
A DNA Regulatory Element Haplotype at Zinc Finger Genes Is Associated with Host Resilience to Small Ruminant Lentivirus in Two Sheep Populations

 , and
, and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. DNA Extractions and Genotyping
2.3. Small Ruminant Lentivirus Phenotypes
2.4. Statistical Analysis
2.5. Linkage Disequilibrium and Haplotype Analyses
2.6. Transcription Factor Binding Analysis
3. Results
3.1. ZNF Gene Region in Detail
3.2. Population One Association Analysis
3.3. Population Two Association Analysis
3.4. Significant Variants in Multiple Populations Were in Two Regulatory Elements
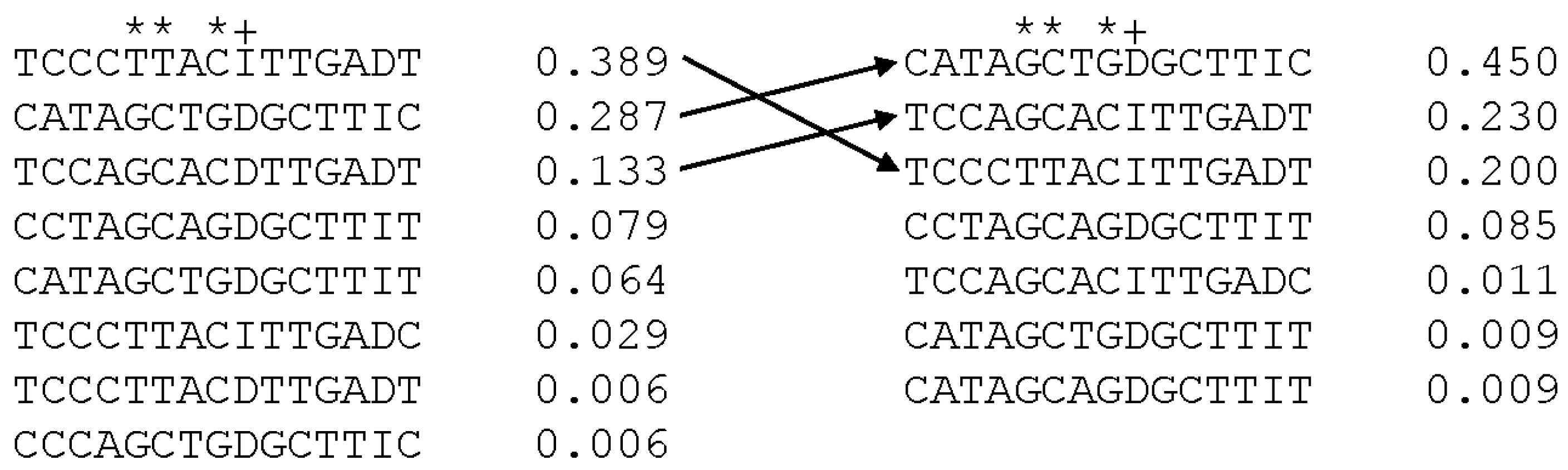
3.5. Haplotypes and Phenotypic Association with Common Haplotypes
3.6. TRANSFAC Predictions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Pablo-Maiso, L.; Doménech, A.; Echeverría, I.; Gómez-Arrebola, C.; De Andrés, D.; Rosati, S.; Gómez-Lucia, E.; Reina, R. Prospects in Innate Immune Responses as Potential Control Strategies against Non-Primate Lentiviruses. Viruses 2018, 10, 435. [Google Scholar] [CrossRef] [Green Version]
- Jáuregui, P.; Crespo, H.; Glaria, I.; Luján, L.; Contreras, A.; Rosati, S.; de Andrés, D.; Amorena, B.; Towers, G.J.; Reina, R. Ovine TRIM5α can restrict visna/maedi virus. J. Virol. 2012, 86, 9504–9509. [Google Scholar] [CrossRef] [Green Version]
- Minguijón, E.; Reina, R.; Pérez, M.; Polledo, L.; Villoria, M.; Ramírez, H.; Leginagoikoa, I.; Badiola, J.J.; García-Marín, J.F.; de Andrés, D.; et al. Small ruminant lentivirus infections and diseases. Vet. Microbiol. 2015, 181, 75–89. [Google Scholar] [CrossRef]
- Blacklaws, B.A. Small ruminant lentiviruses: Immunopathogenesis of visna-maedi and caprine arthritis and encephalitis virus. Comp. Immunol. Microbiol. Infect. Dis. 2012, 35, 259–269. [Google Scholar] [CrossRef]
- White, S.N.; Knowles, D.P. Expanding possibilities for intervention against small ruminant lentiviruses through genetic marker-assisted selective breeding. Viruses 2013, 5, 1466–1499. [Google Scholar] [CrossRef]
- Arsenault, J.; Dubreuil, P.; Girard, C.; Simard, C.; Bélanger, D. Maedi-visna impact on productivity in Quebec sheep flocks (Canada). Prev. Vet. Med. 2003, 59, 125–137. [Google Scholar] [CrossRef]
- Keen, J.E.; Hungerford, L.L.; Littledike, E.T.; Wittum, T.E.; Kwang, J. Effect of ewe ovine lentivirus infection on ewe and lamb productivity. Prev. Vet. Med. 1997, 30, 155–169. [Google Scholar] [CrossRef]
- Dohoo, I.R.; Heaney, D.P.; Stevenson, R.G.; Samagh, B.S.; Rhodes, C.S. The effects of maedi-visna virus infection on productivity in ewes. Prev. Vet. Med. 1987, 4, 471–484. [Google Scholar] [CrossRef]
- Peterhans, E.; Greenland, T.; Badiola, J.; Harkiss, G.; Bertoni, G.; Amorena, B.; Eliaszewicz, M.; Juste, R.A.; Krassnig, R.; Lafont, J.-P.; et al. Routes of transmission and consequences of small ruminant lentiviruses (SRLVs) infection and eradication schemes. Vet. Res. 2004, 35, 257–274. [Google Scholar] [CrossRef] [Green Version]
- Lago, N.; López, C.; Panadero, R.; Cienfuegos, S.; Pato, J.; Prieto, A.; Díaz, P.; Mourazos, N.; Fernández, G. Seroprevalence and risk factors associated with Visna/Maedi virus in semi-intensive lamb-producing flocks in northwestern Spain. Prev. Vet. Med. 2012, 103, 163–169. [Google Scholar] [CrossRef]
