
Genome-Wide Selective Analysis of Boer Goat to Investigate the Dynamic Heredity Evolution under Different Stages


 , ,
, ,  ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals, Genome Sequencing, and Data Acquisition
2.2. Read Filtering, Alignment, and Variant Calling
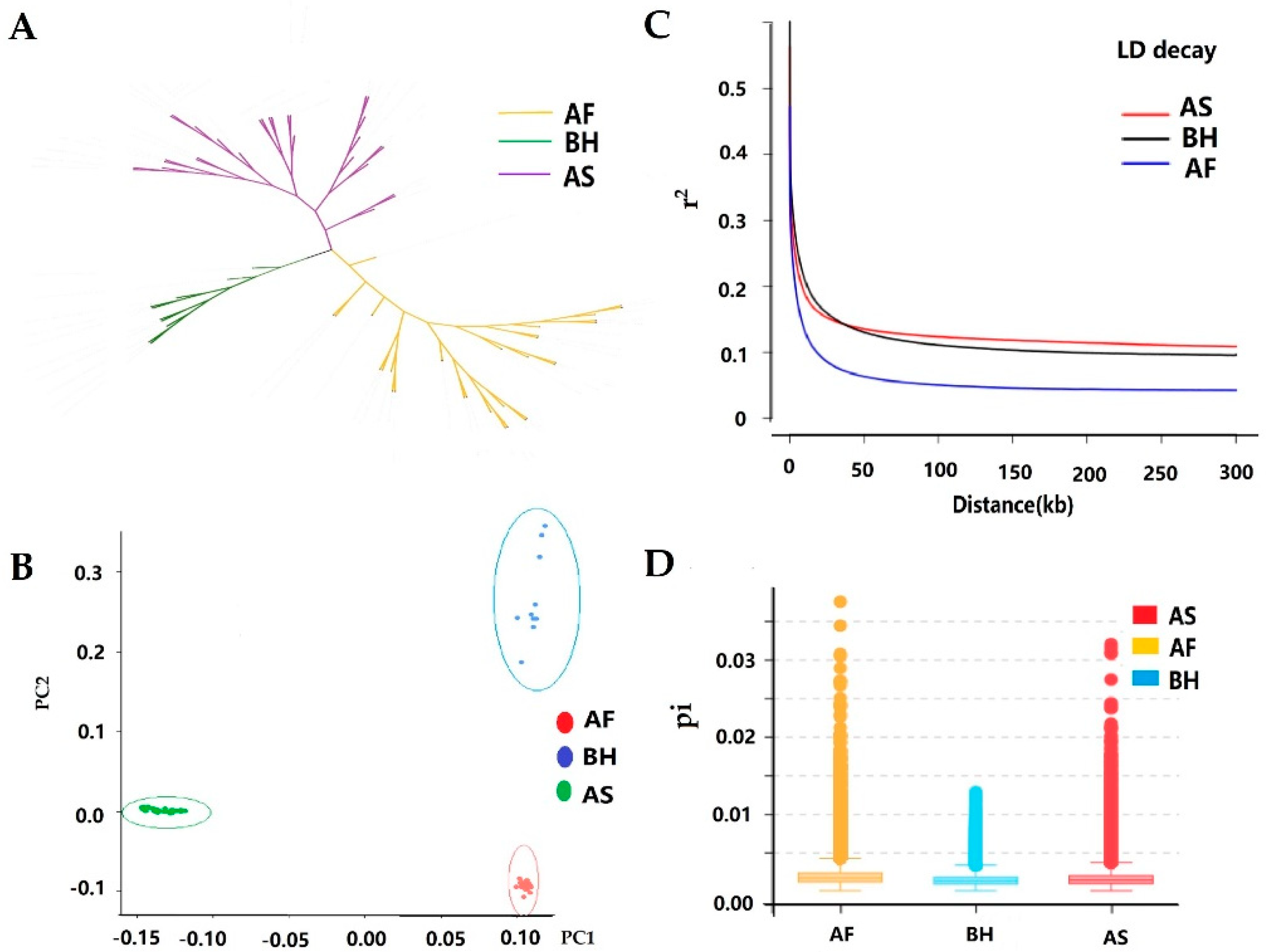
2.3. Diversity, Phylogenetic, and Population Genetic Analyses
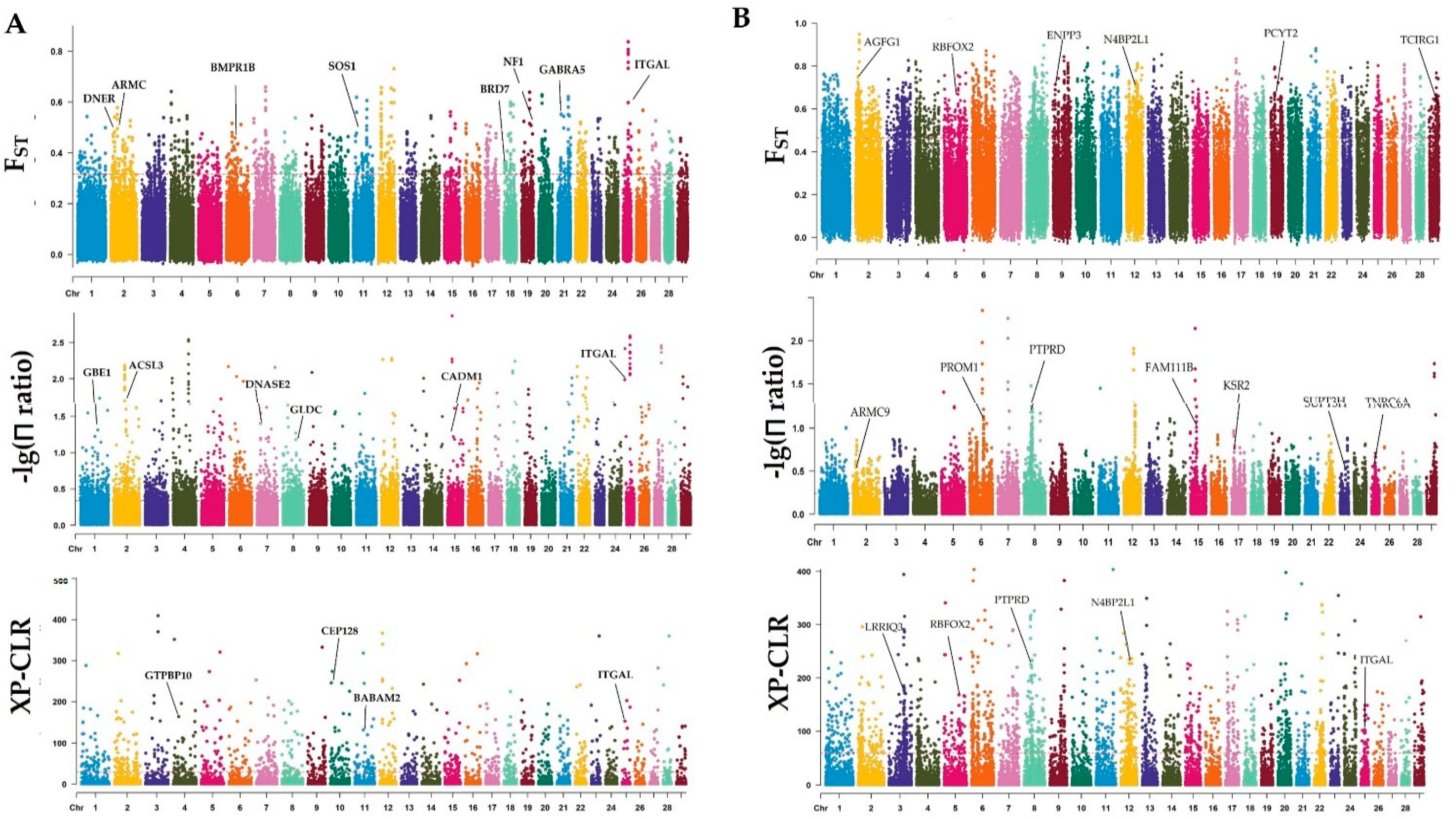
2.4. Genome-Wide Selective Sweep Analysis and Gene Annotation
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Bovo, S.; Ribani, A.; Muñoz, M.; Alves, E.; Araujo, J.P.; Bozzi, R.; Čandek-Potokar, M.; Charneca, R.; Di Palma, F.; Etherington, G.; et al. Whole-Genome Sequencing of European Autochthonous and Commercial Pig Breeds Allows the Detection of Signatures of Selection for Adaptation of Genetic Resources to Different Breeding and Production Systems. Genet. Sel. Evol. 2020, 52, 33. [Google Scholar] [CrossRef] [PubMed]
- Lan, D.; Xiong, X.; Mipam, T.-D.; Fu, C.; Li, Q.; Ai, Y.; Hou, D.; Chai, Z.; Zhong, J.; Li, J. Genetic Diversity, Molecular Phylogeny, and Selection Evidence of Jinchuan Yak Revealed by Whole-Genome Resequencing. Genes Genomes Genetics 2018, 8, 945–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fawcett, J.A.; Sato, F.; Sakamoto, T.; Iwasaki, W.M.; Tozaki, T.; Innan, H. Genome-wide SNP analysis of Japanese Thoroughbred racehorses. PLoS ONE 2019, 14, e0218407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Yang, J.; Shen, M.; Xie, X.-L.; Liu, G.-J.; Xu, Y.-X.; Lv, F.-H.; Yang, H.; Yang, Y.-L.; Liu, C.-B.; et al. Whole-genome resequencing of wild and domestic sheep identifies genes associated with morphological and agronomic traits. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, W.; Yang, B.; Zhang, Z.; Ai, H.; Ren, J.; Huang, L. Signatures of Selection and Interspecies Introgression in the Genome of Chinese Domestic Pigs. Genome Biol. Evol. 2017, 9, 2592–2603. [Google Scholar] [CrossRef] [Green Version]
- Axelsson, E.; Ratnakumar, A.; Arendt, M.L.; Maqbool, K.; Webster, M.T.; Perloski, M.; Liberg, O.; Arnemo, J.M.; Hedhammar, Å.; Lindblad-Toh, K. The genomic signature of dog domestication reveals adaptation to a starch-rich diet. Nature 2013, 495, 360–364. [Google Scholar] [CrossRef]
- Chebii, V.J.; Oyola, S.O.; Kotze, A.; Entfellner, J.-B.D.; Mutuku, J.M.; Agaba, M. Genome-Wide Analysis of Nubian Ibex Reveals Candidate Positively Selected Genes That Contribute to Its Adaptation to the Desert Environment. Animals 2020, 10, 2181. [Google Scholar] [CrossRef]
