorce, D.; Bréchot, C. Hepatitis B virus-related insertional mutagenesis occurs frequently in human liver cancers and recurrently targets human telomerase gene. Oncogene 2003, 22, 3911–3916. [Google Scholar] [CrossRef] [PubMed]
- Gozuacik, D.; Murakami, Y.; Saigo, K.; Chami, M.; Mugnier, C.; Lagorce, D.; Okanoue, T.; Urashima, T.; Bréchot, C.; Paterlini-Bréchot, P. Identification of human cancer-related genes by naturally occurring hepatitis B virus DNA tagging. Oncogene 2001, 20, 6233–6240. [Google Scholar] [CrossRef] [Green Version]
- Razavi-Shearer, D.; Gamkrelidze, I.; Nguyen, M.H.; Chen, D.S.; Van Damme, P.; Abbas, Z.; Abdulla, M.; Abou Rached, A.; Adda, D.; Aho, I.; et al. Global prevalence, treatment, and prevention of hepatitis B virus infection in 2016: A modelling study. Lancet Gastroenterol. Hepatol. 2018, 3, 383–403. [Google Scholar] [CrossRef]
- Waheed, Y.; Siddiq, M.; Jamil, Z.; Najmi, M.H. Hepatitis elimination by 2030: Progress and challenges. World J. Gastroenterol. 2018, 24, 4959–4961. [Google Scholar] [CrossRef]
- Hutin, Y.J.F.; Bulterys, M.; Hirnschall, G.O. How far are we from viral hepatitis elimination service coverage targets? J. Int. AIDS Soc. 2018, 21, e25050. [Google Scholar] [CrossRef] [Green Version]
- Tatemichi, M.; Furuya, H.; Nagahama, S.; Takaya, N.; Shida, Y.; Fukai, K.; Owada, S.; Endo, H.; Kinoue, T.; Korenaga, M. A nationwide cross-sectional survey on hepatitis B and C screening among workers in Japan. Sci. Rep. 2020, 10, 1–8. [Google Scholar] [CrossRef]
- Seeger, C.; Mason, W.S. Molecular biology of hepatitis B virus infection. Virology 2015, 479–480, 672–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Revill, P.A.; Tu, T.; Netter, H.J.; Yuen, L.K.; Locarnini, S.A.; Littlejohn, M. The evolution and clinical impact of hepatitis B virus genome diversity. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 618–634. [Google Scholar] [CrossRef] [PubMed]
- Paraskevis, D.; Magiorkinis, G.; Magiorkinis, E.; Ho, S.Y.; Belshaw, R.; Allain, J.P.; Hatzakis, A. Dating the origin and dispersal of hepatitis B virus infection in humans and primates. Hepatology 2013, 57, 908–916. [Google Scholar] [CrossRef]
- Paraskevis, D.; Angelis, K.; Magiorkinis, G.; Kostaki, E.; Ho, S.Y.; Hatzakis, A. Dating the origin of hepatitis B virus reveals higher substitution rate and adaptation on the branch leading to F/H genotypes. Mol. Phylogenet. Evol. 2015, 93, 44–54. [Google Scholar] [CrossRef]
- Comas, I.; Coscolla, M.; Luo, T.; Borrell, S.; Holt, K.E.; Kato-Maeda, M.; Parkhill, J.; Malla, B.; Berg, S.; Thwaites, G.; et al. Out-of-Africa migration and Neolithic coexpansion of Mycobacterium tuberculosis with modern humans. Nat. Genet. 2013, 45, 1176–1182. [Google Scholar] [CrossRef]
- Linz, B.; Balloux, F.; Moodley, Y.; Manica, A.; Liu, H.; Roumagnac, P.; Falush, D.; Stamer, C.; Prugnolle, F.; van der Merwe, S.W.; et al. An African origin for the intimate association between humans and Helicobacter pylori. Nature 2007, 445, 915–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahila Bar-Gal, G.; Kim, M.J.; Klein, A.; Shin, D.H.; Oh, C.S.; Kim, J.W.; Kim, T.H.; Kim, S.B.; Grant, P.R.; Pappo, O.; et al. Tracing hepatitis B virus to the 16th century in a Korean mummy. Hepatology 2012, 56, 1671–1680. [Google Scholar] [CrossRef]
- Patterson Ross, Z.; Klunk, J.; Fornaciari, G.; Giuffra, V.; Duchêne, S.; Duggan, A.T.; Poinar, D.; Douglas, M.W.; Eden, J.S.; Holmes, E.C.; et al. The paradox of HBV evolution as revealed from a 16th century mummy. PLoS Pathog. 2018, 14, e1006750. [Google Scholar] [CrossRef] [Green Version]
- Mühlemann, B.; Jones, T.C.; Damgaard, P.d.B.; Allentoft, M.E.; Shevnina, I.; Logvin, A.; Usmanova, E.; Panyushkina, I.P.; Boldgiv, B.; Bazartseren, T.; et al. Ancient hepatitis B viruses from the Bronze Age to the Medieval period. Nature 2018, 557, 418–423. [Google Scholar] [CrossRef]
- Krause-Kyora, B.; Susat, J.; Key, F.M.; Kühnert, D.; Bosse, E.; Immel, A.; Rinne, C.; Kornell, S.C.; Yepes, D.; Franzenburg, S.; et al. Neolithic and medieval virus genomes reveal complex evolution of hepatitis B. eLife 2018, 7, 1–15. [Google Scholar] [CrossRef]
- Dill, J.A.; Camus, A.C.; Leary, J.H.; Di Giallonardo, F.; Holmes, E.C.; Ng, T.F.F. Distinct Viral Lineages from Fish and Amphibians Reveal the Complex Evolutionary History of Hepadnaviruses. J. Virol. 2016, 90, 7920–7933. [Google Scholar] [CrossRef] [Green Version]
- Littlejohn, M.; Locarnini, S.; Yuen, L. Origins and Evolution of Hepatitis B Virus and Hepatitis D Virus. Cold Spring Harb. Perspect. Med. 2016, 6, a021360. [Google Scholar] [CrossRef]
- Velkov, S.; Ott, J.J.; Protzer, U.; Michler, T. The global hepatitis B virus genotype distribution approximated from available genotyping data. Genes 2018, 9, 495. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Kramvis, A. Genotypes and genetic variability of hepatitis B virus. Intervirology 2014, 57, 141–150. [Google Scholar] [CrossRef]
- Sunbul, M. Hepatitis B virus genotypes: Global distribution and clinical importance. World J. Gastroenterol. 2014, 20, 5427–5434. [Google Scholar] [CrossRef]
- Domingo, E.; Sheldon, J.; Perales, C. Viral quasispecies evolution. Microbiol. Mol. Biol. Rev. MMBR 2012, 76, 159–216. [Google Scholar] [CrossRef] [Green Version]
- Raihan, R.; Akbar, S.M.F.; Mahtab, M.A.; Takahashi, K.; Masumoto, J.; Tabassum, S.; Tee, K.K.; Mohamed, R.B. Genomic analysis of Hepatitis B virus and its association with disease manifestations in Bangladesh. PLoS ONE 2019, 14, e0218744. [Google Scholar] [CrossRef]
- Sarma, M.P.; Bhattacharjee, M.; Kar, P.; Medhi, S. Detection of HBV Genotype C in Hepatocellular Carcinoma Patients from North East India: A Brief Report. Asian Pac. J. Cancer Prev. APJCP 2018, 19, 1741–1746. [Google Scholar] [CrossRef]
- Livingston, S.E.; Simonetti, J.P.; Bulkow, L.R.; Homan, C.E.; Snowball, M.M.; Cagle, H.H.; Negus, S.E.; McMahon, B.J. Clearance of Hepatitis B e Antigen in Patients with Chronic Hepatitis B and Genotypes A, B, C, D, and F. Gastroenterology 2007, 133, 1452–1457. [Google Scholar] [CrossRef] [PubMed]
- Giovanna, F.; Bortolotti, F.; Francesco, D. Natural history of chronic hepatitis B: Special emphasis on disease progression and prognostic factors. J. Hepatol. 2008, 48, 335–352. [Google Scholar] [CrossRef]
- Guidotti, L.G.; Chisari, F.V. Immunobiology and pathogenesis of viral hepatitis. Annu. Rev. Pathol. 2006, 1, 23–61. [Google Scholar] [CrossRef] [Green Version]
- Kuipery, A.; Gehring, A.J.; Isogawa, M. Mechanisms of HBV immune evasion. Antivir. Res. 2020, 179, 104816. [Google Scholar] [CrossRef]
- McMahon, B.J. The natural history of chronic hepatitis B virus infection. Hepatology 2009, 49, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Di Marco, V.; Lo Iacono, O.; Cammà, C.; Vaccaro, A.; Giunta, M.; Martorana, G.; Fuschi, P.; Almasio, P.L.; Craxì, A. The long-term course of chronic hepatitis B. Hepatology 1999, 30, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Iloeje, U.H.; Yang, H.I.; Su, J.; Jen, C.L.; You, S.L.; Chen, C.J. Predicting cirrhosis risk based on the level of circulating hepatitis B viral load. Gastroenterology 2006, 130, 678–686. [Google Scholar] [CrossRef] [PubMed]
