Identification of Important Proteins and Pathways Affecting Feed Efficiency in DLY Pigs by iTRAQ-Based Proteomic Analysis

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Samples
2.2. Protein Preparation and iTRAQ Labeling
2.3. High pH Reverse Phase Fractionation
2.4. Liquid Chromatography (LC)-Tandem Mass Spectrometry (MS/MS) Analysis
2.5. Data Analysis
2.6. RT-qPCR
3. Results
3.1. Statistics and Analysis of FE-Related Phenotypes
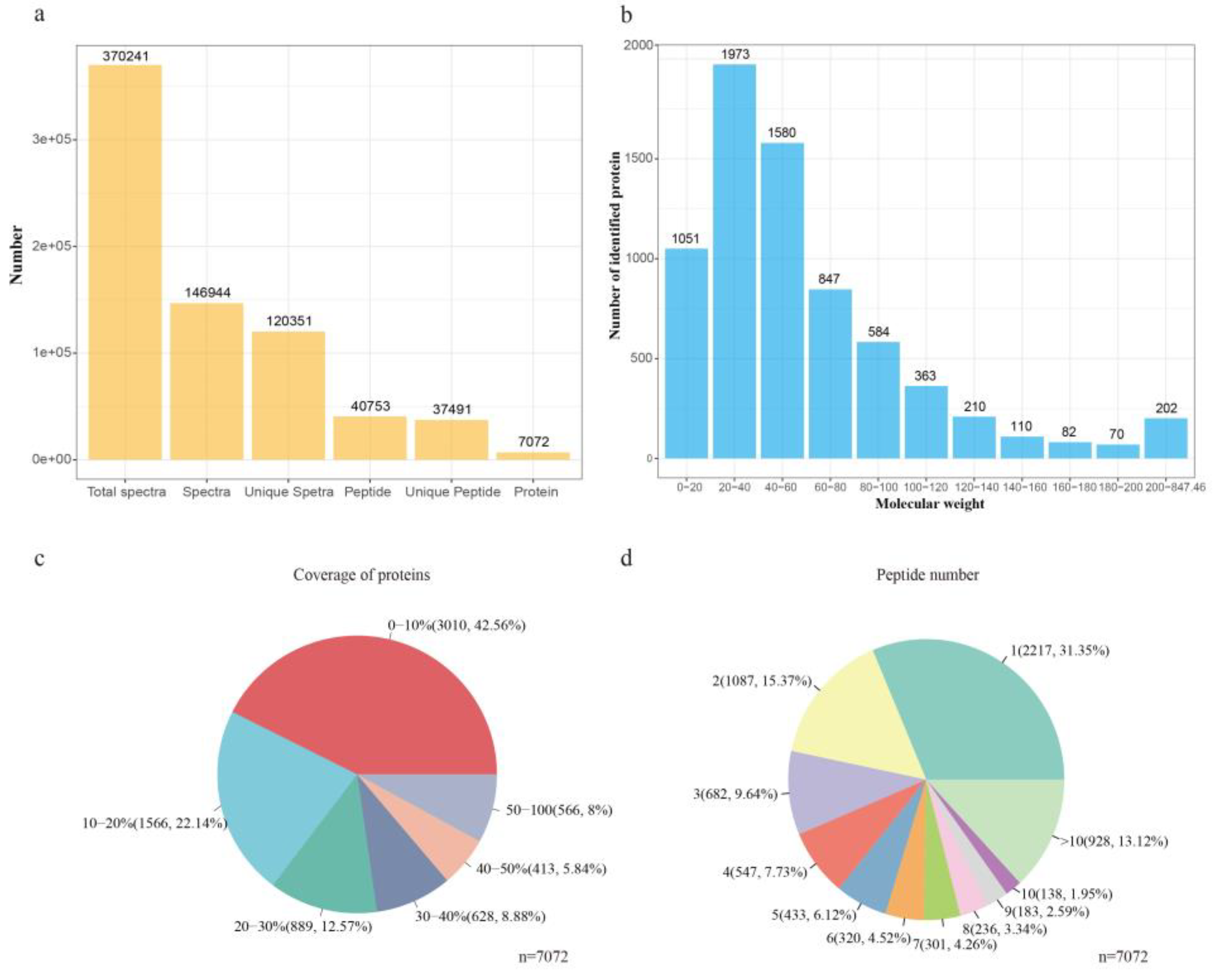
3.2. Protein Identification
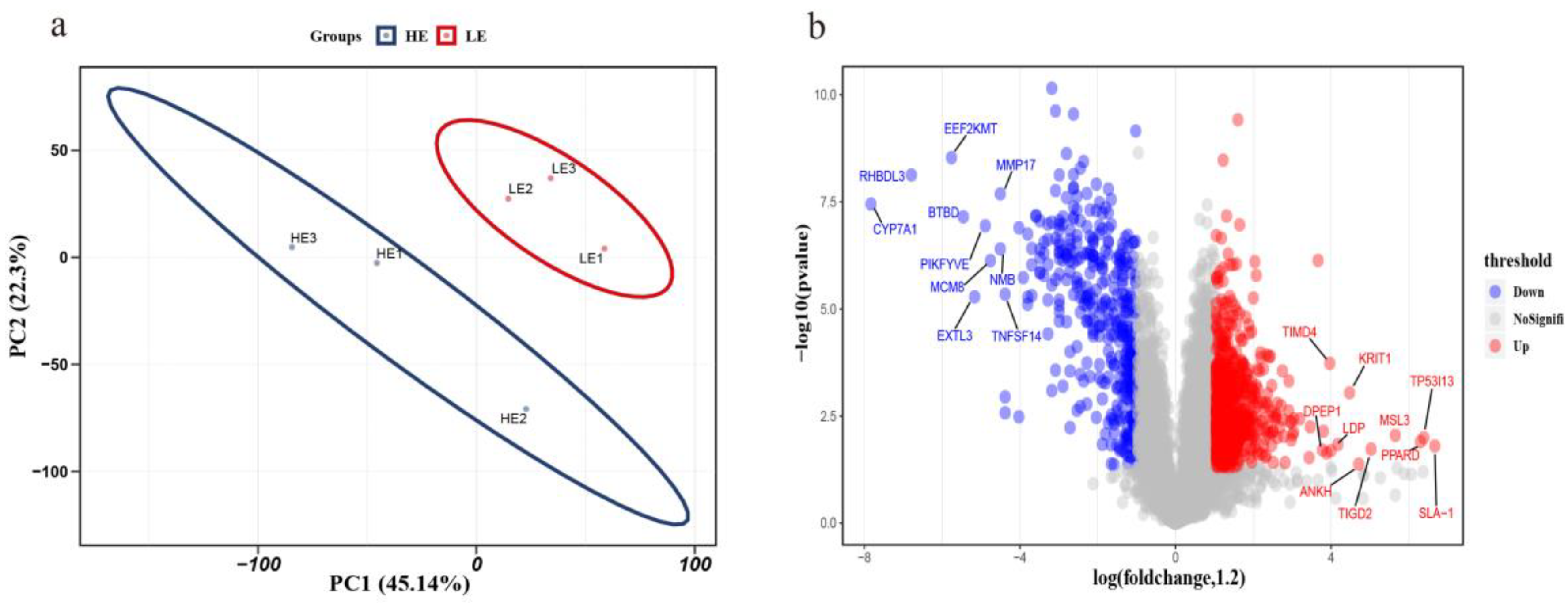
3.3. Protein Quantification by iTRAQ Coupled with LC-MS/MS
3.4. GO Annotation of DEPs
3.5. KEGG and Reactome Pathway Analysis of the DEPs
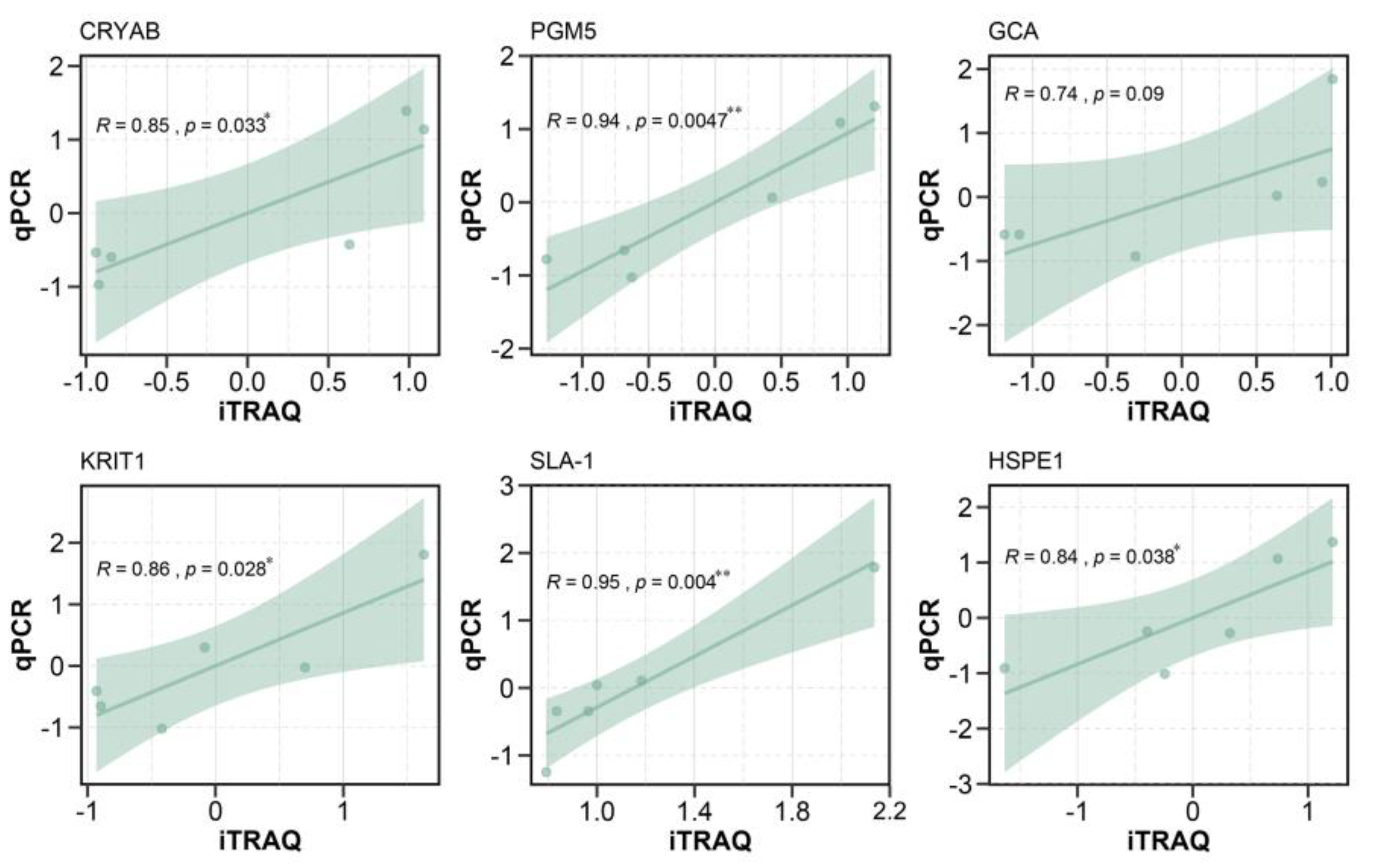
3.6. Real-Time Quantitative Polymerase Chain Reaction
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Patience, J.F.; Rossoni-Serão, M.C.; Gutiérrez, N.A. A review of feed efficiency in swine: Biology and application. J. Anim. Sci. Biotechnol. 2015, 6, 33. [Google Scholar] [CrossRef] [Green Version]
- Case, L.A.; Wood, B.J.; Miller, S.P. The genetic parameters of feed efficiency and its component traits in the turkey (Meleagris gallopavo). Genet. Sel. Evol. 2012, 44, 2. [Google Scholar] [CrossRef] [Green Version]
- Saintilan, R.; Mérour, I.; Schwob, S.; Sellier, P.; Bidanel, J.; Gilbert, H. Genetic parameters and halothane genotype effect for residual feed intake in Piétrain growing pigs. Livest. Sci. 2011, 142, 203–209. [Google Scholar] [CrossRef]
- Gilbert, H.; Bidanel, J.-P.; Billon, Y.; Lagant, H.; Guillouet, P.; Sellier, P.; Noblet, J.; Hermesch, S. Correlated responses in sow appetite, residual feed intake, body composition, and reproduction after divergent selection for residual feed intake in the growing pig. J. Anim. Sci. 2012, 90, 1097–1108. [Google Scholar] [CrossRef] [Green Version]
- Hoque, M.; Suzuki, K.; Kadowaki, H.; Shibata, T.; Oikawa, T. Genetic parameters for feed efficiency traits and their relationships with growth and carcass traits in Duroc pigs. J. Anim. Breed. Genet. 2007, 124, 108–116. [Google Scholar] [CrossRef]
