
Whole-Genome Sequencing and Characterization of Buffalo Genetic Resources: Recent Advances and Future Challenges






Abstract
:Simple Summary
Abstract
1. Introduction
2. Buffalo Genome Architecture
2.1. Chromosomal Array of Asian Buffaloes
2.2. Chromosomal Array of African Buffaloes
2.3. Sex Chromosomes
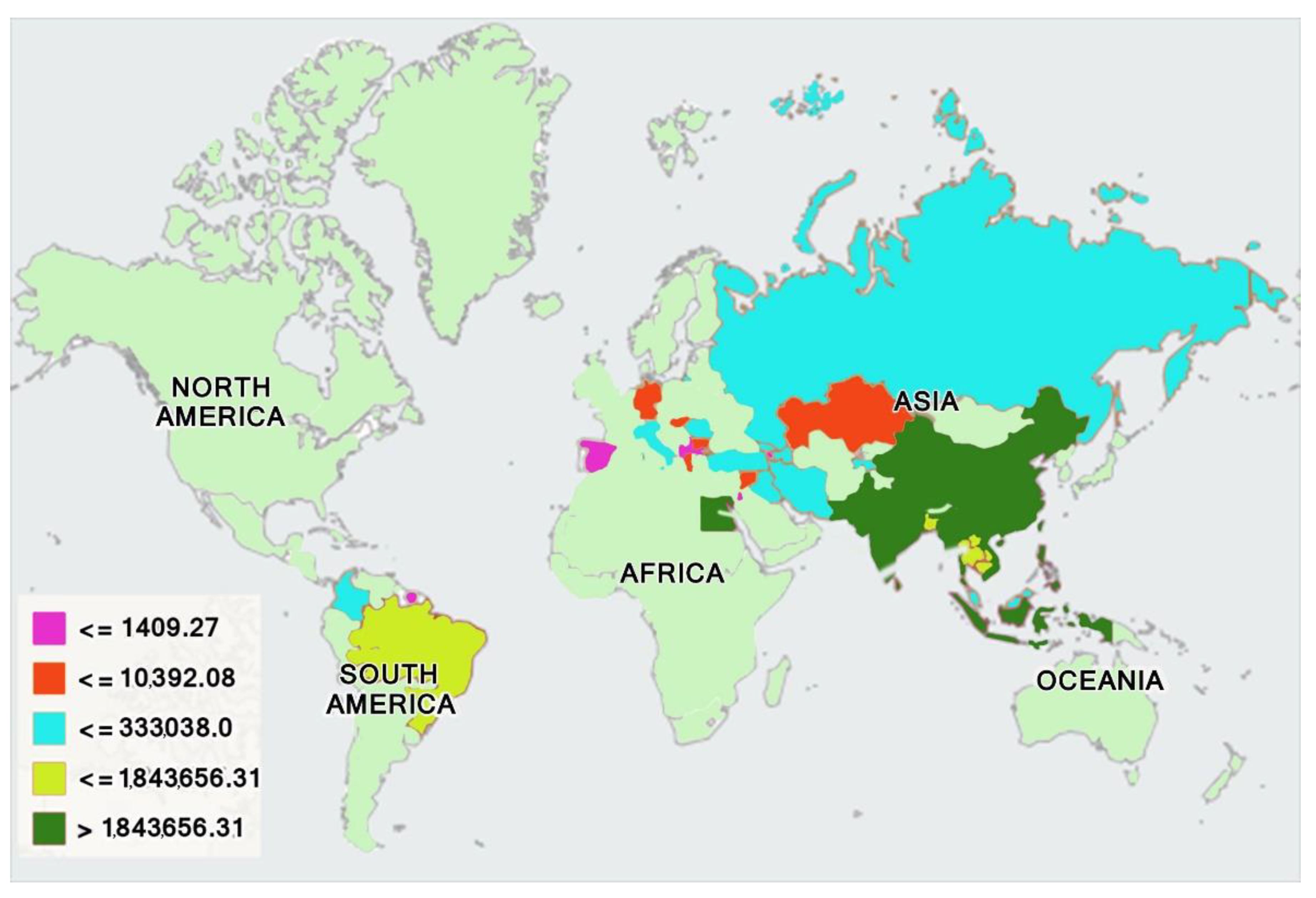
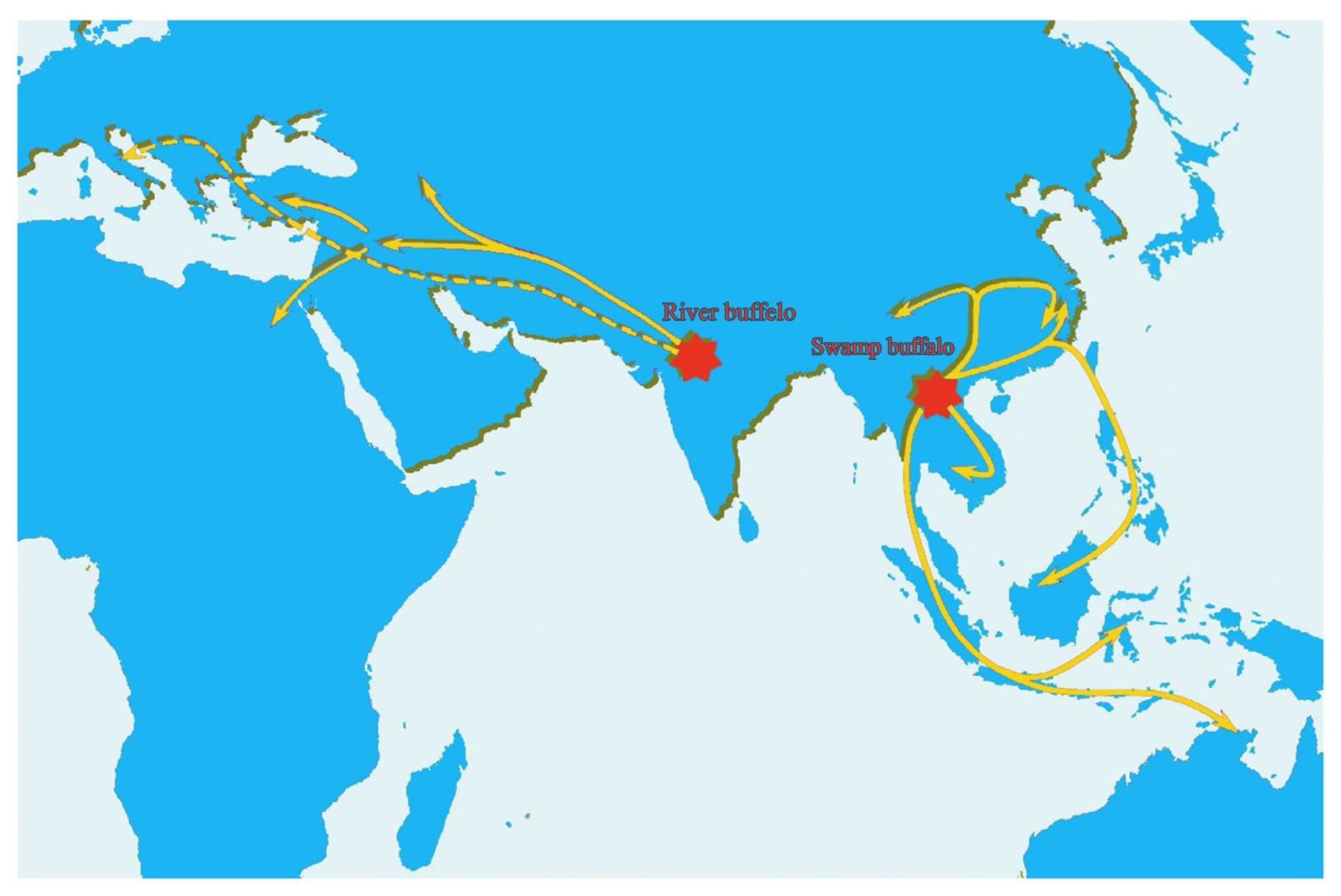
3. Evolution and Domestication of Buffaloes
4. Recent Advances in Whole-Genome Sequencing of the Buffalo Genome
5. Identification of Genes Affecting Important Buffalo Traits
5.1. Candidate Gene Studies
5.1.1. Heat Shock Protein Genes (HSPs)
5.1.2. Reproductive Physiology-Related Genes
5.1.3. Milk Production-Related Genes
5.1.4. Body Coat Color
5.1.5. Disease Resistance
6. The Future Perspective of Buffaloes
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Kierstein, G.; Vallinoto, M.; Silva, A.; Schneider, M.P.; Iannuzzi, L.; Brenig, B. Analysis of mitochondrial D-loop region casts new light on domestic water buffalo (Bubalus bubalis) phylogeny. Mol. Phylogenet. Evol. 2004, 30, 308–324. [Google Scholar] [CrossRef]
- Roth, J.; Myers, P. Bubalus Bubalis 2004. Available online: http://animaldiversity.ummz.umich.edu/site/accounts/information/Bubalus_bubalis.html (accessed on 15 January 2021).
- Yindee, M.; Vlamings, B.H.; Wajjwalku, W.; Techakumphu, M.; Lohachit, C.; Sirivaidyapong, S.; Thitaram, C.; Amarasinghe, A.A.; Alexander, P.A.; Colenbrander, B.; et al. Y-chromosomal variation confirms independent domestications of swamp and river buffalo. Anim. Genet. 2010, 41, 433–435. [Google Scholar] [CrossRef]
- FAO. 2019. Available online: http://www.fao.org/faostat/en/#data/QA/visualize (accessed on 15 January 2021).
- Rehman, S.; Shafique, L.; Yousuf, M.R.; Liu, Q.; Ahmed, J.Z.; Riaz, H. Spectrophotometric calibration and comparison of different semen evaluation methods in Nili-Ravi buffalo bulls. Pak. Vet. J. 2019, 39, 568–572. [Google Scholar] [CrossRef]
- Council, N.R. The Water Buffalo: New Prospects for an Underutilized Animal; National Academy Press: Washington, DC, USA, 1981. [Google Scholar]
- Akhtar, M.S.; Lodhi, L.A.; Ahmad, I.; Qureshi, Z.I.; Muhammad, G. Serum trace mineral variations in Nili-Ravi buffaloes suffering with prepartum vaginal prolapse in two different agro-ecological zones of Punjab, Pakistan. Theriogenology 2012, 77, 1328–1333. [Google Scholar] [CrossRef]
- Warriach, H.M.; McGill, D.M.; Bush, R.D.; Wynn, P.C.; Chohan, K.R. A review of recent developments in buffalo reproduction—A review. Asian-Australas. J. Anim. Sci. 2015, 28, 451. [Google Scholar] [CrossRef] [PubMed]
- Haldar, A.; Prakash, B.S. Peripheral patterns of growth hormone, luteinizing hormone, and progesterone before, at, and after puberty in buffalo heifer. Endocr. Res. 2005, 31, 295–306. [Google Scholar] [CrossRef] [PubMed]
- De Rensis, F.; Lopez-Gatius, F. Protocols for synchronizing estrus and ovulation in buffalo (Bubalus bubalis): A review. Theriogenology 2007, 67, 209–216. [Google Scholar] [CrossRef]
- Hassan, F.; Khan, M.S.; Rehman, M.S.; Sarwar, M.; Bhatti, S.A. Seasonality of calving in Nili-Ravi buffaloes, purebred Sahiwal and crossbred cattle in Pakistan. Ital. J. Anim. Sci. 2007, 6, 1298–1301. [Google Scholar] [CrossRef]
- Perera, B.M.A.O. Reproductive cycles of buffalo. Anim. Reprod. Sci. 2011, 124, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Hussain Shah, S.N. Prolonged calving intervals in the Nili ravi buffalo. Ital. J. Anim. Sci. 2007, 6, 694–696. [Google Scholar] [CrossRef]
- El-Wishy, A.B. The postpartum buffalo: II. Acyclicity and anestrus. Anim. Reprod. Sci. 2007, 97, 216–236. [Google Scholar] [CrossRef] [PubMed]
- Fahimuddin, M. Domestic water buffalo. In Domestic Water Buffalo; Oxford and IBH Publishing Co.: New Delhi, India, 1975. [Google Scholar]
- Berardino, D.D.; Iannuzzi, L. Chromosome banding homologies in Swamp and Murrah buffalo. J. Hered. 1981, 72, 183–188. [Google Scholar] [CrossRef]
- Iannuzzi, L.; Di Meo, G. Water Buffalo. In Genome Mapping and Genomics in Domestic Animals; Cockett, N.E., Kole, C., Eds.; Springer: Berlin/Herdelberg, Germany, 2009. [Google Scholar]
- De Abreu Santos, D.J.; Ferreira de Camargo, G.M.; Cardoso, D.F.; Buzanskas, M.E.; Aspilcueta-Borquis, R.R.; Hurtado-Lugo, N.A.; de Araújo Neto, F.R.; Galvão de Albuquerque, L.; Ma, L.; Tonhati, H. Linkage disequilibrium-based inference of genome homology and chromosomal rearrangements between species. G3 Genes Genomes Genet. 2020, 10, 2327–2343. [Google Scholar] [CrossRef] [PubMed]
