

Optimising Electroporation Condition for CRISPR/Cas-Mediated Knockout in Zona-Intact Buffalo Zygotes

 , ,
, ,
Abstract
:Simple Summary
Abstract
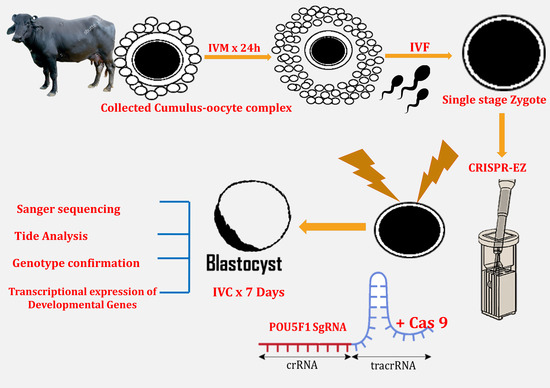
1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Guide RNA Designing and In Vitro Testing
2.3. Buffalo Fibroblasts Isolation and In Vitro Testing of sgRNA
2.4. Oocyte Collection, In Vitro Maturation, In Vitro Fertilization and Zygote Collection
2.5. RNP Complex Formation and Electroporation of Presumptive Zygotes and Culture
2.6. Extraction of Cell Lysate from Single Blastocyst and Genotyping for Validation of Knockout
2.7. Genotype Confirmation for Monoallelic and Biallelic Mutation
2.8. Gene Expression
2.9. Statistical Analysis
3. Results
3.1. Selection of Functional POU5F1 Guide RNAs
3.2. Effect of Post-Insemination Time and Voltage on Buffalo Embryonic Development
3.3. Effect of Different Electroporation Conditions on Knockout Efficiency
3.4. Examination of Mutations and Mosaicism Produced by the Optimum Electroporation Condition (20 V, 5 P, 3 ms)
3.5. Genotype Identification Assay for Monoallelic and Biallelic Mutation
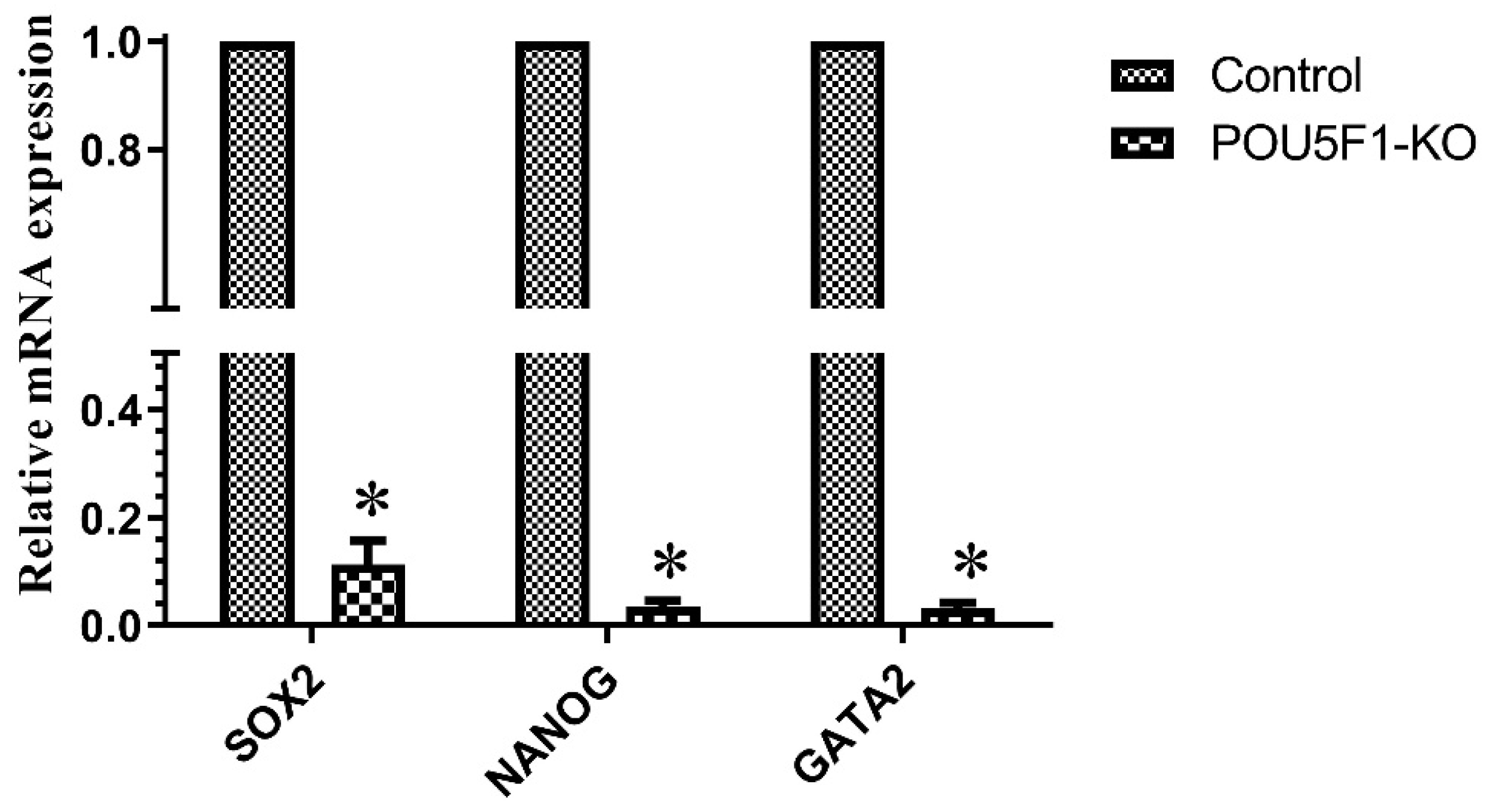
3.6. Effect of CRISPR-Cas9-RNP Induced POU5F1 KO on Blastocyst Formation and the Transcriptional Abundance of Pluripotency-Related Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Menchaca, A.; Dos Santos-Neto, P.C.; Mulet, A.P.; Crispo, M. CRISPR in livestock: From editing to printing. Theriogenology 2020, 150, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Kues, W.A. Genome Engineering in Livestock: Recent Advances and Regulatory Framework. Anim. Reprod. Update 2022, 3, 14–30. [Google Scholar] [CrossRef]
- Wang, S.; Qu, Z.; Huang, Q.; Zhang, J.; Lin, S.; Yang, Y.; Meng, F.; Li, J.; Zhang, K. Application of Gene Editing Technology in Resistance Breeding of Livestock. Life 2022, 12, 1070. [Google Scholar] [CrossRef] [PubMed]
- Lino, C.A.; Harper, J.C.; Carney, J.P.; Timlin, J.A. Delivering CRISPR: A review of the challenges and approaches. Drug Deliv. 2018, 25, 1234–1257. [Google Scholar] [CrossRef]
- Petersen, B. Basics of genome editing technology and its application in livestock species. Reprod. Domest. Anim. 2017, 52 (Suppl. S3), 4–13. [Google Scholar] [CrossRef] [PubMed]
- Dua, S.; Bansal, S.; Gautam, D.; Jose, B.; Singh, P.; Singh, M.K.; De, S.; Kumar, D.; Yadav, P.S.; Kues, W.; et al. Production of MSTN Gene-Edited Embryos of Buffalo Using the CRISPR/Cas9 System and SCNT. Cell Reprogram. 2023, 25, 121–127. [Google Scholar] [CrossRef]