- Redden, R.R.; Schmidt, R.J.; Kirsch, J.D. Ovine Progressive Oneumonia Virus Infection Rate and Incidence of Genetic Susceptibility Diplotype in North Dakota Sheep Flocks. Available online: https://www.ag.ndsu.edu/HettingerREC/sheep/individual-articles-from-2014-sheep-research-report/ovine-progressive-pneumonia-virus-infection-rate-and-incidence-of-genetic-susceptibility-diplotype-in-north-dakota-sheep-flocks (accessed on 8 April 2021).
- APHIS. Info Sheet: Ovine Progressive Pneumonia Awareness, Management, and Seroprevalence. Available online: http://www.aphis.usda.gov/animal_health/nahms/sheep/downloads/sheep01/Sheep01_is_OPP.pdf (accessed on 8 April 2021).
- Cutlip, R.C.; Lehmkuhl, H.D.; Sacks, J.M.; Weaver, A.L. Seroprevalence of ovine progressive pneumonia virus in sheep in the United States as assessed by analyses of voluntarily submitted samples. Am. J. Vet. Res. 1992, 53, 976–979. [Google Scholar] [PubMed]
- Herrmann-Hoesing, L.M.; Lehmkuhl, H.D.; Cutlip, R.C. Minimum intravenous infectious dose of ovine progressive pneumonia virus (OPPV). Res. Vet. Sci. 2009, 87, 329–331. [Google Scholar] [CrossRef] [PubMed]
- Herrmann-Hoesing, L.M.; Noh, S.M.; Snekvik, K.R.; White, S.N.; Schneider, D.A.; Truscott, T.; Knowles, D.P. Ovine progressive pneumonia virus capsid antigen as found in CD163- and CD172a-positive alveolar macrophages of persistently infected sheep. Vet. Pathol. 2010, 47, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Herrmann-Hoesing, L.M.; White, S.N.; Lewis, G.S.; Mousel, M.R.; Knowles, D.P. Development and validation of an ovine progressive pneumonia virus quantitative PCR. Clin. Vaccine Immunol. 2007, 14, 1274–1278. [Google Scholar] [CrossRef] [Green Version]
- Kalogianni, A.I.; Bossis, I.; Ekateriniadou, L.V.; Gelasakis, A.I. Etiology, Epizootiology and Control of Maedi-Visna in Dairy Sheep: A Review. Animals 2020, 10, 616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, S.N.; Mousel, M.R.; Herrmann-Hoesing, L.M.; Reynolds, J.O.; Leymaster, K.A.; Neibergs, H.L.; Lewis, G.S.; Knowles, D.P. Genome-Wide Association Identifies Multiple Genomic Regions Associated with Susceptibility to and Control of Ovine Lentivirus. PLoS ONE 2012, 7, e47829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrmann-Hoesing, L.M.; Noh, S.M.; White, S.N.; Snekvik, K.R.; Truscott, T.; Knowles, D.P. Peripheral ovine progressive pneumonia provirus levels correlate with and predict histological tissue lesion severity in naturally infected sheep. Clin. Vaccine Immunol. 2009, 16, 551–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woldemeskel, B.A.; Kwaa, A.K.; Blankson, J.N. Viral reservoirs in elite controllers of HIV-1 infection: Implications for HIV cure strategies. EBioMedicine 2020, 62, 103118. [Google Scholar] [CrossRef] [PubMed]
- Re, M.C.; Vitone, F.; Biagetti, C.; Schiavone, P.; Alessandrini, F.; Bon, I.; de Crignis, E.; Gibellini, D. HIV-1 DNA proviral load in treated and untreated HIV-1 sero-positive patients. Clin. Microbiol. Infect. 2010, 16, 640–646. [Google Scholar] [CrossRef] [Green Version]