- Amaral, M.E.; Grant, J.R.; Riggs, P.K.; Stafuzza, N.B.; Rodrigues Filho, E.A.; Goldammer, T.; Weikard, R.; Brunner, R.M.; Kochan, K.J.; Greco, A.J.; et al. A first-generation whole genome RH map of the river buffalo with comparison to domestic cattle. BMC Genom. 2008, 9, 631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaral, M.E.; Owens, K.E.; Elliott, J.S.; Fickey, C.; Schäffer, A.A.; Agarwala, R.; Womack, J.E. Construction of a river buffalo (Bubalus bubalis) whole-genome radiation hybrid panel and preliminary RH mapping of chromosomes 3 and 10. Ital. J. Anim. Sci. 2007, 6, 237–245. [Google Scholar] [CrossRef]
- Luo, X.; Zhou, Y.; Zhang, B.; Zhang, Y.; Wang, X.; Feng, T.; Li, Z.; Cui, K.; Wang, Z.; Luo, C.; et al. Understanding divergent domestication traits from the whole-genome sequencing of swamp-and river-buffalo populations. Natl. Sci. Rev. 2020, 7, 686–701. [Google Scholar] [CrossRef] [Green Version]
- Low, W.Y.; Tearle, R.; Bickhart, D.M.; Rosen, B.D.; Kingan, S.B.; Swale, T.; Thibaud-Nissen, F.; Murphy, T.D.; Young, R.; Lefevre, L.; et al. Chromosome-level assembly of the water buffalo genome surpasses human and goat genomes in sequence contiguity. Nat. Commun. 2019, 10, 260. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, D.S., Jr.; Womack, J.E. Chromosome conservation in the Bovidae. J. Hered. 1992, 83, 287–298. [Google Scholar] [CrossRef]
- Iannuzzi, L.; Di Meo, G.P.; Perucatti, A.; Schibler, L.; Incarnato, D.; Cribiu, E.P. Comparative FISH mapping in river buffalo and sheep chromosomes: Assignment of forty autosomal type I loci from sixteen human chromosomes. Cytogenet. Genome Res. 2001, 94, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Iannuzzi, L.; King, W.A.; Di Berardino, D. Chromosome evolution in domestic bovids as revealed by chromosome banding and FISH-mapping techniques. Cytogenet. Genome Res. 2009, 126, 49–62. [Google Scholar] [CrossRef]
- Amano, T.; Miyakoshi, Y.; Takada, T.; Kikkawa, Y.; Suzuki, H. Genetic variants of ribosomal DNA and mitochondrial DNA between swamp and river buffaloes. Anim. Genet. 1994, 25, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Solis, C.D.; Masangkay, J.S.; Maeda, K.; Kawamoto, Y.; Namikawa, T. Phylogenetic relationship among all living species of the genus Bubalus based on DNA sequences of the cytochrome b gene. Biochem. Genet. 1996, 34, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Barker, J.S.; Tan, S.G.; Selvaraj, O.S.; Mukherjee, T.K. Genetic variation within and relationships among populations of Asian water buffalo (Bubalus bubalis). Anim. Genet. 1997, 28, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Barker, J.S.; Moore, S.S.; Hetzel, D.J.; Evans, D.; Byrne, K.; Tan, S.G. Genetic diversity of Asian water buffalo (Bubalus bubalis): Microsatellite variation and a comparison with protein-coding loci. Anim. Genet. 1997, 28, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.H.; Drinkwater, R.D.; Yusoff, K.; Tan, S.G.; Hetzel, D.J.S.; Barker, J.S.F. Genetic diversity of Asian water buffalo (Bubalus bubalis): Mitochondrial DNA D-loop and cytochrome b sequence variation. Anim. Genet. 1998, 29, 253–264. [Google Scholar] [CrossRef] [Green Version]
- Lei, C.Z.; Zhang, W.; Chen, H.; Lu, F.; Liu, R.Y.; Yang, X.Y.; Zhang, H.C.; Liu, Z.G.; Yao, L.B.; Lu, Z.F.; et al. Independent maternal origin of Chinese swamp buffalo (Bubalus bubalis). Anim. Genet. 2007, 38, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Nagarajan, M.; Sandhu, J.S.; Kumar, N.; Behl, V.; Nishanth, G. Mitochondrial DNA analyses of Indian water buffalo support a distinct genetic origin of river and swamp buffalo. Anim. Genet. 2007, 38, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Mishra, B.P.; Dubey, P.K.; Prakash, B.; Kathiravan, P.; Goyal, S.; Sadana, D.K.; Das, G.C.; Goswami, R.N.; Bhasin, V.; Joshi, B.K.; et al. Genetic analysis of river, swamp and hybrid buffaloes of north-east India throw new light on phylogeography of water buffalo (Bubalus bubalis). J. Anim. Breed. Genet. 2015, 132, 454–466. [Google Scholar] [CrossRef]
- Wang, S.; Chen, N.; Capodiferro, M.R.; Zhang, T.; Lancioni, H.; Zhang, H.; Miao, Y.; Chanthakhoun, V.; Wanapat, M.; Yindee, M.; et al. Whole mitogenomes reveal the history of swamp buffalo: Initially shaped by glacial periods and eventually modelled by domestication. Sci. Rep. 2017, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Takahata, N.; Nei, M. Gene genealogy and variance of interpopulational nucleotide differences. Genetics 1985, 110, 325–344. [Google Scholar] [CrossRef]