- Kim, J.-Y.; Jeong, S.; Kim, K.H.; Lim, W.-J.; Lee, H.-Y.; Kim, N. Discovery of Genomic Characteristics and Selection Signatures in Korean Indigenous Goats Through Comparison of 10 Goat Breeds. Front. Genet. 2019, 10, 699. [Google Scholar] [CrossRef] [Green Version]
- Guan, D.; Martínez, A.; Luigi-Sierra, M.G.; Delgado, J.V.; Landi, V.; Castelló, A.; Fernández Álvarez, J.; Such, X.; Jordana, J.; Amills, M. Detecting the Footprint of Selection on the Genomes of Murciano-Granadina Goats. Anim. Genet. 2021, 52, 683–693. [Google Scholar] [CrossRef]
- Bertolini, F.; Servin, B.; Talenti, A.; Rochat, E.; Kim, E.S.; Oget, C.; Palhière, I.; Crisà, A.; Catillo, G.; Steri, R.M.; et al. Signatures of Selection and Environmental Adaptation across the Goat Genome Post-Domestication. Genet Sel. Evol. 2018, 50, 57. [Google Scholar] [CrossRef]
- Bertolini, F.; Cardoso, T.F.; Marras, G.; Nicolazzi, E.L.; Rothschild, M.F.; Amills, M. Genome-wide patterns of homozygosity provide clues about the population history and adaptation of goats. Genet. Sel. Evol. 2018, 50, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Z.; Wang, X.; Li, M.; Li, Y.; Yang, Z.; Wang, X.; Pan, X.; Gong, M.; Zhang, Y.; Guo, Y.; et al. The origin of domestication genes in goats. Sci. Adv. 2020, 6, eaaz5216. [Google Scholar] [CrossRef] [PubMed]
- Mdladla, K.; Dzomba, E.F.; Huson, H.; Muchadeyi, F.C. Population genomic structure and linkage disequilibrium analysis of South African goat breeds using genome-wide SNP data. Anim. Genet. 2016, 47, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Malan, S. The improved Boer goat. Small Rumin. Res. 2000, 36, 165–170. [Google Scholar] [CrossRef]
- Yang, B.; Yuan, Y.; Zhou, D.; Ma, Y.; Mahrous, K.; Wang, S.; He, Y.; Duan, X.; Zhang, W. Genome-wide selection signal analysis of Australian Boer goat reveals artificial selection imprinting on candidate genes related to muscle development. Anim. Genet. 2021, 52, 550–555. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Chen, H.; Patterson, N.; Reich, D. Population differentiation as a test for selective sweeps. Genome Res. 2010, 20, 393–402. [Google Scholar] [CrossRef] [Green Version]
- Gray, M.M.; Granka, J.M.; Bustamante, C.D.; Sutter, N.B.; Boyko, A.R.; Zhu, L.; Ostrander, E.A.; Wayne, R.K. Linkage Disequilibrium and Demographic History of Wild and Domestic Canids. Genetics 2009, 181, 1493–1505. [Google Scholar] [CrossRef] [Green Version]