- European Association For The Study Of The Liver. EASL 2017 Clinical Practice Guidelines on the management of hepatitis B virus infection. J. Hepatol. 2017, 67, 370–398. [Google Scholar] [CrossRef] [Green Version]
- Hui, C.K.; Leung, N.; Yuen, S.T.; Zhang, H.Y.; Leung, K.W.; Lu, L.; Cheung, S.K.; Wong, W.M.; Lau, G.K. Natural history and disease progression in Chinese chronic hepatitis B patients in immune-tolerant phase. Hepatology 2007, 46, 395–401. [Google Scholar] [CrossRef]
- McMahon, B.J. Epidemiology and natural history of hepatitis B. Semin. Liver Dis. 2005, 25, 3–8. [Google Scholar] [CrossRef]
- Lok, A.S.F.; McMahon, B.J. Chronic hepatitis B. Hepatology (Baltimore Md.) 2007, 45, 507–539. [Google Scholar] [CrossRef] [Green Version]
- Gish, R.G.; Given, B.D.; Lai, C.L.; Locarnini, S.A.; Lau, J.Y.; Lewis, D.L.; Schluep, T. Chronic hepatitis B: Virology, natural history, current management and a glimpse at future opportunities. Antivir. Res. 2015, 121, 47–58. [Google Scholar] [CrossRef]
- Thomas, E.; Yoneda, M.; Schiff, E.R. Viral hepatitis: Past and future of HBV and HDV. Cold Spring Harb. Perspect. Med. 2015, 5, a021345. [Google Scholar] [CrossRef] [Green Version]
- Iannacone, M.; Guidotti, L.G. Immunobiology and pathogenesis of hepatitis B virus infection. Nat. Rev. Immunol. 2021. [Google Scholar] [CrossRef]
- Fanning, G.C.; Zoulim, F.; Hou, J.; Bertoletti, A. Therapeutic strategies for hepatitis B virus infection: Towards a cure. Nat. Rev. Drug Discov. 2019, 18, 827–844. [Google Scholar] [CrossRef]
- Hu, J.; Liu, K. Complete and incomplete hepatitis B virus particles: Formation, function, and application. Viruses 2017, 9, 56. [Google Scholar] [CrossRef] [Green Version]
- Tsukuda, S.; Watashi, K. Hepatitis B virus biology and life cycle. Antivir. Res. 2020, 182, 104925. [Google Scholar] [CrossRef]
- Dane, D.S.; Cameron, C.H.; Briggs, M. Virus-like particles in serum of patients with Australia-antigen-associated hepatitis. Lancet 1970, 1, 695–698. [Google Scholar] [CrossRef]
- Luckenbaugh, L.; Kitrinos, K.M.; Delaney, W.E.t.; Hu, J. Genome-free hepatitis B virion levels in patient sera as a potential marker to monitor response to antiviral therapy. J. Viral Hepat. 2015, 22, 561–570. [Google Scholar] [CrossRef] [Green Version]
- Barrera, A.; Guerra, B.; Notvall, L.; Lanford, R.E. Mapping of the Hepatitis B Virus Pre-S1 Domain Involved in Receptor Recognition. J. Virol. 2005, 79, 9786–9798. [Google Scholar] [CrossRef] [Green Version]
- Rabe, B.; Glebe, D.; Kann, M. Lipid-Mediated Introduction of Hepatitis B Virus Capsids into Nonsusceptible Cells Allows Highly Efficient Replication and Facilitates the Study of Early Infection Events. J. Virol. 2006, 80, 5465–5473. [Google Scholar] [CrossRef] [Green Version]
- Blondot, M.L.; Bruss, V.; Kann, M. Intracellular transport and egress of hepatitis B virus. J. Hepatol. 2016, 64, S49–S59. [Google Scholar] [CrossRef]
- KöNiger, C.; Wingert, I.; Marsmann, M.; Rösler, C.; Beck, J.; Nassal, M. Involvement of the host DNA-repair enzyme TDP2 in formation of the covalently closed circular DNA persistence reservoir of hepatitis B viruses. Proc. Natl. Acad. Sci. USA 2014, 111, E4244–E4253. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.; McAllister, R.; Boregowda, R.; Sohn, J.A.; Ledesma, F.C.; Caldecott, K.W.; Seeger, C.; Hu, J. Does tyrosyl DNA phosphodiesterase-2 play a role in hepatitis B virus genome repair? PLoS ONE 2015, 10, e0128401. [Google Scholar] [CrossRef] [Green Version]
- Qi, Y.; Gao, Z.; Xu, G.; Peng, B.; Liu, C.; Yan, H.; Yao, Q.; Sun, G.; Liu, Y.; Tang, D.; et al. DNA Polymerase κ Is a Key Cellular Factor for the Formation of Covalently Closed Circular DNA of Hepatitis B Virus. PLoS Pathog. 2016, 12, e1005893. [Google Scholar] [CrossRef] [Green Version]
- Tang, L.; Sheraz, M.; McGrane, M.; Chang, J.; Guo, J.T. DNA Polymerase alpha is essential for intracellular amplification of hepatitis B virus covalently closed circular DNA. PLoS Pathog. 2019, 15, e1007742. [Google Scholar] [CrossRef]
- Long, Q.; Yan, R.; Hu, J.; Cai, D.; Mitra, B.; Kim, E.S.; Marchetti, A.; Zhang, H.; Wang, S.; Liu, Y.; et al. The role of host DNA ligases in hepadnavirus covalently closed circular DNA formation. PLoS Pathog. 2017, 13, e1006784. [Google Scholar] [CrossRef]
- Kitamura, K.; Que, L.; Shimadu, M.; Koura, M.; Ishihara, Y.; Wakae, K.; Nakamura, T.; Watashi, K.; Wakita, T.; Muramatsu, M. Flap endonuclease 1 is involved in cccDNA formation in the hepatitis B virus. PLoS Pathog. 2018, 14, e1007124. [Google Scholar] [CrossRef]
- Gerlich, W.H.; Robinson, W.S. Hepatitis B virus contains protein attached to the 5’ terminus of its complete DNA strand. Cell 1980, 21, 801–809. [Google Scholar] [CrossRef]
- Guo, H.; Jiang, D.; Zhou, T.; Cuconati, A.; Block, T.M.; Guo, J.T. Characterization of the Intracellular Deproteinized Relaxed Circular DNA of Hepatitis B Virus: An Intermediate of Covalently Closed Circular DNA Formation. J. Virol. 2007, 81, 12472–12484. [Google Scholar] [CrossRef] [Green Version]
- Nassal, M. HBV cccDNA: Viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 2015, 64, 1972–1984. [Google Scholar] [CrossRef] [Green Version]
- Rall, L.B.; Standring, D.N.; Laub, O.; Rutter, W.J. Transcription of hepatitis B virus by RNA polymerase II. Mol. Cell. Biol. 1983, 3, 1766–1773. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Summers, J. Integration of hepadnavirus DNA in infected liver: Evidence for a linear precursor. J. Virol. 1999, 73, 9710–9717. [Google Scholar] [CrossRef] [Green Version]
- Mason, W.S.; Jilbert, A.R.; Summers, J. Clonal expansion of hepatocytes during chronic woodchuck hepatitis virus infection. Proc. Natl. Acad. Sci. USA 2005, 102, 1139–1144. [Google Scholar] [CrossRef] [Green Version]
- Summers, J.; Mason, W.S. Residual integrated viral DNA after hepadnavirus clearance by nucleoside analog therapy. Proc. Natl. Acad. Sci. USA 2004, 101, 638–640. [Google Scholar] [CrossRef] [Green Version]
- Mason, W.S.; Liu, C.; Aldrich, C.E.; Litwin, S.; Yeh, M.M. Clonal expansion of normal-appearing human hepatocytes during chronic hepatitis B virus infection. J. Virol. 2010, 84, 8308–8315. [Google Scholar] [CrossRef] [Green Version]
- Tu, T.; Mason, W.S.; Clouston, A.D.; Shackel, N.A.; McCaughan, G.W.; Yeh, M.M.; Schiff, E.R.; Ruszkiewicz, A.R.; Chen, J.W.; Harley, H.A.; et al. Clonal expansion of hepatocytes with a selective advantage occurs during all stages of chronic hepatitis B virus infection. J. Viral Hepat. 2015, 22, 737–753. [Google Scholar] [CrossRef]