- Nagarajan, M.; Nimisha, K.; Kumar, S. Mitochondrial DNAvariability of domestic river buffalo (Bubalus bubalis) populations: Genetic evidence for domestication of river buffalo in Indian subcontinent. Genome Biol. Evol. 2015, 7, 1252–1259. [Google Scholar] [CrossRef] [Green Version]
- Colli, L.; Milanesi, M.; Vajana, E.; Iamartino, D.; Bomba, L.; Puglisi, F.; Del Corvo, M.; Nicolazzi, E.L.; Ahmed, S.S.; Herrera, J.R.; et al. New insights on water buffalo genomic diversity and post-domestication migration routes from medium density SNP chip data. Front. Genet. 2018, 9, 53. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Nagarajan, M.; Sandhu, J.S.; Kumar, N.; Behl, V. Phylogeography and domestication of Indian river buffalo. BMC Evol. Biol. 2007, 7, 186–193. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Vankan, D.; Zhang, Y.; Barker, J.S. Genetic differentiation of water buffalo (Bubalus bubalis) populations in China, Nepal and south-east Asia: Inferences on the region of domestication of the swamp buffalo. Anim. Genet. 2011, 42, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lu, Y.; Yindee, M.; Li, K.Y.; Kuo, H.Y.; Ju, Y.T.; Ye, S.; Faruque, M.O.; Li, Q.; Wang, Y.; et al. Strong and stable geographic differentiation of swamp buffalo maternal and paternal lineages indicates domestication in the China/Indochina border region. Mol. Ecol. 2016, 25, 1530–1550. [Google Scholar] [CrossRef] [PubMed]
- Finlay, E.K.; Gaillard, C.; Vahidi, S.M.; Mirhoseini, S.Z.; Jianlin, H.; Qi, X.B.; El-Barody, M.A.; Baird, J.F.; Healy, B.C.; Bradley, D.G. Bayesian inference of population expansions in domestic bovines. Biol. Lett. 2007, 3, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Clutton-Brock, J. A natural History of Domesticated Mammals; Cambridge University Press: Cambridge, UK, 1999. [Google Scholar]
- Zeuner, F.E. A History of Domesticated Animals; Hutchinson: London, UK; Harper & Row: New York, NY, USA, 1963. [Google Scholar]
- Unal, E.O.; Soysal, M.I.; Yuncu, E.; Dagtas, N.D.; Togan, I. Microsatellite based genetic diversity among the three-water buffalo (Bubalus bubalis) populations in Turkey. Arch. Anim. Breed. 2014, 57, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Colli, L.; Barker, J.S. Asian water buffalo: Domestication, history and genetics. Anim. Genet. 2020, 51, 177–191. [Google Scholar] [CrossRef]
- Higham, C. Early Cultures of Mainland Southeast Asia; River Books, Bangkok; Cambridge University Press: Cambridge, UK, 2002. [Google Scholar]
- Sun, T.; Shen, J.; Achilli, A.; Chen, N.; Chen, Q.; Dang, R.; Zhang, T. Genomic analyses reveal distinct genetic architectures and selective pressures in buffaloes. GigaScience 2020, 9, giz166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larson, G.; Burger, J. A population genetics view of animal domestication. Trends Genet 2013, 29, 197–205. [Google Scholar] [CrossRef]
- NCBI. Available online: https://www.ncbi.nlm.nih.gov (accessed on 14 March 2021).
- Mintoo, A.A.; Zhang, H.; Chen, C.; Moniruzzaman, M.; Deng, T.; Anam, M.; Han, P. Draft genome of the river water buffalo. Ecol. Evol. 2019, 9, 3378–3388. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J. BLAT–the BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, L.; Su, B.; Li, W.H.; Zhao, Z. CpG island density and its correlations with genomic features in mammalian genomes. Genome Biol. 2008, 9, R79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jobse, C.; Buntjer, J.B.; Haagsma, N.; Breukelman, H.J.; Beintema, J.J.; Lenstral, J.A. Evolution and recombination of bovine DNA repeats. J. Mol. Evol. 1995, 41, 277–283. [Google Scholar] [CrossRef]
- Elsik, C.G.; Tellam, R.L.; Worley, K.C. The genome sequence of taurine cattle: A window to ruminant biology and evolution. Science 2009, 324, 522–528. [Google Scholar]
- Li, J.; Liu, J.; Campanile, G.; Plastow, G.; Zhang, C.; Wang, Z.; Yang, L. Novel insights into the genetic basis of buffalo reproductive performance. BMC Genom. 2018, 19, 814. [Google Scholar] [CrossRef]
- Glanzmann, B.; Möller, M.; Le Roex, N.; Tromp, G.; Hoal, E.G.; Van Helden, P.D. The complete genome sequence of the African buffalo (Syncerus caffer). BMC Genom. 2016, 17, 1001. [Google Scholar] [CrossRef] [Green Version]
- Dutta, P.; Talenti, A.; Young, R.; Jayaraman, S.; Callaby, R.; Jadhav, S.K.; Dhanikachalam, V.; Manikandan, M.; Biswa, B.B.; Low, W.Y.; et al. Whole genome analysis of water buffalo and global cattle breeds highlights convergent signatures of domestication. Nat. Commun. 2020, 11, 1–3. [Google Scholar] [CrossRef]