- Harris, A.; Patience, J.F.; Lonergan, S.M.; JM Dekkers, C.; Gabler, N.K. Improved nutrient digestibility and retention partially explains feed efficiency gains in pigs selected for low residual feed intake. J. Anim. Sci. 2012, 90, 164–166. [Google Scholar] [CrossRef] [Green Version]
- Tan, Z.; Wang, Y.; Yang, T.; Xing, K.; Ao, H.; Chen, S.; Zhang, F.; Zhao, X.; Liu, J.; Wang, C. Differentially expressed genes in the caecal and colonic mucosa of Landrace finishing pigs with high and low food conversion ratios. Sci. Rep. 2017, 7, 14886. [Google Scholar] [CrossRef] [Green Version]
- Vigors, S.; O’Doherty, J.V.; Kelly, A.K.; O’Shea, C.J.; Sweeney, T. The effect of divergence in feed efficiency on the intestinal microbiota and the intestinal immune response in both unchallenged and lipopolysaccharide challenged ileal and colonic explants. PLoS ONE 2016, 11, e0148145. [Google Scholar] [CrossRef]
- Mani, V.; Harris, A.; Keating, A.F.; Weber, T.E.; Dekkers, J.C.; Gabler, N.K. Intestinal integrity, endotoxin transport and detoxification in pigs divergently selected for residual feed intake. J. Anim. Sci. 2013, 91, 2141–2150. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Xiong, X.; Yang, J.; Zhou, L.; Yang, B.; Ai, H.; Ma, H.; Xie, X.; Huang, Y.; Fang, S. Genome-wide association analyses for meat quality traits in Chinese Erhualian pigs and a Western Duroc × (Landrace × Yorkshire) commercial population. Genet. Sel. Evol. 2015, 47, 44. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Ren, J.; Ren, D.; Ma, J.; Guo, Y.; Huang, L. Mapping quantitative trait loci for feed consumption and feeding behaviors in a White Duroc × Chinese Erhualian resource population. J. Anim. Sci. 2009, 87, 3458–3463. [Google Scholar] [CrossRef] [Green Version]
- Do, D.N.; Ostersen, T.; Strathe, A.B.; Mark, T.; Jensen, J.; Kadarmideen, H.N. Genome-wide association and systems genetic analyses of residual feed intake, daily feed consumption, backfat and weight gain in pigs. BMC Genet. 2014, 15, 27. [Google Scholar] [CrossRef] [Green Version]
- Sahana, G.; Kadlecová, V.; Hornshøj, H.; Nielsen, B.; Christensen, O.F. A genome-wide association scan in pig identifies novel regions associated with feed efficiency trait. J. Anim. Sci. 2013, 91, 1041–1050. [Google Scholar] [CrossRef]