- Lathey, J.L.; Hughes, M.D.; Fiscus, S.A.; Pi, T.; Jackson, J.B.; Rasheed, S.; Elbeik, T.; Reichman, R.; Japour, A.; D’Aquila, R.T.; et al. Variability and Prognostic Values of Virologic and CD4 Cell Measures in Human Immunodeficiency Virus Type 1-Infected Patients with 200–500 CD4 Cells/mm3 (ACTG 175). J. Infect. Dis. 1998, 177, 617–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tierney, C.; Lathey, J.L.; Christopherson, C.; Bettendorf, D.M.; D’Aquila, R.T.; Hammer, S.M.; Katzenstein, D.A. Prognostic Value of Baseline Human Immunodeficiency Virus Type 1 DNA Measurement for Disease Progression in Patients Receiving Nucleoside Therapy. J. Infect. Dis. 2003, 187, 144–148. [Google Scholar] [CrossRef]
- Pereyra, F.; Addo, M.M.; Kaufmann, D.E.; Liu, Y.; Miura, T.; Rathod, A.; Baker, B.; Trocha, A.; Rosenberg, R.; Mackey, E.; et al. Genetic and Immunologic Heterogeneity among Persons Who Control HIV Infection in the Absence of Therapy. J. Infect. Dis. 2008, 197, 563–571. [Google Scholar] [CrossRef]
- Rauw, W. Immune response from a resource allocation perspective. Front. Genet. 2012, 3, 267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stear, M.J.; Bishop, S.C.; Mallard, B.A.; Raadsma, H. The sustainability, feasibility and desirability of breeding livestock for disease resistance. Res. Vet. Sci. 2001, 71, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Salavati, M.; Caulton, A.; Clark, R.; Gazova, I.; Smith, T.P.L.; Worley, K.C.; Cockett, N.E.; Archibald, A.L.; Clarke, S.M.; Murdoch, B.M.; et al. Global analysis of transcription start sites in the new ovine reference genome (Oar rambouillet v1.0). Front. Genet. 2020. [Google Scholar] [CrossRef] [PubMed]
- Clark, E.L.; Bush, S.J.; McCulloch, M.E.B.; Farquhar, I.L.; Young, R.; Lefevre, L.; Pridans, C.; Tsang, H.G.; Wu, C.; Afrasiabi, C.; et al. A high resolution atlas of gene expression in the domestic sheep (Ovis aries). PLoS Genet. 2017, 13, e1006997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Xie, M.; Chen, W.; Talbot, R.; Maddox, J.F.; Faraut, T.; Wu, C.; Muzny, D.M.; Li, Y.; Zhang, W.; et al. The sheep genome illuminates biology of the rumen and lipid metabolism. Science 2014, 344, 1168–1173. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Chen, G.; Lv, F.; Wang, X.; Ji, X.; Xu, Y.; Sun, J.; Wu, L.; Zheng, Y.T.; Gao, G. Zinc-finger antiviral protein inhibits HIV-1 infection by selectively targeting multiply spliced viral mRNAs for degradation. Proc. Natl. Acad. Sci. USA 2011, 108, 15834–15839. [Google Scholar] [CrossRef] [Green Version]
- Lukic, S.; Nicolas, J.-C.; Levine, A.J. The diversity of zinc-finger genes on human chromosome 19 provides an evolutionary mechanism for defense against inherited endogenous retroviruses. Cell Death Differ. 2014, 21, 381–387. [Google Scholar] [CrossRef] [Green Version]
- Thomas, J.H.; Schneider, S. Coevolution of retroelements and tandem zinc finger genes. Genome Res. 2011, 21, 1800–1812. [Google Scholar] [CrossRef] [Green Version]