- Mishra, S.R. Significance of molecular chaperones and micro RNAs in acquisition of thermo-tolerance in dairy cattle. Anim. Biotechnol. 2020, 1–11. [Google Scholar] [CrossRef]
- Rehman, S.; Nadeem, A.; Javed, M.; Hassan, F.; Luo, X.; Khalid, R.B.; Liu, Q. Genomic Identification, Evolution and Sequence Analysis of the Heat-Shock Protein Gene Family in Buffalo. Genes 2020, 11, 1388. [Google Scholar] [CrossRef]
- Archana, P.R.; Aleena, J.; Pragna, P.; Vidya, M.K.; Niyas, A.P.; Bagath, M.; Krishnan, G.; Manimaran, A.; Beena, V.; Kurien, E.K.; et al. Role of heat shock proteins in livestock adaptation to heat stress. J. Dairy Vet. Anim. Res. 2017, 5, 00127. [Google Scholar]
- Collier, R.J.; Collier, J.L.; Rhoads, R.P.; Baumgard, L. Invited review: Genes involved in the bovine heat stress response. J. Dairy Sci. 2008, 91, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Hooda, O.K.; Singh, G.; Meur, S.K. Influence of induced heat stress on HSP70 in buffalo lymphocytes. J. Anim. Physiol. Anim. Nutr. 2011, 95, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Cushman, R.A. Physiology and endocrinology symposium: The current status of heat shock in early embryonic survival and reproductive efficiency. J. Anim. Sci. 2013, 91, 1141–1142. [Google Scholar] [CrossRef] [Green Version]
- Dong, C.W.; Zhang, Y.B.; Zhang, Q.Y.; Gui, J.F. Differential expression of three Paralichthys olivaceus Hsp40 genes in responses to virus infection and heat shock. Fish Shellfish Immunol. 2006, 21, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Banerji, U. Heat Shock Protein 90 as a Drug Target: Some Like It Hot. Clin. Cancer Res. 2009, 15, 9–14. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Feng, P.; Liu, T.; Jin, D. Recent advances in machine learning methods for predicting heat shock proteins. Curr. Drug Metab. 2019, 20, 224–228. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.X.; Li, D.Q.; Cui, Q.W.; Shi, H.X.; Wang, G.L. Analysis of HSP70 mRNA level and association between linked microsatellite loci and heat tolerance traits in dairy cows. Yi Chuan Hered. 2010, 32, 935–941. [Google Scholar] [CrossRef]
- Hassan, F.; Nawaz, A.; Rehman, M.S.; Ali, M.A.; Dilshad, S.M.; Yang, C. Prospects of HSP70 as a genetic marker for thermo-tolerance and immuno-modulation in animals under climate change scenario. Anim. Nutr. 2019, 5, 340–350. [Google Scholar] [CrossRef]
- Sodhi, M.; Mukesh, M.; Kishore, A.; Mishra, B.; Kataria, R.; Joshi, B. Novel polymorphisms in UTR and coding region of inducible heat shock protein 70.1 gene in tropically adapted Indian zebu cattle (Bos indicus) and riverine buffalo (Bubalus bubalis). Gene 2013, 527, 606–615. [Google Scholar] [CrossRef]
- Rosenkrans, C., Jr.; Banks, A.; Reiter, S.; Looper, M. Calving traits of crossbred Brahman cows are associated with Heat Shock Protein 70 genetic polymorphisms. Anim. Reprod. Sci. 2010, 119, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Barile, V.L. Improving reproductive efficiency in female buffaloes. Livest. Prod. Sci. 2005, 92, 183–194. [Google Scholar] [CrossRef]
- Shi, W.; Yuan, X.; Cui, K.; Li, H.; Fu, P.; Rehman, S.U.; Shi, D.; Liu, Q.; Li, Z. LC-MS/MS Based Metabolomics Reveal Candidate Biomarkers and Metabolic Changes in Different Buffalo Species. Animals 2021, 11, 560. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Gu, J.; Xue, H.; Li, Q.; Liang, M.; Wang, N.; Wang, G.; Wu, Q.; Liu, S.; Yu, H.; et al. Identification of four SNPs in LHB gene and their associations with sperm qualities of Chinese buffaloes. Anim. Biotechnol. 2017, 28, 168–173. [Google Scholar] [CrossRef]
- Wang, G.; Hao, L.; Cheng, Y.; Li, S.; Zhang, Y.; Lv, C.; Wei, W.; Huang, S.; Shi, H.; Dong, L.; et al. Effects of GnRHR polymorphisms on sperm quality in Chinese water buffalo. Anim. Reprod. Sci. 2017, 186, 37–43. [Google Scholar] [CrossRef] [PubMed]
- El-Bayomi, K.M.; Saleh, A.A.; Awad, A.; El-Tarabany, M.S.; El-Qaliouby, H.S.; Afifi, M.; El-Komy, S.; Essawi, W.M.; Almadaly, E.A.; El-Magd, M.A. Association of CYP19A1 gene polymorphisms with anoestrus in water buffaloes. Reprod. Fertil. Dev. 2018, 30, 487–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, A.K.; Gunwant, P.; Soni, N.; Kumar, S.; Kumar, A.; Magotra, A.; Singh, I.; Phogat, J.B.; Sharma, R.K.; Bangar, Y.; et al. Genotype of MTNR1A gene regulates the conception rate following melatonin treatment in water buffalo. Theriogenology 2019, 128, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Surya, T.; Vineeth, M.R.; Sivalingam, J.; Tantia, M.S.; Dixit, S.P.; Niranjan, S.K.; Gupta, I.D. Genomewide identification and annotation of SNPs in Bubalus bubalis. Genomics 2019, 111, 1695–1698. [Google Scholar] [CrossRef] [PubMed]