- Wang, X.; Li, S.; Wu, J.; Ding, R.; Quan, J.; Zheng, E.; Yang, J.; Wu, Z. A Transcriptome Analysis Identifies Biological Pathways and Candidate Genes for Feed Efficiency in DLY Pigs. Genes 2019, 10, 725. [Google Scholar] [CrossRef] [Green Version]
- Ramayo-Caldas, Y.; Ballester, M.; Sánchez, J.P.; González-Rodríguez, O.; Revilla, M.; Reyer, H.; Wimmers, K.; Torrallardona, D.; Quintanilla, R. Integrative approach using liver and duodenum RNA-Seq data identifies candidate genes and pathways associated with feed efficiency in pigs. Sci. Rep. 2018, 8, 558. [Google Scholar] [CrossRef]
- Xu, Y.; Qi, X.; Hu, M.; Lin, R.; Hou, Y.; Wang, Z.; Zhou, H.; Zhao, Y.; Luan, Y.; Zhao, S. Transcriptome Analysis of Adipose Tissue Indicates That the cAMP Signaling Pathway Affects the Feed Efficiency of Pigs. Genes 2018, 9, 336. [Google Scholar] [CrossRef] [Green Version]
- Ono, T.; Hishigaki, H.; Tanigami, A.; Takagi, T. Automated extraction of information on protein-protein interactions from the biological literature. Bioinformatics 2001, 17, 155–161. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.S.; Chen, Y.C.; Lu, C.H.; Hwang, J.K. Prediction of protein subcellular localization. Proteins 2006, 64, 643–651. [Google Scholar] [CrossRef]
- Seet, B.T.; Dikic, I.; Zhou, M.-M.; Pawson, T. Reading protein modifications with interaction domains. Nat. Rev. Mol. Cell Biol. 2006, 7, 473–483. [Google Scholar] [CrossRef]
- Bassols, A.; Costa, C.; Eckersall, P.D.; Osada, J.; Sabrià, J.; Tibau, J. The pig as an animal model for human pathologies: A proteomics perspective. Proteom. Clin. Appl. 2015, 8, 715–731. [Google Scholar] [CrossRef]
- Wang, Z.; Shang, P.; Li, Q.; Wang, L.; Chamba, Y.; Zhang, B.; Zhang, H.; Wu, C. iTRAQ-based proteomic analysis reveals key proteins affecting muscle growth and lipid deposition in pigs. Sci. Rep. 2017, 7, 46717. [Google Scholar] [CrossRef]
- Pierce, A.; Unwin, R.D.; Evans, C.A.; Griffiths, S.; Carney, L.; Zhang, L.; Jaworska, E.; Lee, C.-F.; Blinco, D.; Okoniewski, M.J. Eight-channel iTRAQ enables comparison of the activity of six leukemogenic tyrosine kinases. Mol. Cell. Proteom. 2008, 7, 853–863. [Google Scholar] [CrossRef] [Green Version]