- White, S.N.; Mousel, M.R.; Reynolds, J.O.; Herrmann-Hoesing, L.M.; Knowles, D.P. Deletion variant near ZNF389 is associated with control of ovine lentivirus in multiple sheep flocks. Anim. Genet. 2014, 45, 297–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, S.N.; Mousel, M.R.; Gonzalez, M.V.; Highland, M.A.; Herrmann-Hoesing, L.M.; Taylor, J.B.; Knowles, D.P. Association analysis of variant near ZNF389 with ewe cumulative production in three sheep breeds. Anim. Genet. 2014, 45, 613–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huntley, S.; Baggott, D.M.; Hamilton, A.T.; Tran-Gyamfi, M.; Yang, S.; Kim, J.; Gordon, L.; Branscomb, E.; Stubbs, L. A comprehensive catalog of human KRAB-associated zinc finger genes: Insights into the evolutionary history of a large family of transcriptional repressors. Genome Res. 2006, 16, 669–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedotova, A.A.; Bonchuk, A.N.; Mogila, V.A.; Georgiev, P.G. C2H2 Zinc Finger Proteins: The Largest but Poorly Explored Family of Higher Eukaryotic Transcription Factors. Acta Nat. 2017, 9, 47–58. [Google Scholar] [CrossRef] [Green Version]
- Consortium, B.G.S.A.; Elsik, C.G.; Tellam, R.L.; Worley, K.C.; Gibbs, R.A.; Muzny, D.M.; Weinstock, G.M.; Adelson, D.L.; Eichler, E.E.; Elnitski, L.; et al. The genome sequence of taurine cattle: A window to ruminant biology and evolution. Science 2009, 324, 522–528. [Google Scholar] [CrossRef] [Green Version]
- Groenen, M.A.M.; Archibald, A.L.; Uenishi, H.; Tuggle, C.K.; Takeuchi, Y.; Rothschild, M.F.; Rogel-Gaillard, C.; Park, C.; Milan, D.; Megens, H.-J.; et al. Analyses of pig genomes provide insight into porcine demography and evolution. Nature 2012, 491, 393–398. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Consortium, I.H.G.S. Finishing the euchromatic sequence of the human genome. Nature 2004, 431, 931–945. [Google Scholar] [CrossRef]
- Massa, A.T.; Mousel, M.R.; Herndon, M.K.; Herndon, D.R.; Murdoch, B.M.; White, S.N. Genome-Wide Histone Modifications and CTCF Enrichment Predict Gene Expression in Sheep Macrophages. Front. Genet. 2021, 11, 1658. [Google Scholar] [CrossRef]
- Won, K.-J.; Chepelev, I.; Ren, B.; Wang, W. Prediction of regulatory elements in mammalian genomes using chromatin signatures. BMC Bioinform. 2008, 9, 547. [Google Scholar] [CrossRef] [Green Version]
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef] [Green Version]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zang, C.; Rosenfeld, J.A.; Schones, D.E.; Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Peng, W.; Zhang, M.Q.; et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008, 40, 897–903. [Google Scholar] [CrossRef] [Green Version]
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.; Brody, J.; et al. Systematic Localization of Common Disease-Associated Variation in Regulatory DNA. Science 2012, 337, 1190–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hindorff, L.A.; Sethupathy, P.; Junkins, H.A.; Ramos, E.M.; Mehta, J.P.; Collins, F.S.; Manolio, T.A. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. USA 2009, 106, 9362–9367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, R.; Berg, I.v.d.; MacLeod, I.M.; Hayes, B.J.; Prowse-Wilkins, C.P.; Wang, M.; Bolormaa, S.; Liu, Z.; Rochfort, S.J.; Reich, C.M.; et al. Quantifying the contribution of sequence variants with regulatory and evolutionary significance to 34 bovine complex traits. Proc. Natl. Acad. Sci. USA 2019, 116, 19398–19408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics 2005, 21, 263–265. [Google Scholar] [CrossRef] [Green Version]