- Rao, Y.S.; Liang, Y.; Na Xia, M.; Shen, X.; Du, Y.J.; Luo, C.G.; Nie, Q.H.; Zeng, H.; Zhang, X.Q. Extent of linkage disequilibrium in wild and domestic chicken populations. Hereditas 2008, 145, 251–257. [Google Scholar] [CrossRef]
- Chai, Z.-X.; Xin, J.-W.; Zhang, C.-F.; Zhang, Q.; Li, C.; Zhu, Y.; Cao, H.-W.; Wang, H.; Han, J.L.; Ji, Q.M.; et al. Whole-genome resequencing provides insights into the evolution and divergence of the native domestic yaks of the Qinghai–Tibet Plateau. BMC Evol. Biol. 2020, 20, 1–10. [Google Scholar] [CrossRef]
- Brand, T.S.; Van Der Merwe, D.A.; Hoffman, L.; Geldenhuys, G. The effect of dietary energy content on quality characteristics of Boer goat meat. Meat Sci. 2018, 139, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Gwaze, F.R.; Chimonyo, M.; Dzama, K. Communal goat production in Southern Africa: A review. Trop. Anim. Health Prod. 2008, 41, 1157–1168. [Google Scholar] [CrossRef] [PubMed]
- Cole, D.B.; Mills, D.B.; Erwin, D.H.; Sperling, E.A.; Porter, S.M.; Reinhard, C.T.; Planavsky, N.J. On the co-evolution of surface oxygen levels and animals. Geobiology 2020, 18, 260–281. [Google Scholar] [CrossRef] [PubMed]
- Ohgushi, T. Eco-evolutionary dynamics of plant–herbivore communities: Incorporating plant phenotypic plasticity. Curr. Opin. Insect Sci. 2016, 14, 40–45. [Google Scholar] [CrossRef]
- Wang, Z.; Ma, T.; Ma, J.; Han, J.; Ding, L.; Qiu, Q. Convergent evolution of SOCS4 between yak and Tibetan antelope in response to high-altitude stress. Gene 2015, 572, 298–302. [Google Scholar] [CrossRef]
- Korekane, H.; Park, J.Y.; Matsumoto, A.; Nakajima, K.; Takamatsu, S.; Ohtsubo, K.; Miyamoto, Y.; Hanashima, S.; Kanekiyo, K.; Kitazume, S.; et al. Identification of Ectonucleotide Pyrophosphatase/Phosphodiesterase 3 (Enpp3) as a Regulator of N-Acetylglucosaminyltransferase Gnt-Ix (Gnt-Vb). J. Biol. Chem. 2013, 288, 27912–27926. [Google Scholar] [CrossRef] [Green Version]
- Kuwajima, M.; Kojima, K.; Osaka, H.; Hamada, Y.; Jimbo, E.; Watanabe, M.; Aoki, S.; Sato-Shirai, I.; Ichimoto, K.; Fushimi, T.; et al. Valine metabolites analysis in ECHS1 deficiency. Mol. Genet. Metab. Rep. 2021, 29, 100809. [Google Scholar] [CrossRef]
- Garcia-Calvo, M.; Lisnock, J.M.; Bull, H.G.; Hawes, B.E.; Burnett, D.A.; Braun, M.P.; Crona, J.H.; Davis, H.R., Jr.; Dean, D.C.; Detmers, P.A.; et al. The target of ezetimibe is Niemann-Pick C1-Like 1 (NPC1L1). Proc. Natl. Acad. Sci. USA 2005, 102, 8132–8137. [Google Scholar] [CrossRef] [Green Version]