- Bajwa, K.K.; Punetha, M.; Kumar, D.; Yadav, P.S.; Long, C.R.; Selokar, N.L. Electroporation-based CRISPR gene editing in adult buffalo fibroblast cells. Anim. Biotechnol. 2023, 1–12. [Google Scholar] [CrossRef]
- Yadav, P.S.; Kumar, D.; Saini, M.; Sharma, R.K.; Dua, S.; Selokar, N.L.; Bansal, S.; Punetha, M.; Gupta, A.; Kumar, R.; et al. Evaluation of postnatal growth, hematology, telomere length and semen attributes of multiple clones and re-clone of superior buffalo breeding bulls. Theriogenology 2023, 213, 24–33. [Google Scholar] [CrossRef]
- Gurdon, J.B.; Melton, D.A. Nuclear reprogramming in cells. Science 2008, 322, 1811–1815. [Google Scholar] [CrossRef]
- Wilmut, I.; Beaujean, N.; de Sousa, P.A.; Dinnyes, A.; King, T.J.; Paterson, L.A.; Wells, D.N.; Young, L.E. Somatic cell nuclear transfer. Nature 2002, 419, 583–586. [Google Scholar] [CrossRef]
- Yang, X.; Smith, S.L.; Tian, X.C.; Lewin, H.A.; Renard, J.P.; Wakayama, T. Nuclear reprogramming of cloned embryos and its implications for therapeutic cloning. Nat. Genet. 2007, 39, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Serna, S.; Vilarino, M.; Park, I.; Gadea, J.; Ross, P.J. Livestock Gene Editing by One-step Embryo Manipulation. J. Equine Vet. Sci. 2020, 89, 103025. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Yamashita, Y.; Takemoto, T. Electroporation of Cas9 protein/sgRNA into early pronuclear zygotes generates non-mosaic mutants in the mouse. Dev. Biol. 2016, 418, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Wu, Y.; Zhang, Y. Efficient delivery of DNA and morpholinos into mouse preimplantation embryos by electroporation. PLoS ONE 2012, 7, e43748. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, T.; Mashimo, T. Simple Genome Editing of Rodent Intact Embryos by Electroporation. PLoS ONE 2015, 10, e0142755. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, T.; Sakuma, T.; Yamamoto, T.; Mashimo, T. Simple knockout by electroporation of engineered endonucleases into intact rat embryos. Sci. Rep. 2014, 4, 6382. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.C.; Van Eenennaam, A.L. Electroporation-Mediated Genome Editing of Livestock Zygotes. Front. Genet. 2021, 12, 648482. [Google Scholar] [CrossRef] [PubMed]
- Troder, S.E.; Ebert, L.K.; Butt, L.; Assenmacher, S.; Schermer, B.; Zevnik, B. An optimized electroporation approach for efficient CRISPR/Cas9 genome editing in murine zygotes. PLoS ONE 2018, 13, e0196891. [Google Scholar] [CrossRef]