- Stephens, M.; Smith, N.J.; Donnelly, P. A New Statistical Method for Haplotype Reconstruction from Population Data. Am. J. Hum. Genet. 2001, 68, 978–989. [Google Scholar] [CrossRef] [Green Version]
- Stephens, M.; Donnelly, P. A Comparison of Bayesian Methods for Haplotype Reconstruction from Population Genotype Data. Am. J. Hum. Genet. 2003, 73, 1162–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kel, A.E.; Gössling, E.; Reuter, I.; Cheremushkin, E.; Kel-Margoulis, O.V.; Wingender, E. MATCH: A tool for searching transcription factor binding sites in DNA sequences. Nucleic Acids Res. 2003, 31, 3576–3579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, J.; Gifford, R.J.; Sato, K. Retroviruses drive the rapid evolution of mammalian APOBEC3 genes. Proc. Natl. Acad. Sci. USA 2020, 117, 610–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cutlip, R.C.; Lehmkuhl, H.D.; Schmerr, M.J.F.; Brogden, K.A. Ovine progressive pneumonia (maedi-visna) in sheep. Vet. Microbiol. 1988, 17, 237–250. [Google Scholar] [CrossRef]
- Najafabadi, H.S.; Mnaimneh, S.; Schmitges, F.W.; Garton, M.; Lam, K.N.; Yang, A.; Albu, M.; Weirauch, M.T.; Radovani, E.; Kim, P.M.; et al. C2H2 zinc finger proteins greatly expand the human regulatory lexicon. Nat. Biotechnol. 2015, 33, 555–562. [Google Scholar] [CrossRef]
- Wang, G.; Zheng, C. Zinc finger proteins in the host-virus interplay: Multifaceted functions based on their nucleic acid-binding property. FEMS Microbiol. Rev. 2020. [Google Scholar] [CrossRef]
- Wolf, G.; Greenberg, D.; Macfarlan, T.S. Spotting the enemy within: Targeted silencing of foreign DNA in mammalian genomes by the Krüppel-associated box zinc finger protein family. Mob. DNA 2015, 6, 17. [Google Scholar] [CrossRef] [Green Version]
- Fu, M.; Blackshear, P.J. RNA-binding proteins in immune regulation: A focus on CCCH zinc finger proteins. Nat. Rev. Immunol. 2017, 17, 130–143. [Google Scholar] [CrossRef] [Green Version]
- Crespo, H.; Bertolotti, L.; Proffiti, M.; Cascio, P.; Cerruti, F.; Acutis, P.L.; de Andrés, D.; Reina, R.; Rosati, S. Low proviral small ruminant lentivirus load as biomarker of natural restriction in goats. Vet. Microbiol. 2016, 192, 152–162. [Google Scholar] [CrossRef]
- Heaton, M.P.; Clawson, M.L.; Chitko-Mckown, C.G.; Leymaster, K.A.; Smith, T.P.; Harhay, G.P.; White, S.N.; Herrmann-Hoesing, L.M.; Mousel, M.R.; Lewis, G.S.; et al. Reduced lentivirus susceptibility in sheep with TMEM154 mutations. PLoS Genet. 2012, 8, e1002467. [Google Scholar] [CrossRef] [Green Version]
- Bowles, D.; Carson, A.; Isaac, P. Genetic distinctiveness of the Herdwick sheep breed and two other locally adapted hill breeds of the UK. PLoS ONE 2014, 9, e87823. [Google Scholar] [CrossRef]
- Alshanbari, F.A.; Mousel, M.R.; Reynolds, J.O.; Herrmann-Hoesing, L.M.; Highland, M.A.; Lewis, G.S.; White, S.N. Mutations in Ovis aries TMEM154 are associated with lower small ruminant lentivirus proviral concentration in one sheep flock. Anim. Genet. 2014, 45, 565–571. [Google Scholar] [CrossRef]