- Takada, T.; Yamanashi, Y.; Konishi, K.; Yamamoto, T.; Toyoda, Y.; Masuo, Y.; Yamamoto, H.; Suzuki, H. NPC1L1 is a key regulator of intestinal vitamin K absorption and a modulator of warfarin therapy. Sci. Transl. Med. 2015, 7, 275ra23. [Google Scholar] [CrossRef]
- Ji, C.; Li, Y.; Yang, K.; Gao, Y.; Sha, Y.; Xiao, D.; Liang, X.; Cheng, Z. Identification of four genes associated with cutaneous metastatic melanoma. Open Med. 2020, 15, 531–539. [Google Scholar] [CrossRef]
- Song, Y.; Pan, Y.; Liu, J. The relevance between the immune response-related gene module and clinical traits in head and neck squamous cell carcinoma. Cancer Manag. Res. 2019, 11, 7455–7472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahlawat, S.; Sharma, R.; Roy, M.; Mandakmale, S.; Prakash, V.; Tantia, M.S. Genotyping of Novel SNPs in BMPR1B, BMP15, and GDF9 Genes for Association with Prolificacy in Seven Indian Goat Breeds. Anim. Biotechnol. 2016, 27, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, X.; Zhao, J.; Ouyang, S.; Li, W.; Zhu, J.; Zhu, Y.; Zhu, X. Molecular cloning of ESR1, BMPR1B, and FOXL2 and differential expressions depend on maternal age and size during breeding season in cultured Asian yellow pond turtle (Mauremys mutica). Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2019, 232, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Hu, W.; Di, R.; Liu, Q.; Wang, X.; Zhang, X.; Zhang, J.; Chu, M. Expression Analysis of the Prolific Candidate Genes, BMPR1B, BMP15, and GDF9 in Small Tail Han Ewes with Three Fecundity (FecB Gene) Genotypes. Animals 2018, 8, 166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, Y.-L.; Guo, X.-F.; Ma, L.; Zhang, X.-S.; Zhang, J.-L.; Zhao, S.-G.; Chu, M.-X. The expression and mutation of BMPR1B and its association with litter size in small-tail Han sheep (Ovis aries). Arch. Anim. Breed. 2021, 64, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Çelikeloğlu, K.; Tekerli, M.; Erdoğan, M.; Koçak, S.; Hacan, Ö.; Bozkurt, Z. An Investigation of the Effects of Bmpr1b, Bmp15, and Gdf9 Genes on Litter Size in Ramlıç and Dağlıç Sheep. Arch. Anim. Breed 2021, 64, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Shokrollahi, B.; Morammazi, S. Polymorphism of GDF9 and BMPR1B genes and their association with litter size in Markhoz goats. Reprod. Domest. Anim. 2018, 53, 971–978. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, L.; Sun, W.; Lang, X.; Wu, J.; Zhu, C.; Jia, J.; Jin, J.; La, Y.; Casper, D.P. Study on the correlation between BMPR1B protein in sheep blood and reproductive performance. J. Anim. Sci. 2020, 98, 100. [Google Scholar] [CrossRef]