- Mason, W.S.; Gill, U.S.; Litwin, S.; Zhou, Y.; Peri, S.; Pop, O.; Hong, M.L.; Naik, S.; Quaglia, A.; Bertoletti, A.; et al. HBV DNA Integration and Clonal Hepatocyte Expansion in Chronic Hepatitis B Patients Considered Immune Tolerant. Gastroenterology 2016, 151, 986–998.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, T.; Budzinska, M.A.; Vondran, F.W.R.; Shackel, N.A.; Urban, S. Hepatitis B Virus DNA Integration Occurs Early in the Viral Life Cycle in an In Vitro Infection Model via Sodium Taurocholate Cotransporting Polypeptide-Dependent Uptake of Enveloped Virus Particles. J. Virol. 2018, 92, e02007-17. [Google Scholar] [CrossRef] [Green Version]
- Sung, W.K.; Zheng, H.; Li, S.; Chen, R.; Liu, X.; Li, Y.; Lee, N.P.; Lee, W.H.; Ariyaratne, P.N.; Tennakoon, C.; et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat. Genet. 2012, 44, 765–769. [Google Scholar] [CrossRef]
- Zhao, L.H.; Liu, X.; Yan, H.X.; Li, W.Y.; Zeng, X.; Yang, Y.; Zhao, J.; Liu, S.P.; Zhuang, X.H.; Lin, C.; et al. Genomic and oncogenic preference of HBV integration in hepatocellular carcinoma. Nat. Commun. 2016, 7, 12992. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Jhunjhunwala, S.; Liu, J.; Haverty, P.M.; Kennemer, M.I.; Guan, Y.; Lee, W.; Carnevali, P.; Stinson, J.; Johnson, S.; et al. The effects of hepatitis B virus integration into the genomes of hepatocellular carcinoma patients. Genome Res. 2012, 22, 593–601. [Google Scholar] [CrossRef] [Green Version]
- Zhao, K.; Liu, A.; Xia, Y. Insights into Hepatitis B Virus DNA Integration-55 Years after Virus Discovery. Innov. States 2020, 1, 100034. [Google Scholar] [CrossRef]
- Dejean, A.; Sonigo, P.; Wain-Hobson, S.; Tiollais, P. Specific hepatitis B virus integration in hepatocellular carcinoma DNA through a viral 11-base-pair direct repeat. Proc. Natl. Acad. Sci. USA 1984, 81, 5350–5354. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.; Yang, Y.; Zhang, L.; Tang, G.; Wang, Y.; Xue, G.; Zhou, W.; Sun, S. Characterization of the genotype and integration patterns of hepatitis B virus in early- and late-onset hepatocellular carcinoma. Hepatology 2015, 61, 1821–1831. [Google Scholar] [CrossRef]
- Chauhan, R.; Churchill, N.D.; Mulrooney-Cousins, P.M.; Michalak, T.I. Initial sites of hepadnavirus integration into host genome in human hepatocytes and in the woodchuck model of hepatitis B-associated hepatocellular carcinoma. Oncogenesis 2017, 6. [Google Scholar] [CrossRef]
- Mason, W.S.; Low, H.C.; Xu, C.; Aldrich, C.E.; Scougall, C.A.; Grosse, A.; Clouston, A.; Chavez, D.; Litwin, S.; Peri, S.; et al. Detection of Clonally Expanded Hepatocytes in Chimpanzees with Chronic Hepatitis B Virus Infection. J. Virol. 2009, 83, 8396–8408. [Google Scholar] [CrossRef] [Green Version]
- Koch, S.; Freytag von Loringhoven, A.; Kahmann, R.; Hofschneider, P.H.; Koshy, R. The genetic organization of integrated hepatitis B virus DNA in the human hepatoma cell line PLC/PRF/5. Nucleic Acids Res. 1984, 12, 6871–6886. [Google Scholar] [CrossRef] [Green Version]
- Shih, C.; Burke, K.; Chou, M.J.; Zeldis, J.B.; Yang, C.S.; Lee, C.S.; Isselbacher, K.J.; Wands, J.R.; Goodman, H.M. Tight clustering of human hepatitis B virus integration sites in hepatomas near a triple-stranded region. J. Virol. 1987, 61, 3491–3498. [Google Scholar] [CrossRef] [Green Version]
- Miller, R.H.; Robinson, W.S. Hepatitis B virus DNA forms in nuclear and cytoplasmic fractions of infected human liver. Virology 1984, 137, 390–399. [Google Scholar] [CrossRef]
- Yaginuma, K.; Kobayashi, M.; Yoshida, E.; Koike, K. Hepatitis B virus integration in hepatocellular carcinoma DNA: Duplication of cellular flanking sequences at the integration site. Proc. Natl. Acad. Sci. USA 1985, 82, 4458–4462. [Google Scholar] [CrossRef] [Green Version]
- Hino, O.; Ohtake, K.; Rogler, C.E. Features of two hepatitis B virus (HBV) DNA integrations suggest mechanisms of HBV integration. J. Virol. 1989, 63, 2638–2643. [Google Scholar] [CrossRef] [Green Version]
- Hino, O.; Shows, T.B.; Rogler, C.E. Hepatitis B virus integration site in hepatocellular carcinoma at chromosome 17;18 translocation. Proc. Natl. Acad. Sci. USA 1986, 83, 8338–8342. [Google Scholar] [CrossRef] [Green Version]
- Lau, C.C.; Sun, T.; Ching, A.K.; He, M.; Li, J.W.; Wong, A.M.; Co, N.N.; Chan, A.W.; Li, P.S.; Lung, R.W.; et al. Viral-human chimeric transcript predisposes risk to liver cancer development and progression. Cancer Cell 2014, 25, 335–349. [Google Scholar] [CrossRef] [Green Version]
- Budzinska, M.A.; Shackel, N.A.; Urban, S.; Tu, T. Sequence analysis of integrated hepatitis B virus DNA during HBeAg-seroconversion. Emerg. Microbes Infect. 2018, 7. [Google Scholar] [CrossRef]
- Mcvey, M.; Lee, S.E.; Avenue, H.; Antonio, S. MMEJ repair of double-strand breaks: Deleted sequences and alternative endings. Trends Genet. 2017, 24, 529–538. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Lau, S.H.; Sham, J.S.T.; Wu, M.C.; Wang, T.; Guan, X.Y. Characterization of HBV integrants in 14 hepatocellular carcinomas: Association of truncated X gene and hepatocellular carcinogenesis. Oncogene 2004, 23, 142–148. [Google Scholar] [CrossRef] [Green Version]
- Tarocchi, M.; Polvani, S.; Marroncini, G.; Galli, A. Molecular mechanism of hepatitis B virus-induced hepatocarcinogenesis. World J. Gastroenterol. 2014, 20, 11630–11640. [Google Scholar] [CrossRef]
- Bouchard, M.J.; Schneider, R.J. The enigmatic X gene of hepatitis B virus. J. Virol. 2004, 78, 12725–12734. [Google Scholar] [CrossRef] [Green Version]
- Akinyemiju, T.; Abera, S.; Ahmed, M.; Alam, N.; Alemayohu, M.A.; Allen, C.; Al-Raddadi, R.; Alvis-Guzman, N.; Amoako, Y.; Artaman, A.; et al. The Burden of Primary Liver Cancer and Underlying Etiologies From 1990 to 2015 at the Global, Regional, and National Level: Results From the Global Burden of Disease Study 2015. JAMA Oncol. 2017, 3, 1683–1691. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.M.; Hwang, L.Y.; Hatten, C.J.; Swaim, M.; Li, D.; Abbruzzese, J.L.; Beasley, P.; Patt, Y.Z. Risk factors for hepatocellular carcinoma: Synergism of alcohol with viral hepatitis and diabetes mellitus. Hepatology 2002, 36, 1206–1213. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.A.; Wu, D.M.; Lin, C.C.; Lu, S.N.; You, S.L.; Wang, L.Y.; Wu, M.H.; Chen, C.J. Incidence and cofactors of hepatitis C virus-related hepatocellular carcinoma: A prospective study of 12,008 men in Taiwan. Am. J. Epidemiol. 2003, 157, 674–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, S.; Yang, Z.; Li, W.; Li, X.; Wang, Y.; Zhang, J.; Xu, C.; Chen, P.J.; Hou, J.; McCrae, M.A.; et al. Re-evaluation of the Carcinogenic Significance of Hepatitis B Virus Integration in Hepatocarcinogenesis. PLoS ONE 2012, 7, e40363. [Google Scholar] [CrossRef] [Green Version]
- Zhong, S.; Chan, J.Y.; Yeo, W.; Tam, J.S.; Johnson, P.J. Frequent integration of precore/core mutants of hepatitis B virus in human hepatocellular carcinoma tissues. J. Viral Hepat. 2000, 7, 115–123. [Google Scholar] [CrossRef]