- Alghadban, S.; Bouchareb, A.; Hinch, R.; Hernandez-Pliego, P.; Biggs, D.; Preece, C.; Davies, B. Electroporation and genetic supply of Cas9 increase the generation efficiency of CRISPR/Cas9 knock-in alleles in C57BL/6J mouse zygotes. Sci. Rep. 2020, 10, 17912. [Google Scholar] [CrossRef]
- Jose, B.; Punetha, M.; Tripathi, M.K.; Khanna, S.; Yadav, V.; Singh, A.K.; Kumar, B.; Singh, K.; Chouhan, V.S.; Sarkar, M.J.T. CRISPR/Cas mediated disruption of BMPR-1B gene and introduction of FecB mutation into the Caprine embryos using Easi-CRISPR strategy. Theriogenology 2023, 211, 125–133. [Google Scholar] [CrossRef]
- Modzelewski, A.J.; Chen, S.; Willis, B.J.; Lloyd, K.C.K.; Wood, J.A.; He, L. Efficient mouse genome engineering by CRISPR-EZ technology. Nat. Protoc. 2018, 13, 1253–1274. [Google Scholar] [CrossRef] [PubMed]
- Jerabek, S.; Merino, F.; Scholer, H.R.; Cojocaru, V. OCT4: Dynamic DNA binding pioneers stem cell pluripotency. Biochim. Biophys. Acta 2014, 1839, 138–154. [Google Scholar] [CrossRef] [PubMed]
- Frum, T.; Halbisen, M.A.; Wang, C.; Amiri, H.; Robson, P.; Ralston, A. Oct4 cell-autonomously promotes primitive endoderm development in the mouse blastocyst. Dev. Cell 2013, 25, 610–622. [Google Scholar] [CrossRef] [PubMed]
- Daigneault, B.W.; Rajput, S.; Smith, G.W.; Ross, P.J. Embryonic POU5F1 is Required for Expanded Bovine Blastocyst Formation. Sci. Rep. 2018, 8, 7753. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, N.; Fujii, T.; Hashizume, T.; Sawai, K. Effects of downregulating oct-4 transcript by RNA interference on early development of porcine embryos. J. Reprod. Dev. 2013, 59, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Haeussler, M.; Schonig, K.; Eckert, H.; Eschstruth, A.; Mianne, J.; Renaud, J.B.; Schneider-Maunoury, S.; Shkumatava, A.; Teboul, L.; Kent, J.; et al. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 2016, 17, 148. [Google Scholar] [CrossRef] [PubMed]
- Dua, S.; Sharma, P.; Saini, M.; Rawat, N.; Rajendran, R.; Bansal, S.; Wakil, A.M.; Beniwal, M.; Parashar, A.; Bajwa, K.K.; et al. Cryobanking of primary somatic cells of elite farm animals—A pilot study in domesticated water buffalo (Bubalus bubalis). Cryobiology 2021, 98, 139–145. [Google Scholar] [CrossRef]
- Bhattacharya, D.; Van Meir, E.G. A simple genotyping method to detect small CRISPR-Cas9 induced indels by agarose gel electrophoresis. Sci. Rep. 2019, 9, 4437. [Google Scholar] [CrossRef]
- Cheng, Q.; Xia, J.; Wang, K.; Zhang, Y.; Chen, Y.; Zhong, Q.; Wang, X.; Wu, Q. CRISPR/Cas9 ribonucleoprotein (RNP) complex enables higher viability of transfected cells in genome editing of acute myeloid cells. Ann. Transl. Med. 2022, 10, 862. [Google Scholar] [CrossRef]