- Cai, W.; Casey, D.; Dekkers, J. Selection response and genetic parameters for residual feed intake in Yorkshire swine. J. Anim. Sci. 2008, 86, 287–298. [Google Scholar] [CrossRef]
- Wen, B.; Zhou, R.; Feng, Q.; Wang, Q.; Wang, J.; Liu, S. IQuant: An automated pipeline for quantitative proteomics based upon isobaric tags. Proteomics 2014, 14, 2280–2285. [Google Scholar] [CrossRef]
- Hakimov, H.A.; Walters, S.; Wright, T.C.; Meidinger, R.G.; Verschoor, C.P.; Gadish, M.; Chiu, D.K.; Strömvik, M.V.; Forsberg, C.W.; Golovan, S.P. Application of iTRAQ to catalogue the skeletal muscle proteome in pigs and assessment of effects of gender and diet dephytinization. Proteomics 2009, 9, 4000–4016. [Google Scholar] [CrossRef]
- Yu, G.; He, Q.-Y. ReactomePA: An R/Bioconductor package for reactome pathway analysis and visualization. Mol. Biosyst. 2016, 12, 477–479. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C T method. Nat. Protoc. 2008, 3, 1101. [Google Scholar] [CrossRef]
- Nasiri Foomani, N.; Zerehdaran, S.; Ahani Azari, M.; Lotfi, E. Genetic parameters for feed efficiency and body weight traits in Japanese quail. Br. Poult. Sci. 2014, 55, 298–304. [Google Scholar] [CrossRef]
- Saintilan, R.; Merour, I.; Brossard, L.; Tribout, T.; Dourmad, J.-Y.; Sellier, P.; Bidanel, J.; Van Milgen, J.; Gilbert, H. Genetics of residual feed intake in growing pigs: Relationships with production traits, and nitrogen and phosphorus excretion traits. J. Anim. Sci. 2013, 91, 2542–2554. [Google Scholar] [CrossRef] [Green Version]
- Vigors, S.; Sweeney, T.; O’shea, C.; Kelly, A.; O’Doherty, J. Pigs that are divergent in feed efficiency, differ in intestinal enzyme and nutrient transporter gene expression, nutrient digestibility and microbial activity. Animal 2016, 10, 1848–1855. [Google Scholar] [CrossRef] [Green Version]
- Dokladny, K.; Zuhl, M.N.; Moseley, P.L. Intestinal epithelial barrier function and tight junction proteins with heat and exercise. J. Appl. Physiol. 2015, 120, 692–701. [Google Scholar] [CrossRef]