- Chethan, S.G.; Singh, S.K.; Nongsiej, J.; Rakesh, H.B.; Singh, R.P.; Kumar, N.; Agarwal, S.K. IFN-τ Acts in a Dose-Dependent Manner on Prostaglandin Production by Buffalo Endometrial Stromal Cells Cultured in vitro. Reprod. Domest. Anim. 2014, 49, 403–408. [Google Scholar] [CrossRef]
- Fortes, M.R.S.; Lehnert, S.A.; Bolormaa, S.; Reich, C.; Fordyce, G.; Corbet, N.J.; Whan, V.; Hawken, R.J.; Reverter, A. Finding genes for economically important traits: Brahman cattle puberty. Anim. Prod Sci. 2012, 52, 143–150. [Google Scholar] [CrossRef]
- McDaneld, T.G.; Kuehn, L.A.; Thomas, M.G.; Snelling, W.M.; Smith, T.P.L.; Pollak, E.J.; Cole, J.B.; Keele, J.W. Genomewide association study of reproductive efficiency in female cattle. J. Anim. Sci. 2014, 92, 1945–1957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Camargo, G.M.; Porto-Neto, L.R.; Kelly, M.J.; Bunch, R.J.; McWilliam, S.M.; Tonhati, H.; Lehnert, S.A.; Fortes, M.R.; Moore, S.S. Non-synonymous mutations mapped to chromosome X associated with andrological and growth traits in beef cattle. BMC Genom. 2015, 16, 384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arndt, L.; Castonguay, J.; Arlt, E.; Meyer, D.; Hassan, S.; Borth, H.; Zierler, S.; Wennemuth, G.; Breit, A.; Biel, M.; et al. NAADP and the two-pore channel protein 1 participate in the acrosome reaction in mammalian spermatozoa. Mol. Biol. Cell 2014, 25, 948–964. [Google Scholar] [CrossRef] [PubMed]
- Spencer, T.E.; Forde, N.; Dorniak, P.; Hansen, T.R.; Romero, J.J.; Lonergan, P. Conceptus-derived prostaglandins regulate gene expression in the endometrium prior to pregnancy recognition in ruminants. Reproduction 2013, 146, 377–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, H.; Choi, Y.; Shim, J.; Yoo, I.; Ka, H. Prostaglandin transporters ABCC4 and SLCO2A1 in the uterine endometrium and conceptus during pregnancy in pigs. Biol. Reprod. 2014, 90, 100–101. [Google Scholar] [CrossRef] [PubMed]
- Refsdal, A.O. To treat or not to treat: A proper use of hormones and antibiotics. Anim. Reprod. Sci. 2000, 60, 109–119. [Google Scholar] [CrossRef]
- Khaitan, D.; Sankpal, U.T.; Weksler, B.; Meister, E.A.; Romero, I.A.; Couraud, P.O.; Ningaraj, S.N. Role of KCNMA1 gene in breast cancer invasion and metastasis to brain. BMC Cancer 2009, 9, 258. [Google Scholar] [CrossRef] [Green Version]
- Peng, L.; Zhang, S.D.; Yuen, H.F.; Mccrudden, C.M.; Wen, Q.; Chan, K.W.; Hang, F.K. Identification of TWIST-interacting genes in prostate cancer. Sci. China Life Sci. 2017, 60, 386–396. [Google Scholar]
- An, C.H.; Je, E.M.; Yoo, N.J.; Lee, S.H. Frameshift mutations of cadherin genes DCHS2, CDH10 and CDH24 genes in gastric and colorectal cancers with high microsatellite instability. Pathol. Oncol. Res. 2015, 21, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Işık, E.; Beck, P.P.; Campi, I.; Özon, A.; Alikaşifoğlu, A.; Gönç, N.; Kandemir, N. Thyroid hormone resistance: A novel mutation in thyroid hormone receptor beta (THRB) gene—Case report. Turk. J. Pediatr. 2013, 55, 322–327. [Google Scholar]
- Barrett, P.; Ebling, F.J.; Schuhler, S.; Wilson, D.; Ross, A.W.; Warner, A.; Jethwa, P.; Boelen, A.; Visser, T.J.; Ozanne, D.M.; et al. Hypothalamic thyroid hormone catabolism acts as a gatekeeper for the seasonal control of body weight and reproduction. Endocrinology 2007, 148, 3608–3617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Castelãn, J.; Anaya-Hernãndez, A.; Mendez-Tepepa, M.; Martinez-Gomez, M.; Castelãn, F.; Cuevas-Romero, E. Distribution of thyroid hormoneand thyrotropin receptors in reproductive tissues of adult female rabbits. Endocr. Res. Commun. 2016, 42, 59–70. [Google Scholar] [CrossRef]