- Ding, D.; Lou, X.; Hua, D.; Yu, W.; Li, L.; Wang, J.; Gao, F.; Zhao, N.; Ren, G.; Li, L.; et al. Recurrent Targeted Genes of Hepatitis B Virus in the Liver Cancer Genomes Identified by a Next-Generation Sequencing-Based Approach. PLoS Genet. 2012, 8, e1003065. [Google Scholar] [CrossRef] [Green Version]
- Minami, M.; Daimon, Y.; Mori, K.; Takashima, H.; Nakajima, T.; Itoh, Y.; Okanoue, T. Hepatitis B virus-related insertional mutagenesis in chronic hepatitis B patients as an early drastic genetic change leading to hepatocarcinogenesis. Oncogene 2005, 24, 4340–4348. [Google Scholar] [CrossRef] [Green Version]
- Tokino, T.; Matsubara, K. Chromosomal sites for hepatitis B virus integration in human hepatocellular carcinoma. J. Virol. 1991, 65, 6761–6764. [Google Scholar] [CrossRef] [Green Version]
- Summers, J.; Jilbert, A.R.; Yang, W.; Aldrich, C.E.; Saputelli, J.; Litwin, S.; Toll, E.; Mason, W.S. Hepatocyte turnover during resolution of a transient hepadnaviral infection. Proc. Natl. Acad. Sci. USA 2003, 100, 11652–11659. [Google Scholar] [CrossRef] [Green Version]
- Feitelson, M.A.; Lee, J. Hepatitis B virus integration, fragile sites, and hepatocarcinogenesis. Cancer Lett. 2007, 252, 157–170. [Google Scholar] [CrossRef]
- Tang, D.; Li, B.; Xu, T.; Hu, R.; Tan, D.; Song, X.; Jia, P.; Zhao, Z. VISDB: A manually curated database of viral integration sites in the human genome. Nucleic Acids Res. 2019, 48, D633–D641. [Google Scholar] [CrossRef]
- Horikawa, I.; Barrett, J.C. Transcriptional regulation of the telomerase hTERT gene as a target for cellular and viral oncogenic mechanisms. Carcinogenesis 2003, 24, 1167–1176. [Google Scholar] [CrossRef]
- Fujimoto, A.; Totoki, Y.; Abe, T.; Boroevich, K.A.; Hosoda, F.; Nguyen, H.H.; Aoki, M.; Hosono, N.; Kubo, M.; Miya, F.; et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat. Genet. 2012, 44, 760–764. [Google Scholar] [CrossRef] [PubMed]
- Khoury, J.D.; Tannir, N.M.; Williams, M.D.; Chen, Y.; Yao, H.; Zhang, J.; Thompson, E.J.; Meric-Bernstam, F.; Medeiros, L.J.; Weinstein, J.N.; et al. Landscape of DNA Virus Associations across Human Malignant Cancers: Analysis of 3775 Cases Using RNA-Seq. J. Virol. 2013, 87, 8916–8926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Zeng, X.; Lee, N.P.; Liu, X.; Chen, S.; Guo, B.; Yi, S.; Zhuang, X.; Chen, F.; Wang, G.; et al. HIVID: An efficient method to detect HBV integration using low coverage sequencing. Genomics 2013, 102, 338–344. [Google Scholar] [CrossRef]
- Kawai-Kitahata, F.; Asahina, Y.; Tanaka, S.; Kakinuma, S.; Murakawa, M.; Nitta, S.; Watanabe, T.; Otani, S.; Taniguchi, M.; Goto, F.; et al. Comprehensive analyses of mutations and hepatitis B virus integration in hepatocellular carcinoma with clinicopathological features. J. Gastroenterol. 2016, 51, 473–486. [Google Scholar] [CrossRef]
- Chiu, Y.T.; Wong, J.K.; Choi, S.W.; Sze, K.M.; Ho, D.W.; Chan, L.K.; Lee, J.M.; Man, K.; Cherny, S.; Yang, W.L.; et al. Novel pre-mRNA splicing of intronically integrated HBV generates oncogenic chimera in hepatocellular carcinoma. J. Hepatol. 2016, 64, 1256–1264. [Google Scholar] [CrossRef] [Green Version]
- Fujimoto, A.; Furuta, M.; Totoki, Y.; Tsunoda, T.; Kato, M.; Shiraishi, Y.; Tanaka, H.; Taniguchi, H.; Kawakami, Y.; Ueno, M.; et al. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat. Genet. 2016, 48, 500–509. [Google Scholar] [CrossRef]
- Gao, Q.; Wang, Z.C.; Duan, M.; Lin, Y.H.; Zhou, X.Y.; Worthley, D.L.; Wang, X.Y.; Niu, G.; Xia, Y.; Deng, M.; et al. Cell Culture System for Analysis of Genetic Heterogeneity Within Hepatocellular Carcinomas and Response to Pharmacologic Agents; Elsevier: Amsterdam, The Netherlands, 2017; pp. 232–242.e4. Volume 152. [Google Scholar] [CrossRef] [Green Version]
- Ally, A.; Balasundaram, M.; Carlsen, R.; Chuah, E.; Clarke, A.; Dhalla, N.; Holt, R.A.; Jones, S.J.; Lee, D.; Ma, Y.; et al. Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341.e23. [Google Scholar] [CrossRef] [Green Version]
- Yoo, S.; Wang, W.; Wang, Q.; Fiel, M.I.; Lee, E.; Hiotis, S.P.; Zhu, J. A pilot systematic genomic comparison of recurrence risks of hepatitis B virus-associated hepatocellular carcinoma with low- and high-degree liver fibrosis. BMC Med. 2017, 15, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuta, M.; Tanaka, H.; Shiraishi, Y.; Unida, T.; Imamura, M.; Fujimoto, A.; Fujita, M.; Sasaki-Oku, A.; Maejima, K.; Nakano, K.; et al. Characterization of HBV integration patterns and timing in liver cancer and HBV-infected livers. Oncotarget 2018, 9, 25075–25088. [Google Scholar] [CrossRef] [Green Version]
- Li, C.L.; Li, C.Y.; Lin, Y.Y.; Ho, M.C.; Chen, D.S.; Chen, P.J.; Yeh, S.H. Androgen Receptor Enhances Hepatic Telomerase Reverse Transcriptase Gene Transcription After Hepatitis B Virus Integration or Point Mutation in Promoter Region. Hepatology 2019, 69, 498–512. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Ye, S.; Zhao, X.; Ji, L.; Zhang, Y.; Zhou, P.; Sun, J.; Guan, Y.; Han, Y.; Ni, C.; et al. Molecular characterization of HBV DNA integration in patients with hepatitis and hepatocellular carcinoma. J. Cancer 2018, 9, 3225–3235. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.W.; Alaei-Mahabadi, B.; Samuelsson, T.; Lindh, M.; Larsson, E. The landscape of viral expression and host gene fusion and adaptation in human cancer. Nat. Commun. 2013, 4, 2513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, H.; Zhang, L.; Qian, Z.; Zhu, X.; Zhu, G.; Chen, Y.; Xie, X.; Ye, Q.; Zang, J.; Ren, Z.; et al. Identification of HBV-MLL4 integration and its molecular basis in Chinese hepatocellular carcinoma. PLoS ONE 2015, 10, e0123175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Liu, S.; Shen, C.; Wu, Y.; Zhang, L.; Chen, X.; Lu, F. DNA methylation consistency implicates the primary tumor cell origin of recurrent hepatocellular carcinoma. Epigenomics 2015, 7, 581–592. [Google Scholar] [CrossRef] [Green Version]
- Cao, S.; Wendl, M.C.; Wyczalkowski, M.A.; Wylie, K.; Ye, K.; Jayasinghe, R.; Xie, M.; Wu, S.; Niu, B.; Grubb, R.; et al. Divergent viral presentation among human tumors and adjacent normal tissues. Sci. Rep. 2016, 6, 28294. [Google Scholar] [CrossRef] [Green Version]
- Bayard, Q.; Meunier, L.; Peneau, C.; Renault, V.; Shinde, J.; Nault, J.C.; Mami, I.; Couchy, G.; Amaddeo, G.; Tubacher, E.; et al. Cyclin A2/E1 activation defines a hepatocellular carcinoma subclass with a rearrangement signature of replication stress. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Bertoli, C.; Skotheim, J.M.; De Bruin, R.A. Control of cell cycle transcription during G1 and S phases. Nat. Rev. Mol. Cell Biol. 2013, 14, 518–528. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Deng, Q.; Wang, Q.; Li, K.Y.; Dai, J.H.; Li, N.; Zhu, Z.D.; Zhou, B.; Liu, X.Y.; Liu, R.F.; et al. Exome sequencing of hepatitis B virus-associated hepatocellular carcinoma. Nat. Genet. 2012, 44, 1117–1121. [Google Scholar] [CrossRef]