- Ramírez, H.; Echeverría, I.; Benito, A.A.; Glaria, I.; Benavides, J.; Pérez, V.; de Andrés, D.; Reina, R. Accurate Diagnosis of Small Ruminant Lentivirus Infection Is Needed for Selection of Resistant Sheep through TMEM154 E35K Genotyping. Pathogens 2021, 10, 83. [Google Scholar] [CrossRef] [PubMed]
- Naval-Sanchez, M.; Nguyen, Q.; McWilliam, S.; Porto-Neto, L.R.; Tellam, R.; Vuocolo, T.; Reverter, A.; Perez-Enciso, M.; Brauning, R.; Clarke, S.; et al. Sheep genome functional annotation reveals proximal regulatory elements contributed to the evolution of modern breeds. Nat. Commun. 2018, 9, 859. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Liu, S.; Liu, M.; Kang, X.; Lin, S.; Li, B.; Connor, E.E.; Baldwin, R.L.; Tenesa, A.; Ma, L.; et al. Functional annotation of the cattle genome through systematic discovery and characterization of chromatin states and butyrate-induced variations. BMC Biol. 2019, 17, 68. [Google Scholar] [CrossRef] [PubMed]
- Xiang, R.; MacLeod, I.M.; Daetwyler, H.D.; de Jong, G.; O’Connor, E.; Schrooten, C.; Chamberlain, A.J.; Goddard, M.E. Genome-wide fine-mapping identifies pleiotropic and functional variants that predict many traits across global cattle populations. Nat. Commun. 2021, 12, 860. [Google Scholar] [CrossRef] [PubMed]
- Schuster, F.; Aldag, P.; Frenzel, A.; Hadeler, K.-G.; Lucas-Hahn, A.; Niemann, H.; Petersen, B. CRISPR/Cas12a mediated knock-in of the Polled Celtic variant to produce a polled genotype in dairy cattle. Sci. Rep. 2020, 10, 13570. [Google Scholar] [CrossRef]
- Albert, F.W.; Kruglyak, L. The role of regulatory variation in complex traits and disease. Nat. Rev. Genet. 2015, 16, 197. [Google Scholar] [CrossRef] [PubMed]
- The ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. Available online: https://www.nature.com/articles/nature11247#supplementary-information (accessed on 20 February 2021). [CrossRef] [PubMed]
- Fishilevich, S.; Nudel, R.; Rappaport, N.; Hadar, R.; Plaschkes, I.; Iny Stein, T.; Rosen, N.; Kohn, A.; Twik, M.; Safran, M.; et al. GeneHancer: Genome-wide integration of enhancers and target genes in GeneCards. Database 2017, 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, D.E.; Patel, Z.H.; Lu, X.; Lynch, A.T.; Weirauch, M.T.; Kottyan, L.C. Screening for Functional Non-coding Genetic Variants Using Electrophoretic Mobility Shift Assay (EMSA) and DNA-affinity Precipitation Assay (DAPA). JoVE 2016, e54093. [Google Scholar] [CrossRef] [Green Version]
- Mathelier, A.; Shi, W.; Wasserman, W.W. Identification of altered cis-regulatory elements in human disease. Trends Genet. 2015, 31, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, P.L.; Sheline, C.T.; Cannon, K.; Voz, M.L.; Pazin, M.J.; Kadonaga, J.T.; Jones, K.A. Activation of the HIV-1 enhancer by the LEF-1 HMG protein on nucleosome-assembled DNA in vitro. Genes Dev. 1995, 9, 2090–2104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, F.; Kawai, E.; Martinez, N.E.; Omura, S.; Park, A.-M.; Takahashi, S.; Yoh, K.; Tsunoda, I. T-bet, but not Gata3, overexpression is detrimental in a neurotropic viral infection. Sci. Rep. 2017, 7, 10496. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Marker | Population 1 n = 164 Minor Allele Freq | Population 2 n = 379 Minor Allele Freq | ||