- Christ, G.J.; Andersson, K.E.; Williams, K.; Zhao, W.; D’Agostino, R., Jr.; Kaplan, J.; Aboushwareb, T.; Yoo, J.; Calenda, G.; Davies, K.P.; et al. Smooth-muscle–specific gene transfer with the human Maxi-K channel improves erectile function and enhances sexual behavior in atherosclerotic cynomolgus monkeys. Eur. Urol. 2009, 56, 1055–1066. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, J.R.; Wiltbank, M.C.; Ginther, O.J. Relationship between follicular development and the decline in the follicle-stimulating hormone surge in heifers. Biol. Reprod. 1999, 60, 72–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Bickhart, D.M.; Ramunno, L.; Iamartino, D.; Williams, J.L.; Liu, G.E. Comparative sequence alignment reveals River Buffalo genomic structural differences compared with cattle. Genomics 2018, 19, 236–239. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.J.; Song, L.J.; Wu, F.J.; Liang, X.W.; Yang, B.Z.; Wathes, D.C.; Pollott, G.E.; Cheng, Z.; Shi, D.S.; Liu, Q.Y.; et al. Investigation of transferability of BovineSNP50 BeadChip from cattle to water buffalo for genome wide association study. Mol. Biol. Rep. 2013, 40, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Shahi, P.; Slorach, E.M.; Wang, C.Y.; Chou, J.; Lu, A.; Ruderisch, A.; Werb, Z. The transcriptional repressor ZNF503/Zeppo2 promotes mammary epithelial cell proliferation and enhances cell invasion. J. Biol. Chem. 2015, 290, 3803–3813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, S.L.; Liu, Y.C.; Chen, S.Y.; Huang, T.H.; Liu, P.T.; Liu, F.C. Identification of two evolutionarily conserved 5′cis-elements involved in regulating spatiotemporal expression of Nolz-1 during mouse embryogenesis. PLoS ONE 2013, 8, e54485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, F.R.; Hinds, D.A.; Tung, J.Y.; Stolk, L.; Styrkarsdottir, U.; Saxena, R.; Bjonnes, A.; Broer, L.; Dunger, D.B.; Halldorsson, B.V.; et al. Causal mechanisms and balancing selection inferred from genetic associations with polycystic ovary syndrome. Nat. Commun. 2015, 6, 8464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makina, S.O.; Muchadeyi, F.C.; van Marle-Koster, E.; Taylor, J.F.; Makgahlela, M.L.; Maiwashe, A. Genome-wide scan for selection signatures in six cattle breeds in South Africa. Genet. Sel. Evol. 2015, 47, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Wang, Y. Molecular characterization, expression patterns and subcellular localization of Myotrophin (MTPN) gene in porcine skeletal muscle. Mol. Biol. Rep. 2012, 39, 2733–2738. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.V. A secreted tumor-suppressor, mac25, with activin-binding activity. Mol. Med. 2000, 6, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Ueno, N.; Ling, N.; Ying, S.Y.; Esch, F.; Shimasaki, S.; Guillemin, R. Isolation and partial characterization of follistatin: A single-chain Mr 35,000 monomeric protein that inhibits the release of follicle-stimulating hormone. Proc. Natl. Acad. Sci. USA 1987, 84, 8282–8286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, D.M.; Klein, R.; de Vos, F.L.; Mclachlan, R.I.; Wettenhall, R.E.; Hearn, M.T.; Burger, H.G.; de Kretser, D.M. The isolation of polypeptides with FSH suppressing activity from bovine follicular fluid which are structurally different to inhibin. Biochem. Biophys. Res. Commun. 1987, 149, 744–749. [Google Scholar] [CrossRef]
- Jorgez, C.J.; Klysik, M.; Jamin, S.P.; Behringer, R.R.; Matzuk, M.M. Granulosa cellspecific inactivation of follistatin causes female fertility defects. Mol. Endocrinol. 2004, 18, 953–967. [Google Scholar] [CrossRef] [PubMed]
- Muttukrishna, S.; Tannetta, D.; Groome, N.; Sargent, I. Activin and follistatin in female reproduction. Mol. Cell. Endocrinol. 2004, 225, 45–56. [Google Scholar] [CrossRef]
- Okamura, H.; Yamamura, T.; Wakabayashi, Y. Kisspeptin as a master player in the central control of reproduction in mammals: An overview of kisspeptin research in domestic animals. Anim. Sci. J. 2013, 84, 369–381. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Deng, T.; Zhou, Y.; Ye, T.; Zhou, Z.; Zhang, S.; Shao, B.; Wei, P.; Sun, H.; Khan, F.A.; et al. Systematic analyses for candidate genes of milk production traits in water buffalo (Bubalus Bubalis). Anim. Genet. 2019, 50, 207–216. [Google Scholar] [CrossRef]
- Venturini, G.C.; Cardoso, D.F.; Baldi, F.; Freitas, A.C.; Aspilcuetaborquis, R.R.; Santos, D.J.; Camargo, G.M.; Stafuzza, N.B.; Albuquerque, L.G.; Tonhati, H. Association between single-nucleotide polymorphisms and milk production traits in buffalo. Genet. Mol. Res. 2014, 13, 10256–10268. [Google Scholar] [CrossRef]
- Michelizzi, V.N.; Dodson, M.V.; Pan, Z.; Amaral, M.E.J.; Michal, J.J.; Mclean, D.J.; Womack, J.E.; Jiang, Z. Water buffalo genome science comes of age. Int. J. Biol. Sci. 2010, 6, 333–349. [Google Scholar] [CrossRef]