- Renault, L.; Patiño, L.C.; Magnin, F.; Delemer, B.; Young, J.; Laissue, P.; Binart, N.; Beau, I. BMPR1A and BMPR1B Missense Mutations Cause Primary Ovarian Insufficiency. J. Clin. Endocrinol. Metab. 2019, 105, e1449–e1457. [Google Scholar] [CrossRef]
- Moniruzzaman, M.; Sakamaki, K.; Akazawa, Y.; Miyano, T. Oocyte growth and follicular development in KIT-deficient Fas-knockout mice. Reproduction 2007, 133, 117–125. [Google Scholar] [CrossRef] [Green Version]
- Sakata, S.; Sakamaki, K.; Watanabe, K.; Nakamura, N.; Toyokuni, S.; Nishimune, Y.; Mori, C.; Yonehara, S. Involvement of death receptor Fas in germ cell degeneration in gonads of Kit-deficient Wv/Wv mutant mice. Cell Death Differ. 2003, 10, 676–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, S.; Chen, Y.; Zhao, B.; Yang, N.; Chen, S.; Shen, J.; Bao, G.; Wu, X. KIT is involved in melanocyte proliferation, apoptosis and melanogenesis in the Rex Rabbit. PeerJ 2020, 8, e9402. [Google Scholar] [CrossRef] [PubMed]
- Meyermans, R.; Gorssen, W.; Buys, N.; Janssens, S. Genomics Confirm an Alarming Status of the Genetic Diversity of Belgian Red and Belgian White Red Cattle. Animals 2021, 11, 3574. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, W.; Wu, S.; Ma, T.; Jiang, H.; Zhang, Q. Differential expression of MC1R gene in Liaoning Cashmere goats with different coat colors. Anim. Biotechnol. 2019, 30, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Xia, Z.; Bland, C.S.; Kalsotra, A.; Scavuzzo, M.; Curk, T.; Ule, J.; Li, W.; Cooper, T.A. Rbfox2-Coordinated Alternative Splicing of Mef2d and Rock2 Controls Myoblast Fusion during Myogenesis. Mol. Cell 2014, 55, 592–603. [Google Scholar] [CrossRef] [Green Version]
- Scimè, A.; Desrosiers, J.; Trensz, F.; Palidwor, G.A.; Caron, A.Z.; Andrade-Navarro, M.A.; Grenier, G. Transcriptional profiling of skeletal muscle reveals factors that are necessary to maintain satellite cell integrity during ageing. Mech. Ageing Dev. 2010, 131, 9–20. [Google Scholar] [CrossRef]
- Clark, D.L.; Boler, D.D.; Kutzler, L.W.; Jones, K.A.; McKeith, F.K.; Killefer, J.; Carr, T.R.; Dilger, A.C. Muscle Gene Expression Associated with Increased Marbling in Beef Cattle. Anim. Biotechnol. 2011, 22, 51–63. [Google Scholar] [CrossRef]