- Toh, S.T.; Jin, Y.; Liu, L.; Wang, J.; Babrzadeh, F.; Gharizadeh, B.; Ronaghi, M.; Toh, H.C.; Chow, P.K.H.; Chung, A.Y.; et al. Deep sequencing of the hepatitis B virus in hepatocellular carcinoma patients reveals enriched integration events, structural alterations and sequence variations. Carcinogenesis 2013, 34, 787–798. [Google Scholar] [CrossRef] [Green Version]
- Hunter, T.; Pines, J. Cyclins and cancer. II: Cyclin D and CDK inhibitors come of age. Cell 1994, 79, 573–582. [Google Scholar] [CrossRef]
- Fujimoto, A.; Furuta, M.; Shiraishi, Y.; Gotoh, K.; Kawakami, Y.; Arihiro, K.; Nakamura, T.; Ueno, M.; Ariizumi, S.I.; Nguyen, H.H.; et al. Whole-genome mutational landscape of liver cancers displaying biliary phenotype reveals hepatitis impact and molecular diversity. Nat. Commun. 2015, 6, 6–13. [Google Scholar] [CrossRef]
- Furler, R.L.; Uittenbogaart, C.H. GLI2 regulates TGF-β1 in human CD4+ T cells: Implications in cancer and HIV pathogenesis. PLoS ONE 2012, 7, e40874. [Google Scholar] [CrossRef] [PubMed]
- Swartling, F.J.; Savov, V.; Persson, A.I.; Chen, J.; Hackett, C.S.; Northcott, P.A.; Grimmer, M.R.; Lau, J.; Chesler, L.; Perry, A.; et al. Distinct neural stem cell populations give rise to disparate brain tumors in response to N-MYC. Cancer Cell 2012, 21, 601–613. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Wang, L.; Nandy, D.; Zhang, Y.; Basu, A.; Radisky, D.; Mukhopadhyay, D. Neuropilin-1 upholds dedifferentiation and propagation phenotypes of renal cell carcinoma cells by activating Akt and sonic hedgehog axes. Cancer Res. 2008, 68, 8667–8672. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Joon, W.Y.; Xiao, X.; Dean, N.M.; Monia, B.P.; Marcusson, E.G. Selective down-regulation of glioma-associated oncogene 2 inhibits the proliferation of hepatocellular carcinoma cells. Cancer Res. 2007, 67, 3583–3593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Han, C.; Lu, L.; Magliato, S.; Wu, T. Hedgehog signaling pathway regulates autophagy in human hepatocellular carcinoma cells. Hepatology 2013, 58, 995–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Cao, L.; Li, Y.; Lu, H.; Yang, X.; Xue, P. Expression of glioma-associated oncogene 2 (Gli 2) is correlated with poor prognosis in patients with hepatocellular carcinoma undergoing hepatectomy. World J. Surg. Oncol. 2013, 11, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, C.; Chen, L.H.; Huang, Y.; Chang, C.C.; Wang, P.; Pirozzi, C.J.; Qin, X.; Bao, X.; Greer, P.K.; McLendon, R.E.; et al. KMT2D maintains neoplastic cell proliferation and global histone H3 lysine 4 monomethylation. Oncotarget 2013, 4, 2144–2153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otsubo, T.; Akiyama, Y.; Hashimoto, Y.; Shimada, S.; Goto, K.; Yuasa, Y. Microrna-126 inhibits sox2 expression and contributes to gastric carcinogenesis. PLoS ONE 2011, 6, e16617. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Han, S.; Wang, X.; Peng, R.; Li, X. SOX5 promotes epithelial–mesenchymal transition and cell invasion via regulation of Twist1 in hepatocellular carcinoma. Med. Oncol. 2015, 32, 10. [Google Scholar] [CrossRef]
- You, J.; Zhao, Q.; Fan, X.; Wang, J. SOX5 promotes cell invasion and metastasis via activation of twist-mediated epithelial– mesenchymal transition in gastric cancer. OncoTargets Ther. 2019, 12, 2465–2476. [Google Scholar] [CrossRef] [Green Version]
- Shiraishi, Y.; Fujimoto, A.; Furuta, M.; Tanaka, H.; Chiba, K.I.; Boroevich, K.A.; Abe, T.; Kawakami, Y.; Ueno, M.; Gotoh, K.; et al. Integrated analysis of whole genome and transcriptome sequencing reveals diverse transcriptomic aberrations driven by somatic genomic changes in liver cancers. PLoS ONE 2014, 9, e114263. [Google Scholar] [CrossRef]
- Tamkun, J.W.; Hynes, R.O. Plasma fibronectin is synthesized and secreted by hepatocytes. J. Biol. Chem. 1983, 258, 4641–4647. [Google Scholar] [CrossRef]
- Wang, F.; Denison, S.; Lai, J.P.; Philips, L.A.; Montoya, D.; Kock, N.; Schüle, B.; Klein, C.; Shridhar, V.; Roberts, L.R.; et al. Parkin Gene Alterations in Hepatocellular Carcinoma. Genes Chromosom. Cancer 2004, 40, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Kähkönen, M. Population cytogenetics of folate-sensitive fragile sites—I. Common fragile sites. Hum. Genet. 1988, 80, 344–348. [Google Scholar] [CrossRef]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Iny Stein, T.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards suite: From gene data mining to disease genome sequence analyses. Curr. Protoc. Bioinform. 2016, 2016, 1.30.1–1.30.33. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.L.; Du, H.; Gregor, P.D.; Novina, C.D.; Martinez, E.; Roeder, R.G. Cloning of an inr- and E-box-binding protein, TFII-I, that interacts physically and functionally with USF1. EMBO J. 1997, 16, 7091–7104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarrini, G.; Ciabatti, E.; Pacini, S.; Galimberti, S.; Petrini, I. GTF2I mutations are common in thymic epithelial tumors but not in hematological malignancies. Anticancer Res. 2017, 37, 5459–5462. [Google Scholar] [CrossRef] [PubMed]
- Müllauer, L. GTF2I gene mutation—A driver of thymoma pathogenesis. Mediastinum 2017, 1, 18. [Google Scholar] [CrossRef]
- Petrini, I.; Meltzer, P.S.; Kim, I.K.; Lucchi, M.; Park, K.S.; Fontanini, G.; Gao, J.; Zucali, P.A.; Calabrese, F.; Favaretto, A.; et al. A specific missense mutation in GTF2I occurs at high frequency in thymic epithelial tumors. Nat. Genet. 2014, 46, 844–849. [Google Scholar] [CrossRef] [Green Version]
- Higuchi, R.; Goto, T.; Hirotsu, Y.; Yokoyama, Y.; Nakagomi, T.; Otake, S.; Amemiya, K.; Oyama, T.; Mochizuki, H.; Omata, M. Primary Driver Mutations in GTF2I Specific to the Development of Thymomas. Cancers 2020, 12, 2032. [Google Scholar] [CrossRef]
- Meacci, E.; Taira, M.; Moos, M.; Smith, C.J.; Movsesian, M.A.; Degerman, E.; Belfrage, P.; Manganiello, V. Molecular cloning and expression of human myocardial cGMP-inhibited cAMP phosphodiesterase. Proc. Natl. Acad. Sci. USA 1992, 89, 3721–3725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, N.; Shen, W.; Du, R.; Jiang, S.; Zhu, J.; Chen, Y.; Huang, C.; Shi, Y.; Xiang, R.; Luo, Y. Phosphodiesterase 3A Represents a Therapeutic Target that Drives Stem Cell-like Property and Metastasis in Breast Cancer. Mol. Cancer Ther. 2020, 19, 868–881. [Google Scholar] [CrossRef] [PubMed]
- De Waal, L.; Lewis, T.A.; Rees, M.G.; Tsherniak, A.; Wu, X.; Choi, P.S.; Gechijian, L.; Hartigan, C.; Faloon, P.W.; Hickey, M.J.; et al. Identification of cancer-cytotoxic modulators of PDE3A by predictive chemogenomics. Nat. Chem. Biol. 2016, 12, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.G.; Nguyen, L.; Samadzadeh, S.; Masouminia, M.; Mendoza, A.; Sweeney, O.; Tillman, B.; Afifyan, N.; Morgan, T.; French, B.A.; et al. Expression of proteins upregulated in hepatocellular carcinoma in patients with alcoholic hepatitis (AH) compared to non-alcoholic steatohepatitis (NASH): An immunohistochemical analysis of candidate proteins. Exp. Mol. Pathol. 2018, 104, 125–129. [Google Scholar] [CrossRef]