- Athman, R.; Louvard, D.; Robine, S. III. How is villin involved in the actin cytoskeleton dynamics in intestinal cells? Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 283, G496–G502. [Google Scholar] [CrossRef] [Green Version]
- Bruewer, M.; Hopkins, A.M.; Hobert, M.E.; Nusrat, A.; Madara, J.L. RhoA, Rac1, and Cdc42 exert distinct effects on epithelial barrier via selective structural and biochemical modulation of junctional proteins and F-actin. Am. J. Physiol. Cell Physiol. 2004, 287, C327–C335. [Google Scholar] [CrossRef] [Green Version]
- Anderson, J.M.; Van Itallie, C.M. Physiology and function of the tight junction. Cold Spring Harb. Perspect. Biol. 2009, 1, a002584. [Google Scholar] [CrossRef]
- Pollard, T.D.; Mooseker, M.S. Direct measurement of actin polymerization rate constants by electron microscopy of actin filaments nucleated by isolated microvillus cores. J. Cell Biol. 1981, 88, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Mooseker, M.S.; Tilney, L.G. Organization of an actin filament-membrane complex. Filament polarity and membrane attachment in the microvilli of intestinal epithelial cells. J. Cell Biol. 1975, 67, 725–743. [Google Scholar] [CrossRef] [Green Version]
- Uruno, T.; Zhang, P.; Liu, J.; Hao, J.J.; Zhan, X. Haematopoietic lineage cell-specific protein 1 (HS1) promotes actin-related protein (Arp) 2/3 complex-mediated actin polymerization. Biochem. J. 2003, 371, 485–493. [Google Scholar] [CrossRef] [Green Version]
- Oshiro, N.; Fukata, Y.; Kaibuchi, K. Phosphorylation of moesin by rho-associated kinase (Rho-kinase) plays a crucial role in the formation of microvilli-like structures. J. Biol. Chem. 1998, 273, 34663–34666. [Google Scholar] [CrossRef] [Green Version]
- Pertz, O.; Hodgson, L.; Klemke, R.L.; Hahn, K.M. Spatiotemporal dynamics of RhoA activity in migrating cells. Nature 2006, 440, 1069. [Google Scholar] [CrossRef]
- Nakamura, N.; Oshiro, N.; Fukata, Y.; Amano, M.; Fukata, M.; Kuroda, S.; Matsuura, Y.; Leung, T.; Lim, L.; Kaibuchi, K. Phosphorylation of ERM proteins at filopodia induced by Cdc42. Genes cells 2000, 5, 571–581. [Google Scholar] [CrossRef]