- Tanihara, F.; Hirata, M.; Thi Nguyen, N.; Anh Le, Q.; Hirano, T.; Otoi, T. Generation of viable PDX1 gene-edited founder pigs as providers of nonmosaics. Mol. Reprod. Dev. 2020, 87, 471–481. [Google Scholar] [CrossRef]
- Miskel, D.; Poirier, M.; Beunink, L.; Rings, F.; Held, E.; Tholen, E.; Tesfaye, D.; Schellander, K.; Salilew-Wondim, D.; Blaschka, C.; et al. Author Correction: The cell cycle stage of bovine zygotes electroporated with CRISPR/Cas9-RNP affects frequency of Loss-of-heterozygosity editing events. Sci. Rep. 2022, 12, 21303. [Google Scholar] [CrossRef] [PubMed]
- Comizzoli, P.; Marquant-Le Guienne, B.; Heyman, Y.; Renard, J.P. Onset of the first S-phase is determined by a paternal effect during the G1-phase in bovine zygotes. Biol. Reprod. 2000, 62, 1677–1684. [Google Scholar] [CrossRef] [PubMed]
- Ward, F.; Enright, B.; Rizos, D.; Boland, M.; Lonergan, P. Optimization of in vitro bovine embryo production: Effect of duration of maturation, length of gamete co-incubation, sperm concentration and sire. Theriogenology 2002, 57, 2105–2117. [Google Scholar] [CrossRef] [PubMed]
- Gasparrini, B.; De Rosa, A.; Attanasio, L.; Boccia, L.; Di Palo, R.; Campanile, G.; Zicarelli, L. Influence of the duration of in vitro maturation and gamete co-incubation on the efficiency of in vitro embryo development in Italian Mediterranean buffalo (Bubalus bubalis). Anim. Reprod. Sci. 2008, 105, 354–364. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Anand, T. In Vitro Embryo Production in Buffalo: Basic Concepts. J. Buffalo Sci. 2012, 1, 50–54. [Google Scholar] [CrossRef]
- Namula, Z.; Wittayarat, M.; Hirata, M.; Hirano, T.; Nguyen, N.T.; Le, Q.A.; Fahrudin, M.; Tanihara, F.; Otoi, T. Genome mutation after the introduction of the gene editing by electroporation of Cas9 protein (GEEP) system into bovine putative zygotes. In Vitro Cell Dev. Biol. Anim. 2019, 55, 598–603. [Google Scholar] [CrossRef] [PubMed]
- Miao, D.; Giassetti, M.I.; Ciccarelli, M.; Lopez-Biladeau, B.; Oatley, J.M. Simplified pipelines for genetic engineering of mammalian embryos by CRISPR-Cas9 electroporationdagger. Biol. Reprod. 2019, 101, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Guo, Z.; Liu, Y.; Tang, B.; Wang, Y.; Yang, L.; Du, J.; Zhang, Y. Oct4 and the small molecule inhibitor, SC1, regulates Tet2 expression in mouse embryonic stem cells. Mol. Biol. Rep. 2013, 40, 2897–2906. [Google Scholar] [CrossRef]
- Nichols, J.; Zevnik, B.; Anastassiadis, K.; Niwa, H.; Klewe-Nebenius, D.; Chambers, I.; Scholer, H.; Smith, A. Formation of pluripotent stem cells in the mammalian embryo depends on the POU transcription factor Oct4. Cell 1998, 95, 379–391. [Google Scholar] [CrossRef]