- Wang, Y.; Li, J.; Wen, S.; Yang, X.; Zhang, Y.; Wang, Z.; Zhang, Z. CHRM3 is a novel prognostic factor of poor prognosis in patients with endometrial carcinoma. Am. J. Transl. Res. 2015, 7, 902–911. [Google Scholar]
- Felton, J.; Hu, S.; Raufman, J.P. Targeting M3 Muscarinic Receptors for Colon Cancer Therapy. Curr. Mol. Pharmacol. 2018, 11, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Cheng, K.; Shant, J.; Raufman, J.P. Acetylcholine-induced activation of M3 muscarinic receptors stimulates robust matrix metalloproteinase gene expression in human colon cancer cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, 755–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Shin, V.Y.; Cheuk, I.; Siu, J.; Kwong, A. Abstract P1-07-01: Cholinergic receptor muscarinic 3 (CHRM3) contributes to breast cancer tumorigenesis through angiogenesis regulation. Cancer Res. 2020, 80. [Google Scholar] [CrossRef]
- Yin, Q.Q.; Xu, L.H.; Zhang, M.; Xu, C. Muscarinic acetylcholine receptor M1 mediates prostate cancer cell migration and invasion through hedgehog signaling. Asian J. Androl. 2018, 20, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhou, J.; Yao, L.; Lang, Y.; Liang, Y.; Chen, L.; Zhang, J.; Wang, F.; Wang, Y.; Chen, H.; et al. High expression of M3 muscarinic acetylcholine receptor is a novel biomarker of poor prognostic in patients with non-small cell lung cancer. Tumor Biol. 2013, 34, 3939–3944. [Google Scholar] [CrossRef]
- Kodaira, M.; Kajimura, M.; Takeuchi, K.; Lin, S.; Hanai, H.; Kaneko, E. Functional muscarinic m3 receptor expressed in gastric cancer cells stimulates tyrosine phosphorylation and MAP kinase. J. Gastroenterol. 1999, 34, 163–171. [Google Scholar] [CrossRef]
- Celiktas, M.; Tanaka, I.; Tripathi, S.C.; Fahrmann, J.F.; Aguilar-Bonavides, C.; Villalobos, P.; Delgado, O.; Dhillon, D.; Dennison, J.B.; Ostrin, E.J.; et al. Role of CPS1 in cell growth, metabolism, and prognosis in LKB1-inactivated lung adenocarcinoma. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kärkkäinen, S.; Hiipakka, M.; Wang, J.H.; Kleino, I.; Vähä-Jaakkola, M.; Renkema, G.H.; Liss, M.; Wagner, R.; Saksela, K. Identification of preferred protein interactions by phage-display of the human Src homology-3 proteome. EMBO Rep. 2006, 7, 186–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Mi, J.Q.; Debernardi, A.; Vitte, A.L.; Emadali, A.; Meyer, J.A.; Charmpi, K.; Ycart, B.; Callanan, M.B.; Carroll, W.L.; et al. A six gene expression signature defines aggressive subtypes and predicts outcome in childhood and adult acute lymphoblastic leukemia. Oncotarget 2015, 6, 16527–16542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, P.; Zhao, Z.; Hulgan, T.; Bush, W.S.; Samuels, D.C.; Bloss, C.S.; Heaton, R.K.; Ellis, R.J.; Schork, N.; Marra, C.M.; et al. Genome-wide association study of HIV-associated neurocognitive disorder (HAND): A CHARTER group study. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. Off. Publ. Int. Soc. Psychiatr. Genet. 2017, 174, 413–426. [Google Scholar] [CrossRef]
- Lee, J.H.; Cheng, R.; Vardarajan, B.; Lantigua, R.; Reyes-Dumeyer, D.; Ortmann, W.; Graham, R.R.; Bhangale, T.; Behrens, T.W.; Medrano, M.; et al. Genetic Modifiers of Age at Onset in Carriers of the G206A Mutation in PSEN1 with Familial Alzheimer Disease Among Caribbean Hispanics. JAMA Neurol. 2015, 72, 1043–1051. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Liu, Y.; Lian, C.; Cao, X.; Wang, Y.; Li, X.; Cong, M.; Tian, P.; Zhang, X.; Wei, G.; et al. SH3RF3 promotes breast cancer stem-like properties via JNK activation and PTX3 upregulation. Nat. Commun. 2020, 11, 2487. [Google Scholar] [CrossRef]
- Mao, X.; Bruneau, N.; Gao, Q.; Becq, H.; Jia, Z.; Xi, H.; Shu, L.; Wang, H.; Szepetowski, P.; Aniksztejn, L. The Epilepsy of Infancy with Migrating Focal Seizures: Identification of de novo Mutations of the KCNT2 Gene That Exert Inhibitory Effects on the Corresponding Heteromeric KNa1.1/KNa1.2 Potassium Channel. Front. Cell. Neurosci. 2020, 14. [Google Scholar] [CrossRef]
- Hines, I.N.; Hartwell, H.J.; Feng, Y.; Theve, E.J.; Hall, G.A.; Hashway, S.; Connolly, J.; Fecteau, M.; Fox, J.G.; Rogers, A.B. Insulin resistance and metabolic hepatocarcinogenesis with parent-of-origin effects in A×B mice. Am. J. Pathol. 2011, 179, 2855–2865. [Google Scholar] [CrossRef] [Green Version]
- Nault, J.C.; Couchy, G.; Balabaud, C.; Morcrette, G.; Caruso, S.; Blanc, J.F.; Bacq, Y.; Calderaro, J.; Paradis, V.; Ramos, J.; et al. Molecular Classification of Hepatocellular Adenoma Associates with Risk Factors, Bleeding, and Malignant Transformation. Gastroenterology 2017, 152, 880–894.e6. [Google Scholar] [CrossRef] [Green Version]
- Bérubé, J.C.; Gaudreault, N.; Lavoie-Charland, E.; Sbarra, L.; Henry, C.; Madore, A.M.; Paré, P.D.; Van Den Berge, M.; Nickle, D.; Laviolette, M.; et al. Identification of Susceptibility Genes of Adult Asthma in French Canadian Women. Can. Respir. J. 2016, 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansel, N.N.; Pare, P.D.; Rafaels, N.; Sin, D.D.; Sandford, A.; Daley, D.; Vergara, C.; Huang, L.; Mark Elliott, W.; Pascoe, C.D.; et al. Genome-wide association study identification of novel loci associated with airway responsiveness in chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2015, 53, 226–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, C.A.; Boucher, G.; Lees, C.W.; Franke, A.; D’Amato, M.; Taylor, K.D.; Lee, J.C.; Goyette, P.; Imielinski, M.; Latiano, A.; et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat. Genet. 2011, 43, 246–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.Q.; Pfeiffer, R.M.; Hyland, P.L.; Shi, J.; Gu, F.; Wang, Z.; Bhattacharjee, S.; Luo, J.; Xiong, X.; Yeager, M.; et al. Genetic polymorphisms in the 9p21 region associated with risk of multiple cancers. Carcinogenesis 2014, 35, 2698–2705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, J.H.; Park, S.B.; Park, S.; Lee, H.S.; Kim, C.; Jung, D.E.; Song, S.Y. Novel gastric cancer stem cell-related marker LINGO2 is associated with cancer cell phenotype and patient outcome. Int. J. Mol. Sci. 2019, 20, 555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belle, N.M.; Ji, Y.; Herbine, K.; Wei, Y.; Park, J.H.; Zullo, K.; Hung, L.Y.; Srivatsa, S.; Young, T.; Oniskey, T.; et al. TFF3 interacts with LINGO2 to regulate EGFR activation for protection against colitis and gastrointestinal helminths. Nat. Commun. 2019, 10, 4408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhry, H.; Albukhari, A.; Morotti, M.; Haider, S.; Moralli, D.; Smythies, J.; Schödel, J.; Green, C.M.; Camps, C.; Buffa, F.; et al. Tumor hypoxia induces nuclear paraspeckle formation through HIF-2α dependent transcriptional activation of NEAT1 leading to cancer cell survival. Oncogene 2015, 34, 4482–4490. [Google Scholar] [CrossRef] [Green Version]