|---|---|---|---|---|
| rs193645606 | C | 0.44 | T | 0.45 |
| rs418789060 | A | 0.35 | A | 0.47 |
| rs161334222 | T | 0.44 | C | 0.44 |
| rs398476053 | C | 0.42 | C | 0.20 |
| rs414155747&rs425583788 | TT | 0.43 | TT | 0.20 |
| rs427575002 | T | 0.36 | T | 0.47 |
| rs407355422 | G | 0.44 | C | 0.44 |
| rs397514112 * | D | 0.44 | I | 0.44 |
| rs599110985 | G | 0.44 | T | 0.44 |
| rs411076283 | C | 0.44 | T | 0.44 |
| rs407841455 | T | 0.44 | G | 0.44 |
| rs161334287 | T | 0.44 | A | 0.44 |
| rs598937573 | I | 0.44 | D | 0.44 |
| rs406431156 | C | 0.33 | C | 0.47 |
| Marker | Resilient Genotype | Susceptible Genotype | Genotypic Log10 Conc. Diff. | p-Value | LD * Pop. 1 (r2) | LD * Pop. 2 (r2) |
|---|---|---|---|---|---|---|
| rs193645606 | T/T | C/C | 0.857 | 8.95 × 10−5 | 0.950 | 0.989 |
| rs418789060 | C/C | A/A | 0.537 | 4.06 × 10−2 | 0.664 | 0.705 |
| rs161334222 | C/C | T/T | 0.830 | 2.57 × 10−4 | 0.926 | 0.989 |
| rs398476053 | C/C | A/A | 0.434 | 1.64 × 10−2 | 0.537 | 0.312 |
| rs414155747&rs425583788 | TT/TT | GC/GC | 0.445 | 1.46 × 10−2 | 0.556 | 0.313 |
| rs427575002 | A/A | T/T | 0.572 | 2.46 × 10−2 | 0.690 | 0.678 |
| rs407355422 | C/C | G/G | 0.859 | 8.70 × 10−5 | 0.950 | 0.989 |
| rs397514112 * | I/I 1 | D/D | 0.803 | 6.00 × 10−4 | - | - |
| rs599110985 | T/T | G/G | 0.857 | 8.39 × 10−5 | 0.950 | 0.989 |
| rs411076283 | T/T | C/C | 0.856 | 9.20 × 10−5 | 0.950 | 0.989 |
| rs407841455 | G/G | T/T | 0.857 | 8.39 × 10−5 | 0.950 | 0.989 |
| rs161334287 | A/A | T/T | 0.857 | 8.72 × 10−5 | 0.950 | 0.989 |
| rs598937573 | D/D 2 | I/I | 0.838 | 1.62 × 10−4 | 0.950 | 0.989 |
| rs406431156 | T/T | C/C | 0.739 | 2.50 × 10−2 | 0.389 | 0.604 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Massa, A.T.; Mousel, M.R.; Durfee, C.J.; Herndon, M.K.; Hemmerling, K.M.; Taylor, J.B.; Neibergs, H.L.; White, S.N. A DNA Regulatory Element Haplotype at Zinc Finger Genes Is Associated with Host Resilience to Small Ruminant Lentivirus in Two Sheep Populations. Animals 2021, 11, 1907. https://doi.org/10.3390/ani11071907
Massa AT, Mousel MR, Durfee CJ, Herndon MK, Hemmerling KM, Taylor JB, Neibergs HL, White SN. A DNA Regulatory Element Haplotype at Zinc Finger Genes Is Associated with Host Resilience to Small Ruminant Lentivirus in Two Sheep Populations. Animals. 2021; 11(7):1907. https://doi.org/10.3390/ani11071907
Chicago/Turabian StyleMassa, Alisha T., Michelle R. Mousel, Codie J. Durfee, Maria K. Herndon, Kaneesha M. Hemmerling, J. Bret Taylor, Holly L. Neibergs, and Stephen N. White. 2021. "A DNA Regulatory Element Haplotype at Zinc Finger Genes Is Associated with Host Resilience to Small Ruminant Lentivirus in Two Sheep Populations" Animals 11, no. 7: 1907. https://doi.org/10.3390/ani11071907
APA StyleMassa, A. T., Mousel, M. R., Durfee, C. J., Herndon, M. K., Hemmerling, K. M., Taylor, J. B., Neibergs, H. L., & White, S. N. (2021). A DNA Regulatory Element Haplotype at Zinc Finger Genes Is Associated with Host Resilience to Small Ruminant Lentivirus in Two Sheep Populations. Animals, 11(7), 1907. https://doi.org/10.3390/ani11071907