- Ralston, A.; Cox, B.J.; Nishioka, N.; Sasaki, H.; Chea, E.; Rugg-Gunn, P.; Guo, G.; Robson, P.; Draper, J.S.; Rossant, J. Gata3 regulates trophoblast development downstream of Tead4 and in parallel to Cdx2. Development 2010, 137, 395–403. [Google Scholar] [CrossRef]
- Simmet, K.; Kurome, M.; Zakhartchenko, V.; Reichenbach, H.D.; Springer, C.; Bahr, A.; Blum, H.; Philippou-Massier, J.; Wolf, E. OCT4/POU5F1 is indispensable for the lineage differentiation of the inner cell mass in bovine embryos. FASEB J. 2022, 36, e22337. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, N.; Takahashi, K.; Emura, N.; Fujii, T.; Hirayama, H.; Kageyama, S.; Hashizume, T.; Sawai, K. The Necessity of OCT-4 and CDX2 for Early Development and Gene Expression Involved in Differentiation of Inner Cell Mass and Trophectoderm Lineages in Bovine Embryos. Cell Reprogram. 2016, 18, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Nichols, J.; Smith, A. Pluripotency in the embryo and in culture. Cold Spring Harb. Perspect. Biol. 2012, 4, a008128. [Google Scholar] [CrossRef] [PubMed]
- Chew, J.L.; Loh, Y.H.; Zhang, W.; Chen, X.; Tam, W.L.; Yeap, L.S.; Li, P.; Ang, Y.S.; Lim, B.; Robson, P.; et al. Reciprocal transcriptional regulation of Pou5f1 and Sox2 via the Oct4/Sox2 complex in embryonic stem cells. Mol. Cell Biol. 2005, 25, 6031–6046. [Google Scholar] [CrossRef] [PubMed]
- Simmet, K.; Zakhartchenko, V.; Philippou-Massier, J.; Blum, H.; Klymiuk, N.; Wolf, E. OCT4/POU5F1 is required for NANOG expression in bovine blastocysts. Proc. Natl. Acad. Sci. USA 2018, 115, 2770–2775. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Scholer, H.R. Role of Oct4 in the early embryo development. Cell Regen. 2014, 3, 7. [Google Scholar] [CrossRef]
- Strumpf, D.; Mao, C.A.; Yamanaka, Y.; Ralston, A.; Chawengsaksophak, K.; Beck, F.; Rossant, J. Cdx2 is required for correct cell fate specification and differentiation of trophectoderm in the mouse blastocyst. Development 2005, 132, 2093–2102. [Google Scholar] [CrossRef]
- Wicklow, E.; Blij, S.; Frum, T.; Hirate, Y.; Lang, R.A.; Sasaki, H.; Ralston, A. HIPPO pathway members restrict SOX2 to the inner cell mass where it promotes ICM fates in the mouse blastocyst. PLoS Genet. 2014, 10, e1004618. [Google Scholar] [CrossRef]
- Ng, Y.K.; George, K.M.; Engel, J.D.; Linzer, D.I. GATA factor activity is required for the trophoblast-specific transcriptional regulation of the mouse placental lactogen I gene. Development 1994, 120, 3257–3266. [Google Scholar] [CrossRef]