- Cesarani, A.; Gaspa, G.; Pauciullo, A.; Degano, L.; Vicario, D.; Macciotta, N.P. Genome-wide analysis of homozygosity regions in european simmental bulls. J. Anim. Breed. Genet. 2021, 138, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Camargo, G.D.; Aspilcuetaborquis, R.R.; Fortes, M.; Portoneto, R.; Cardoso, D.F.; Santos, D.; Lehnert, S.A.; Reverter, A.; Moore, S.S.; Tonhati, H. Prospecting major genes in dairy buffaloes. BMC Genom. 2015, 16, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Halawany, N.; Abdel-Shafy, H.; Shawky, A.E.M.A.; Abdel-Latif, M.A.; Al-Tohamy, A.F.M.; El-Moneim, O.M.A. Genomewide association study for milk production in Egyptian buffalo. Livest. Sci. 2017, 198, 10–16. [Google Scholar] [CrossRef]
- Iamartino, D.; Nicolazzi, E.L.; Van, C.T.; Reecy, J.M.; Fritzwaters, E.R.; Koltes, J.E.; Biffani, S.; Sonstegard, T.S.; Schroeder, S.G.; Ajmonemarsan, P. Design and validation of a 90K SNP genotyping assay for the water buffalo (Bubalus bubalis). PLoS ONE 2017, 12, e0185220. [Google Scholar] [CrossRef]
- Liu, J.J.; Liang, A.X.; Campanile, G.; Plastow, G.; Zhang, C.; Wang, Z.; Salzano, A.; Gasparrini, B.; Cassandro, M.; Yang, L.G. Genome-wide association studies to identify quantitative trait loci affecting milk production traits in water buffalo. J. Dairy Sci. 2017, 101, 433–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, M.; Cosenza, G.; Gaspa, G.; Iannaccone, M.; Macciotta, N.P.; Chemello, G.; Di Stasio, L.; Pauciullo, A. Sequencing of lipoprotein lipase gene in the Mediterranean river buffalo identified novel variants affecting gene expression. J. Dairy Sci. 2020, 103, 6374–6382. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Bao, H.; Ishikawa, Z.; Wang, W.; Lim, H.Y. Cardiomyocyte regulation of systemic lipid metabolism by the apolipoprotein B-containing lipoproteins in drosophila. PLoS Genet. 2017, 13, e1006555. [Google Scholar] [CrossRef] [Green Version]
- Abo-Al-Ela, H.G.; El-Magd, M.A.; El-Nahas, A.F.; Mansour, A.A. Association of a novel SNP in exon 10 of the IGF2 gene with growth traits in Egyptian water buffalo (Bubalus bubalis). Trop. Anim. Health Prod. 2014, 46, 947–952. [Google Scholar] [CrossRef] [PubMed]
- Coizet, B.; Frattini, S.; Nicoloso, L.; Iannuzzi, L.; Coletta, A.; Talenti, A.; Minozzi, G.; Pagnacco, G.; Crepaldi, P. Polymorphism of the STAT5A, MTNR1A and TNFα genes and their effect on dairy production in Bubalus bubalis. Ital. J. Anim. Sci. 2018, 17, 31–37. [Google Scholar] [CrossRef] [Green Version]
- El-Komy, S.M.; Saleh, A.A.; Abdel-Hamid, T.M.; El-Magd, M.A. Association of GHR Polymorphisms with Milk Production in Buffaloes. Animals 2020, 10, 1203. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Gao, S.; Fu, L.; Qiu, L.; Miao, Y. Polymorphism and molecular characteristics of the CSN1S2 gene in river and swamp buffalo. Arch. Anim. Breed. 2020, 63, 345–354. [Google Scholar] [CrossRef]
- Rehman, S.U.; Feng, T.; Wu, S.; Luo, X.; Lei, A.; Luobu, B.; Hassan, F.; Liu, Q. Comparative genomics, evolutionary and gene regulatory regions analysis of casein gene family in Bubalus bubalis. Front. Genet. 2021, 21, 287–307. [Google Scholar] [CrossRef]
- Liu, J.; Wang, Z.; Li, J.; Li, H.; Yang, L. Genome-wide identification of Diacylglycerol Acyltransferases (DGAT) family genes influencing Milk production in Buffalo. BMC Genet. 2020, 21, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, D.; Zhao, P.; Si, J.; Fang, L.; Pairo-Castineira, E.; Hu, X.; Tian, B. Genomic analysis revealed a convergent evolution of LINE-1 in coat color: A case study in water buffaloes (Bubalus bubalis). Mol. Biol. Evol. 2020, 38, 1122–1136. [Google Scholar] [CrossRef] [PubMed]
- Rife, D.C. Color and horn variations in water buffalo: The inheritance of coat color, eye color and shape of horns. J. Hered. 1962, 53, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Rife, D.C.; Buranamanas, P. Inheritance of white coat color in the water buffalo of Thailand. J. Hered. 1959, 50, 269–272. [Google Scholar] [CrossRef]
- Miao, Y.; Wu, G.; Wang, L.; Li, D.; Tang, S.; Liang, J.; Mao, H.; Luo, H.; Zhang, Y. The role of MC1R gene in buffalo coat color. Sci. China Life Sci. 2010, 53, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Yusnizar, Y.; Wilbe, M.; Herlino, A.O.; Sumantri, C.; Noor, R.R.; Boediono, A.; Andersson, L.; Andersson, G. Microphthalmia-associated transcription factor mutations are associated with white-spotted coat color in swamp buffalo. Anim. Genet. 2015, 46, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Iannaccone, M.; Cosenza, G.; Pauciullo, A.; Martino, G.; Capparelli, R. The SNP g.4667G>A at 30-UTR of IFNG gene is associated with susceptibility to bovine tuberculosis in Mediterranean water buffalo (Bubalus bubalis). Anim. Genet. 2018, 49, 496–497. [Google Scholar] [CrossRef] [PubMed]