- Hassan, M.H.; Salama, S.A.; Arafa, H.M.M.; Hamada, F.M.A.; Al-Hendy, A. Adenovirus-Mediated Delivery of a Dominant-Negative Estrogen Receptor Gene in Uterine Leiomyoma Cells Abrogates Estrogen- and Progesterone-Regulated Gene Expression. J. Clin. Endocrinol. Metab. 2007, 92, 3949–3957. [Google Scholar] [CrossRef] [Green Version]
- Brooker, R.M.; Feeney, W.E. Animal Domesticators. Curr. Biol. 2019, 29, R1168–R1169. [Google Scholar] [CrossRef]
- Lord, K.A.; Larson, G.; Coppinger, R.P.; Karlsson, E.K. The History of Farm Foxes Undermines the Animal Domestication Syndrome. Trends Ecol. Evol. 2020, 35, 125–136. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Qu, K.; Jia, P.; Zhang, J.; Liu, J.; Lei, C.; Huang, B. Assessing Genomic Diversity and Productivity Signatures in Dianzhong Cattle by Whole-Genome Scanning. Front. Genet. 2021, 12, 719215. [Google Scholar] [CrossRef] [PubMed]
- Henkel, J.; Saif, R.; Jagannathan, V.; Schmocker, C.; Zeindler, F.; Bangerter, E.; Herren, U.; Posantzis, D.; Bulut, Z.; Ammann, P.; et al. Selection signatures in goats reveal copy number variants underlying breed-defining coat color phenotypes. PLoS Genet. 2019, 15, e1008536. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Liu, X.; Qi, T.; Hui, Y.; Yan, H.; Qu, L.; Lan, X.; Pan, C. Whole-genome sequencing to identify candidate genes for litter size and to uncover the variant function in goats (Capra hircus). Genomics 2020, 113, 142–150. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| BH vs. AF | AS vs. BH | |
|---|---|---|
| Digestive system | Gastric acid secretion (ADCY6, ADCY7) | Protein digestion and absorption (COL5A3, KCNN4, COL4A4) |
| Salivary secretion (LPO, ADCY6, ADCY7) | Pancreatic secretion (SLC4A4, PRKACA, KCNQ1) | |
| Pancreatic secretion (ADCY6, CPA1, ADCY7) | Bile secretion (SLC4A4, ABCG2) | |
| Bile secretion (ADCY6, ADCY7, LDLRAD4) | Gastric acid secretion (PRKACA, KCNQ1) | |
| Mineral absorption (SLC30A1) | Vitamin digestion and absorption (SLC19A3) | |
| Vitamin digestion and absorption (FOLH1) | Salivary secretion (KCNN4, PRKACA) | |
| Fat digestion and absorption (NPC1L1) | ||
| Cholesterol metabolism (LDLR) | ||
| Protein digestion and absorption (CPA1) | ||
| Amino acid metabolism | Lysine degradation (SETD7) | Tryptophan metabolism (KMO, ECHS1) |
| Alanine, aspartate and glutamate metabolism (ASPA, FOLH1) | Valine, leucine and isoleucine degradation (ECHS1, HMGCLL1) | |