- Roberts, R.E.; Hallett, M.B. Neutrophil cell shape change: Mechanism and signalling during cell spreading and phagocytosis. Int. J. Mol. Sci. 2019, 20, 1383. [Google Scholar] [CrossRef] [Green Version]
- Yonemura, S.; Tsukita, S.; Tsukita, S. Direct involvement of ezrin/radixin/moesin (ERM)-binding membrane proteins in the organization of microvilli in collaboration with activated ERM proteins. J. Cell Biol. 1999, 145, 1497–1509. [Google Scholar] [CrossRef] [Green Version]
- Hall, A. Rho GTPases and the actin cytoskeleton. Science 1998, 279, 509–514. [Google Scholar] [CrossRef] [Green Version]
- Lamarche, N.; Tapon, N.; Stowers, L.; Burbelo, P.D.; Aspenström, P.; Bridges, T.; Chant, J.; Hall, A. Rac and Cdc42 induce actin polymerization and G1 cell cycle progression independently of p65PAK and the JNK/SAPK MAP kinase cascade. Cell 1996, 87, 519–529. [Google Scholar] [CrossRef] [Green Version]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, J.; Wang, X.; Ding, R.; Quan, J.; Ye, Y.; Gu, T.; Xu, Z.; Zheng, E.; Cai, G.; Wu, Z.; et al. Identification of Important Proteins and Pathways Affecting Feed Efficiency in DLY Pigs by iTRAQ-Based Proteomic Analysis. Animals 2020, 10, 189. https://doi.org/10.3390/ani10020189
Wu J, Wang X, Ding R, Quan J, Ye Y, Gu T, Xu Z, Zheng E, Cai G, Wu Z, et al. Identification of Important Proteins and Pathways Affecting Feed Efficiency in DLY Pigs by iTRAQ-Based Proteomic Analysis. Animals. 2020; 10(2):189. https://doi.org/10.3390/ani10020189
Chicago/Turabian StyleWu, Jie, Xingwang Wang, Rongrong Ding, Jianping Quan, Yong Ye, Ting Gu, Zheng Xu, Enqin Zheng, Gengyuan Cai, Zhenfang Wu, and et al. 2020. "Identification of Important Proteins and Pathways Affecting Feed Efficiency in DLY Pigs by iTRAQ-Based Proteomic Analysis" Animals 10, no. 2: 189. https://doi.org/10.3390/ani10020189
APA StyleWu, J., Wang, X., Ding, R., Quan, J., Ye, Y., Gu, T., Xu, Z., Zheng, E., Cai, G., Wu, Z., Yang, M., & Yang, J. (2020). Identification of Important Proteins and Pathways Affecting Feed Efficiency in DLY Pigs by iTRAQ-Based Proteomic Analysis. Animals, 10(2), 189. https://doi.org/10.3390/ani10020189