- Ma, G.T.; Linzer, D.I. GATA-2 restricts prolactin-like protein A expression to secondary trophoblast giant cells in the mouse. Biol. Reprod. 2000, 63, 570–574. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Forward | Reverse | |
|---|---|---|
| POU5F1 guide | TAATACGACTCACTATAGGATATACCCAGGCCGATGTG | TTCTAGCTCTAAAACCACATCGGCCTGGGTATATC |
| POU5F1 for T7E1 assay | GCCCTATGACTTGTGTGG | GGAAGGAAAACCCAGACTCC |
| POU5F1 for sequencing and genotype confirmation | CCCCCTTCCTAACCTGACAT | GTCGTTTGGCTGAACACCTT |
| SOX2 | GTTCATCGACGAGGCCAA | CCCGGCAGTGTGTACTTATC |
| NANOG | GGGAAGGGTAATGAGTCCAA | AGCCTCCCTATCCCAGAAAA |
| GATA2 | GCACAGCCGGACTAACTTAT | GGAATAGGAAGAGCGCATACA |
| Time of Electroporation Post Insemination (h) | Voltage (V/mm) | Zygotes (N) | Zygotes Kept for IVC, N | Cleaved, N (%) | Blastocyst, N (%) |
|---|---|---|---|---|---|
| 10 | 0 | 468 | 468 | 355 (75.85 ± 1.84) a | 91 (25.86 ± 1.09) a |
| 15 | 510 | 431 | 319 (74.2 ± 2.2) a | 90 (28.22 ± 2.49) a | |
| 20 | 479 | 435 | 298 (68.63 ± 3.22) a | 91 (30.57 ± 3.82) a | |
| 8 | 0 | 295 | 295 | 136 (45.9 ± 1.89) b | 2 (1.67 ± 0.68) b |
| 15 | 465 | 361 | 145 (40.03 ± 1.12) b | 3 (1.8 ± 0.84) b | |
| 20 | 459 | 420 | 157 (37.05 ± 2.40) b | 1 (0.49 ± 0.406) b |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Punetha, M.; Kumar, D.; Saini, S.; Chaudhary, S.; Bajwa, K.K.; Sharma, S.; Mangal, M.; Yadav, P.S.; Green, J.A.; Whitworth, K.; et al. Optimising Electroporation Condition for CRISPR/Cas-Mediated Knockout in Zona-Intact Buffalo Zygotes. Animals 2024, 14, 134. https://doi.org/10.3390/ani14010134
Punetha M, Kumar D, Saini S, Chaudhary S, Bajwa KK, Sharma S, Mangal M, Yadav PS, Green JA, Whitworth K, et al. Optimising Electroporation Condition for CRISPR/Cas-Mediated Knockout in Zona-Intact Buffalo Zygotes. Animals. 2024; 14(1):134. https://doi.org/10.3390/ani14010134
Chicago/Turabian StylePunetha, Meeti, Dharmendra Kumar, Sheetal Saini, Suman Chaudhary, Kamlesh Kumari Bajwa, Surabhi Sharma, Manu Mangal, Prem S. Yadav, Jonathan A. Green, Kristin Whitworth, and et al. 2024. "Optimising Electroporation Condition for CRISPR/Cas-Mediated Knockout in Zona-Intact Buffalo Zygotes" Animals 14, no. 1: 134. https://doi.org/10.3390/ani14010134
APA StylePunetha, M., Kumar, D., Saini, S., Chaudhary, S., Bajwa, K. K., Sharma, S., Mangal, M., Yadav, P. S., Green, J. A., Whitworth, K., & Datta, T. K. (2024). Optimising Electroporation Condition for CRISPR/Cas-Mediated Knockout in Zona-Intact Buffalo Zygotes. Animals, 14(1), 134. https://doi.org/10.3390/ani14010134