- El-Halawany, N.; Abd-el-monsif, A.S.; Al-Tohamy, A.F.; Hegazy, L.; Abdel-Shafy, H.; Abdel-Latif, M.A.; Ghazi, Y.A.; Neuhoff, C.; Salilew-Wondim, D.; Schellander, K. Complement component 3: Characterization and association with mastitis resistance in Egyptian water buffalo and cattle. J. Genet. 2017, 96, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Othman, O.E.; Khodary, M.G.; El-Deeb, A.H.; Hussein, H.A. Five BoLA-DRB3 genotypes detected in Egyptian buffalo infected with Foot and Mouth disease virus serotype O. J. Genet. Eng. Biotechnol. 2018, 16, 513–518. [Google Scholar] [CrossRef]
- Abdi, Z.; Rafat, S.A.; Moghaddam, G.; Hajikolaii, M. A Review on the Polymorphism of Genes/Markers Responsible for Genetically Brucellosis Disease Resistance in Water Buffalo; Asian Buffalo Congress: Istanbul, Turkey, 2015. [Google Scholar]
- Ganguly, I.; Sharma, A.; Singh, R.; Deb, S.M.; Singh, D.K.; Mitra, A. Association of microsatellite (GT) n polymorphism at 3′ UTR of NRAMP1 with the macrophage function following challenge with Brucella LPS in buffalo (Bubalus bubalis). Vet. Microbiol. 2008, 129, 188–196. [Google Scholar] [CrossRef]
- Garry, A.L.; Schutta, C. Natural resistance against brucellosis: A review. Open Vet. Sci. J. 2010, 4, 61–71. [Google Scholar] [CrossRef] [Green Version]
- Di Stasio, L.; Albera, A.; Pauciullo, A.; Cesarani, A.; Macciotta, N.P.; Gaspa, G. Genetics of Arthrogryposis and Macroglossia in Piemontese Cattle Breed. Animals 2020, 10, 1732. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Lu, S.; Cui, K.; Shafique, L.; Rehman, S.U.; Luo, C.; Wang, Z.; Ruan, J.; Qian, Q.; Liu, Q. Fatty acid biosynthesis and transcriptional regulation of Stearoyl-CoA Desaturase 1 (SCD1) in buffalo milk. BMC Genet. 2020, 21, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meuwissen, T.; Hayes, B.; Goddard, M. Accelerating Improvement of Livestock with Genomic Selection. Annu. Rev. Anim. Biosci. 2013, 1, 221–237. [Google Scholar] [CrossRef] [PubMed]
- Moaeen-ud-Din, M.; Bilal, G. Genomic selection of Nili-Ravi buffalo: A choice for buffalo breeders. Buffalo Bull. 2016, 35, 595–605. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Swamp Buffalo | River Buffalo | Mediterranean Buffalo | ||
|---|---|---|---|---|
| Genome | Total genome size (Mb) | 2631 | 2646 | 2654 |
| Chromosome number | 48 | 50 | 50 | |
| Scaffold number | 24 + 1510 | 25 + 2279 | 25 + 506 | |
| Scaffold N50 (Mb) | 117.3 | 116.1 | 117.2 | |
| Total contig size (Mb) | 2609 | 2626 | 2622 | |
| Contig N50 (Mb) | 8.8 | 3.1 | 18.8 | |
| Annotation | Total genes | 19,272 | 20,202 | 24,014 |
| Average CDS length (bp) | 1764.5 | 1662.2 | - | |
| BUSCO assessment | 96.80% | 96.00% | 93.6% | |
| Repeat content | 47.26% | 47.31 | 45.89% | |
| Population | Sample number | 132 | 98 | - |
| Number of SNPs | 18,737,564 | 23,722,820 | - | |
| Genetic diversity θ (4 Nμ) | 0.001805 | 0.002743 | - | |
| Population differentiation (Fst) | 0.27 | - |
| No. | Trait | Candidate Genes |
|---|---|---|
| 1 | Milk yield | STAT1, STAT5A, LEP, MC4R, OXT, INSIG2, LALBA, BTN1A1, PRL, SCD, and SREBF1 |
| 2 | Milk fat yield | GHRL and A2M |
| 3 | Milk fat (%) | STAT1, TG, A2M, DGAT1, GHRL, LEP, MC4R, PRL, SCD, and SREBF1 |
| 4 | Milk protein (%) | CSN1S1, DGAT1, GHRL, ADRA1A, A2M MTNR1A, PRL and SPP1, INSIG2, and MC4R |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rehman, S.u.; Hassan, F.-u.; Luo, X.; Li, Z.; Liu, Q. Whole-Genome Sequencing and Characterization of Buffalo Genetic Resources: Recent Advances and Future Challenges. Animals 2021, 11, 904. https://doi.org/10.3390/ani11030904
Rehman Su, Hassan F-u, Luo X, Li Z, Liu Q. Whole-Genome Sequencing and Characterization of Buffalo Genetic Resources: Recent Advances and Future Challenges. Animals. 2021; 11(3):904. https://doi.org/10.3390/ani11030904
Chicago/Turabian StyleRehman, Saif ur, Faiz-ul Hassan, Xier Luo, Zhipeng Li, and Qingyou Liu. 2021. "Whole-Genome Sequencing and Characterization of Buffalo Genetic Resources: Recent Advances and Future Challenges" Animals 11, no. 3: 904. https://doi.org/10.3390/ani11030904
APA StyleRehman, S. u., Hassan, F. -u., Luo, X., Li, Z., & Liu, Q. (2021). Whole-Genome Sequencing and Characterization of Buffalo Genetic Resources: Recent Advances and Future Challenges. Animals, 11(3), 904. https://doi.org/10.3390/ani11030904