| Glycine, serine and threonine metabolism (GLDC) | Lysine degradation (ECHS1, BBOX1) | |
| Cysteine and methionine metabolism (DNMT1, MRI1) | ||
| Histidine metabolism (ASPA) | ||
| Valine, leucine and isoleucine degradation (MCCC1) | ||
| Metabolism of cofactors and vitamins | Thiamine metabolism (DDX31) | Pantothenate and CoA biosynthesis (ENPP3) |
| Pantothenate and CoA biosynthesis (DPYS) | Riboflavin metabolism (ENPP3) | |
| Folate biosynthesis (GPHN) | Nicotinate and nicotinamide metabolism (ENPP3) | |
| beta-Alanine metabolism (DPYS) | Retinol metabolism (RPE65) | |
| Lipid metabolism | Steroid biosynthesis (MSMO1) | Fatty acid degradation (ECHS1, ACOX1) |
| Ether lipid metabolism (PLD1) | Synthesis and degradation of ketone bodies (HMGCLL1) | |
| Steroid hormone biosynthesis (DHRS11) | Glycerolipid metabolism (MBOAT2, AGK) | |
| Glycerophospholipid metabolism (PLD1) | Fatty acid biosynthesis (FASN) | |
| alpha-Linolenic acid metabolism (ACOX1) | ||
| Biosynthesis of unsaturated fatty acids (ACOX1) | ||
| Fatty acid elongation (ECHS1) | ||
| Sphingolipid metabolism (SPHK1) | ||
| Steroid hormone biosynthesis (HSD17B3) | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, Y.; Zhang, W.; Liu, C.; He, Y.; Zhang, H.; Xu, L.; Yang, B.; Zhao, Y.; Ma, Y.; Chu, M.; et al. Genome-Wide Selective Analysis of Boer Goat to Investigate the Dynamic Heredity Evolution under Different Stages. Animals 2022, 12, 1356. https://doi.org/10.3390/ani12111356
Yuan Y, Zhang W, Liu C, He Y, Zhang H, Xu L, Yang B, Zhao Y, Ma Y, Chu M, et al. Genome-Wide Selective Analysis of Boer Goat to Investigate the Dynamic Heredity Evolution under Different Stages. Animals. 2022; 12(11):1356. https://doi.org/10.3390/ani12111356
Chicago/Turabian StyleYuan, Ying, Weiyi Zhang, Chengli Liu, Yongmeng He, Haoyuan Zhang, Lu Xu, Baigao Yang, Yongju Zhao, Yuehui Ma, Mingxing Chu, and et al. 2022. "Genome-Wide Selective Analysis of Boer Goat to Investigate the Dynamic Heredity Evolution under Different Stages" Animals 12, no. 11: 1356. https://doi.org/10.3390/ani12111356
APA StyleYuan, Y., Zhang, W., Liu, C., He, Y., Zhang, H., Xu, L., Yang, B., Zhao, Y., Ma, Y., Chu, M., Zhao, Z., Huang, Y., Han, Y., Zeng, Y., Ren, H., Wang, G., & E, G. (2022). Genome-Wide Selective Analysis of Boer Goat to Investigate the Dynamic Heredity Evolution under Different Stages. Animals, 12(11), 1356. https://doi.org/10.3390/ani12111356