- Chakravarty, D.; Sboner, A.; Nair, S.S.; Giannopoulou, E.; Li, R.; Hennig, S.; Mosquera, J.M.; Pauwels, J.; Park, K.; Kossai, M.; et al. The oestrogen receptor alpha-regulated lncRNA NEAT1 is a critical modulator of prostate cancer. Nat. Commun. 2014, 5, 5383. [Google Scholar] [CrossRef]
- Kawaguchi, T.; Tanigawa, A.; Naganuma, T.; Ohkawa, Y.; Souquere, S.; Pierron, G.; Hirose, T.; Steitz, J.A. SWI/SNF chromatin-remodeling complexes function in noncoding RNA-dependent assembly of nuclear bodies. Proc. Natl. Acad. Sci. USA 2015, 112, 4304–4309. [Google Scholar] [CrossRef] [Green Version]
- Skalsky, R.L.; Cullen, B.R. Viruses, microRNAs, and host interactions. Annu. Rev. Microbiol. 2010, 64, 123–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Jiang, L.; Ji, X.; Yang, B.; Zhang, Y.; Fu, X.D. Hepatitis B viral RNA directly mediates down-regulation of the tumor suppressor microRNA miR-15a/miR-16-1 in hepatocytes. J. Biol. Chem. 2013, 288, 18484–18493. [Google Scholar] [CrossRef] [Green Version]
- Gel, B.; Serra, E. karyoploteR: An R/Bioconductor package to plot customizable genomes displaying arbitrary data. Bioinformatics 2017, 33, 3088–3090. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.; Li, Q.J.; Feng, Y.; Zhang, Y.; Markowitz, G.J.; Ning, S.; Deng, Y.; Zhao, J.; Jiang, S.; Yuan, Y.; et al. TGF-β-miR-34a-CCL22 Signaling-Induced Treg Cell Recruitment Promotes Venous Metastases of HBV-Positive Hepatocellular Carcinoma. Cancer Cell 2012, 22, 291–303. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.H.; Mei, M.H.; Fei, R.; Liu, F.; Wang, J.H.; Liao, W.J.; Qin, L.L.; Wei, L.; Chen, H.S. Regulatory T cells in chronic hepatitis B patients affect the immunopathogenesis of hepatocellular carcinoma by suppressing the anti-tumour immune responses. J. Viral Hepat. 2010, 17, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Fujita, N.; Sugimoto, R.; Ma, N.; Tanaka, H.; Iwasa, M.; Kobayashi, Y.; Kawanishi, S.; Watanabe, S.; Kaito, M.; Takei, Y. Comparison of hepatic oxidative DNA damage in patients with chronic hepatitis B and C. J. Viral Hepat. 2008, 15, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Ha, H.L.; Shin, H.J.; Feitelson, M.A.; Yu, D.Y. Oxidative stress and antioxidants in hepatic pathogenesis. World J. Gastroenterol. 2010, 16, 6035–6043. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, M.J.; Navas-Martin, S. Hepatitis B and C virus hepatocarcinogenesis: Lessons learned and future challenges. Cancer Lett. 2011, 305, 123–143. [Google Scholar] [CrossRef]
- Chun, E.; Lee, J.; Cheong, H.S.; Lee, K.Y. Tumor Eradication by Hepatitis B Virus X Antigen-Specific CD8 + T Cells in Xenografted Nude Mice. J. Immunol. 2003, 170, 1183–1190. [Google Scholar] [CrossRef] [Green Version]
- Malmassari, S.L.; Deng, Q.; Fontaine, H.; Houitte, D.; Rimlinger, F.; Thiers, V.; Maillere, B.; Pol, S.; Michel, M.L. Impact of hepatitis B virus basic core promoter mutations on T cell response to an immunodominant HBx-derived epitope. Hepatology 2007, 45, 1199–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, G.Y.; Lin, C.Y.; Huang, L.M.; Wang, Y.H.; Wang, J.C.; Hsu, C.T.; Yang, S.S.; Wu, C.C. Detection of the Hepatitis B Virus X Protein (HBx) Antigen and Anti-HBx Antibodies in Cases of Human Hepatocellular Carcinoma. J. Clin. Microbiol. 2003, 41, 5598–5603. [Google Scholar] [CrossRef] [Green Version]
- Martín-Vílchez, S.; Sanz-Cameno, P.; Rodríguez-Muñoz, Y.; Majano, P.L.; Molina-Jiménez, F.; López-Cabrera, M.; Moreno-Otero, R.; Lara-Pezzi, E. The hepatitis B virus X protein induces paracrine activation of human hepatic stellate cells. Hepatology 2008, 47, 1872–1883. [Google Scholar] [CrossRef]
- Calvisi, D.F.; Ladu, S.; Gorden, A.; Farina, M.; Lee, J.S.; Conner, E.A.; Schroeder, I.; Factor, V.M.; Thorgeirsson, S.S. Mechanistic and prognostic significance of aberrant methylation in the molecular pathogenesis of human hepatocellular carcinoma. J. Clin. Investig. 2007, 117, 2713–2722. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Wu, G.; Bu, F.; Lu, B.; Liang, A.; Cao, L.; Tong, X.; Lu, X.; Wu, M.; Guo, Y. Epigenetic silence of ankyrin-repeat-containing, SH3-domain-containing, and proline-rich-region-containing protein 1 (ASPP1) and ASPP2 genes promotes tumor growth in hepatitis B virus-positive hepatocellular carcinoma. Hepatology 2010, 51, 142–153. [Google Scholar] [CrossRef]
- Zhang, Z.Z.; Liu, X.; Wang, D.Q.; Teng, M.K.; Niu, L.W.; Huang, A.L.; Liang, Z. Hepatitis B virus and hepatocellular carcinoma at the miRNA level. World J. Gastroenterol. 2011, 17, 3353–3358. [Google Scholar] [CrossRef]
- Wei, X.; Tan, C.; Tang, C.; Ren, G.; Xiang, T.; Qiu, Z.; Liu, R.; Wu, Z. Epigenetic repression of miR-132 expression by the hepatitis B virus x protein in hepatitis B virus-related hepatocellular carcinoma. Cell. Signal. 2013, 25, 1037–1043. [Google Scholar] [CrossRef]
- Chisari, F.; Ferrari, C. Hepatitis B virus immunopathogenesis. Annu. Rev. Immunol. 1995, 13, 29–60. [Google Scholar] [CrossRef]
- Guidotti, L.G.; Isogawa, M.; Chisari, F.V. Host–virus interactions in hepatitis B virus infection. Curr. Opin. Immunol. 2015, 36, 61–66. [Google Scholar] [CrossRef] [Green Version]
- Bertoletti, A.; Ferrari, C. Adaptive immunity in HBV infection. J. Hepatol. 2016, 64, S71–S83. [Google Scholar] [CrossRef]
- Meuleman, P.; Libbrecht, L.; Wieland, S.; De Vos, R.; Habib, N.; Kramvis, A.; Roskams, T.; Leroux-Roels, G. Immune suppression uncovers endogenous cytopathic effects of the hepatitis B virus. J. Virol. 2006, 80, 2797–2807. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.Y.; Hu, K.Q. Rethinking the pathogenesis of hepatitis B virus (HBV) infection. J. Med. Virol. 2015, 87, 1989–1999. [Google Scholar] [CrossRef]
- Raihan, R.; Akbar, S.M.F.; Al Mahtab, M.; Khan, M.S.I.; Tabassum, S.; Tee, K.K.; Mohamed, R.B. Increased Proinflammatory Cytokine Production by Chronic Hepatitis B Patients with Mutant Hepatitis B Virus: Plausible Mechanisms Underlying Severe Liver Diseases in These Patients. Viral Immunol. 2020, 33, 530–534. [Google Scholar] [CrossRef]
- Kao, J.H.; Chen, P.J.; Lai, M.Y.; Chen, D.S. Basal core promoter mutations of hepatitis B virus increase the risk of hepatocellular carcinoma in hepatitis B carriers. Gastroenterology 2003, 124, 327–334. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, H.; Gu, C.; Yin, J.; He, Y.; Xie, J.; Cao, G. Associations between hepatitis B virus mutations and the risk of hepatocellular carcinoma: A meta-analysis. J. Natl. Cancer Inst. 2009, 101, 1066–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elpek, G.O. Molecular pathways in viral hepatitis-associated liver carcinogenesis: An update. World J. Clin. Cases 2021, 9, 4890–4917. [Google Scholar] [CrossRef]
- Enrietto, P.J.; Wyke, J.A. The pathogenesis of oncogenic avian retroviruses. Adv. Cancer Res. 1983, 39, 269–314. [Google Scholar] [CrossRef]
- Bruni, R.; D’Ugo, E.; Giuseppetti, R.; Argentini, C.; Rapicetta, M. Activation of the N-myc2 oncogene by woodchuck hepatitis virus integration in the linked downstream b3n locus in woodchuck hepatocellular carcinoma. Virology 1999, 257, 483–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacob, J.R.; Sterczer, A.; Toshkov, I.A.; Yeager, A.E.; Korba, B.E.; Cote, P.J.; Buendia, M.A.; Gerin, J.L.; Tennant, B.C. Integration of Woodchuck Hepatitis and N-myc Rearrangement Determine Size and Histologic Grade of Hepatic Tumors. Hepatology 2004, 39, 1008–1016. [Google Scholar] [CrossRef]
- Nakajima, T.; Moriguchi, M.; Mitsumoto, Y.; Sekoguchi, S.; Nishikawa, T.; Takashima, H.; Watanabe, T.; Katagishi, T.; Kimura, H.; Okanoue, T.; et al. Centrosome aberration accompanied with p53 mutation can induce genetic instability in hepatocellular carcinoma. Mod. Pathol. 2004, 17, 722–727. [Google Scholar] [CrossRef] [Green Version]
- Wilkens, L.; Bredt, M.; Flemming, P.; Kubicka, S.; Klempnauer, J.; Kreipe, H. Cytogenetic aberrations in primary and recurrent fibrolamellar hepatocellular carcinoma detected by comparative genomic hybridization. Am. J. Clin. Pathol. 2000, 114, 867–874. [Google Scholar] [CrossRef]
- Wooddell, C.I.; Yuen, M.F.; Chan, H.L.Y.; Gish, R.G.; Locarnini, S.A.; Chavez, D.; Ferrari, C.; Given, B.D.; Hamilton, J.; Kanner, S.B.; et al. RNAi-based treatment of chronically infected patients and chimpanzees reveals that integrated hepatitis B virus DNA is a source of HBsAg. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thi Vo, T.; Poovorawan, K.; Charoen, P.; Soonthornworasiri, N.; Nontprasert, A.; Kittitrakul, C.; Phumratanaprapin, W.; Tangkijvanich, P. Association between Hepatitis B Surface Antigen Levels and the Risk of Hepatocellular Carcinoma in Patients with Chronic Hepatitis B Infection: Systematic Review and Meta-Analysis. Asian Pac. J. Cancer Prev. APJCP 2019, 20, 2239–2246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boni, C.; Fisicaro, P.; Valdatta, C.; Amadei, B.; Di Vincenzo, P.; Giuberti, T.; Laccabue, D.; Zerbini, A.; Cavalli, A.; Missale, G.; et al. Characterization of hepatitis B virus (HBV)-specific T-cell dysfunction in chronic HBV infection. J. Virol. 2007, 81, 4215–4225. [Google Scholar] [CrossRef] [Green Version]
- Bertoletti, A.; Ferrari, C. Innate and adaptive immune responses in chronic hepatitis B virus infections: Towards restoration of immune control of viral infection. Postgrad. Med. J. 2013, 89, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Evan, G.I. A matter of life and death. Cancer Cell 2002, 1, 19–30. [Google Scholar] [CrossRef] [Green Version]
- Cox, A.L.; El-Sayed, M.H.; Kao, J.H.; Lazarus, J.V.; Lemoine, M.; Lok, A.S.; Zoulim, F. Progress towards elimination goals for viral hepatitis. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 533–542. [Google Scholar] [CrossRef]
- Cornberg, M.; Lok, A.S.F.; Terrault, N.A.; Zoulim, F.; Berg, T.; Brunetto, M.R.; Buchholz, S.; Buti, M.; Chan, H.L.; Chang, K.M.; et al. Guidance for Design and Endpoints of Clinical Trials in Chronic Hepatitis B—Report From the 2019 EASL-AASLD HBV Treatment Endpoints Conference. Hepatology 2020, 71, 1070–1092. [Google Scholar] [CrossRef]
- Lin, C.L.; Kao, J.H. Perspectives and control of hepatitis B virus infection in Taiwan. J. Formos. Med. Assoc. Taiwan Yi Zhi 2015, 114, 901–909. [Google Scholar] [CrossRef] [Green Version]
- Kao, J.H. Hepatitis B: From control to cure. J. Formos. Med. Assoc. 2018, 117, 868–870. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Gene Full Name | Gene Type | External Link | Genomic Location | Times Reported | References |
|---|---|---|---|---|---|---|
| TERT | Telomerase reverse transciptase | protein coding | HGNC:11730 | 5p15.33 | 457 | [82,83,87,96,105,107,114,115,116,117,118,119,120,121,122,123,124,125] |
| FN1 | Fibronectin 1 | protein coding | HGNC:3778 | 2q35 | 283 | [82,83,84,87,105,107,115,118,119,121,122,123,125,126,127] |
| KMT2B | Lysine methyltransferase 2B | protein coding | HGNC:15840 | 19q13.12 | 243 | [82,83,84,87,107,115,117,118,119,121,122,123,124,125,126,128,129] |
| ALB | Albumin | protein coding | HGNC:399 | 4q13.3 | 70 | [83,84,87,107,117,118,119,122,123,125,129] |
| CCNA2 | Cyclin A2 | protein coding | HGNC:1578 | 4q27 | 36 | [83,107,118,119,122,123,124,125,130] |
| LINC00486 | Long intergenic non-protein coding RNA 486 | ncRNA | HGNC:42946 | 2p22.3 | 36 | [82,83,118,125] |
| CPS1 | Carbamoyl-phosphate synthase 1 | protein coding | HGNC:2323 | 2q34 | 34 | [82,83,107,118,119,121,122,123,125,126,129] |
| SH3RF3 | SH3 domain containing ring finger 3 | protein coding | HGNC:24699 | 2q13 | 29 | [84,125] |
| CCNE1 | Cyclin E1 | protein coding | HGNC:1589 | 19q12 | 27 | [82,83,119,121,124,127] |
| GLI2 | GLI family zinc finger 2 | protein coding | HGNC:4318 | 2q14.2 | 27 | [84,115,119,121,126] |
| KCNT2 | Potassium sodium-activated channel subfamily T member 2 | protein coding | HGNC:18866 | 1q31.3 | 26 | [118,119,121,122,123,125] |
| LINGO2 | Leucine rich repeat and Ig domain containing 2 | protein coding | HGNC:21207 | 9p21.1 | 25 | [83,105,107,123,125] |
| PRKN | Parkin RBR E3 ubiquitin protein ligase | protein coding | HGNC:8607 | 6q26 | 24 | [81,119,123,125] |
| FAM157A | Family with sequence similarity 157 member A | ncRNA | HGNC:34079 | 3q29 | 23 | [82,83,87,122,125] |
| SOX5 | SRY-box transcription factor 5 | protein coding | HGNC:11201 | 12p12.1 | 23 | [81,83,105,107,119,122,123,124,125] |
| GTF2I | General transcription factor IIi | protein coding | HGNC:4659 | 7q11.23 | 22 | [84,105,118,122,123,125,127] |
| PDE3A | Phosphodiesterase 3A | protein coding | HGNC:8778 | 12q12.2 | 22 | [82,84,118,122,125] |
| CHRM3 | Cholinergic receptor muscarinic 3 | protein coding | HGNC:1952 | 1q43 | 21 | [83,122,125,129] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bousali, M.; Papatheodoridis, G.; Paraskevis, D.; Karamitros, T. Hepatitis B Virus DNA Integration, Chronic Infections and Hepatocellular Carcinoma. Microorganisms 2021, 9, 1787. https://doi.org/10.3390/microorganisms9081787
Bousali M, Papatheodoridis G, Paraskevis D, Karamitros T. Hepatitis B Virus DNA Integration, Chronic Infections and Hepatocellular Carcinoma. Microorganisms. 2021; 9(8):1787. https://doi.org/10.3390/microorganisms9081787
Chicago/Turabian StyleBousali, Maria, George Papatheodoridis, Dimitrios Paraskevis, and Timokratis Karamitros. 2021. "Hepatitis B Virus DNA Integration, Chronic Infections and Hepatocellular Carcinoma" Microorganisms 9, no. 8: 1787. https://doi.org/10.3390/microorganisms9081787
APA StyleBousali, M., Papatheodoridis, G., Paraskevis, D., & Karamitros, T. (2021). Hepatitis B Virus DNA Integration, Chronic Infections and Hepatocellular Carcinoma. Microorganisms, 9(8), 1787. https://doi.org/10.3390/microorganisms9081787